
Abstract
Oncolytic viruses (OVs) represent a new class of multi-modal immunotherapies for cancer, with OV-elicited antitumor immunity being key to their overall therapeutic efficacy. Currently, the clinical effectiveness of OV as monotherapy remains limited, and thus investigators have been exploring various combinations with other anti-cancer agents and demonstrated improved therapeutic efficacy. As cancer cells have evolved to alter key signaling pathways for enhanced cell proliferation, cancer progression and metastasis, these cellular and molecular changes offer promising targets for rational cancer therapy design. In this regard, key molecules in relevant signaling pathways for cancer cells or/and immune cells, such as EGFR-KRAS (e.g., KRASG12C), PI3K-AKT-mTOR, ERK-MEK, JAK-STAT, p53, PD-1-PD-L1, and epigenetic, or immune pathways (e.g., histone deacetylases, cGAS-STING) are currently under investigation and have the potential to synergize with OV to modulate the immune milieu of the tumor microenvironment (TME), thereby improving and sustaining antitumor immunity. As many small molecule modulators of these signaling pathways have been developed and have shown strong therapeutic potential, here we review key findings related to both OV-mediated immunotherapy and the utility of small molecule modulators of signaling pathways in immuno-oncology. Then, we focus on discussion of the rationales and potential strategies for combining OV with selected modulators targeting key cellular signaling pathways in cancer or/and immune cells to modulate the TME and enhance antitumor immunity and therapeutic efficacy. Finally, we provide perspectives and viewpoints on the application of novel experimental systems and technologies that can propel this exciting branch of medicine into a bright future.
Similar content being viewed by others

Introduction
The mammalian immune system comprises a network of innate and adaptive cell subsets which collectively discriminate invading or arising “non-self” elements from healthy “self” components of the body to eliminate pathogens such as viruses, bacteria, parasites, or pathogenic cellular changes or features, as arise in cancer. Evolving pathogens have developed multiple sophisticated mechanisms to antagonize or even exploit host immunity to their own advantage [1, 2]. Recent developments have improved our understanding of the molecular and cellular interplay between viruses and the immune system and have led to the design of new strategies to turn viruses from stealth pathogens into finely tuned therapeutic vehicles that can promote both direct viral-mediated and secondary immune-mediated attack against cancer cells. Oncolytic viruses (OVs) represent a key example of such approaches [3].
OVs are a diverse collection of viruses being developed as versatile therapeutic platforms for treating cancer. OVs preferentially infect and replicate in cancer cells and cancer-associated stromal cells and can be engineered to express transgenes that augment their cytotoxic and immunostimulatory activities [4,5,6,7,8,9]. Importantly, in addition to direct lytic function, OVs modulate the tumor microenvironment (TME) and enhance loco-regional inflammation and immune cell-mediated tumor eradication, while also enhancing systemic cancer immunity [10, 11]. In this way, OVs have the potential to be utilized as a comprehensive and potent therapeutic platform in combination regimens [4,5,6,7,8,9, 12]. They have also undergone through clinical trials [13,14,15].
Small-molecule drugs targeting key cellular signaling pathways are becoming an important class of drugs for cancer therapy [16,17,18]. Importantly, this approach has uncovered a previously underappreciated crosstalk between tumors and the immune cells present within the TME. First, some of these modulators targeting key signaling pathways for cancer cell survival and proliferation have been shown to induce immunogenic cell death (ICD) of cancer cells, enhancing cancer immunogenicity and subsequent antitumor immunity [19,20,21,22]. Second, in addition to the expected effects of inducing death of cancer cells, targeting tumors with kinase inhibitors has been shown to reverse the immunosuppressive TME [23, 24]. Third, as both cancer cells and immune cells undergo metabolic and epigenetic reprogramming in the TME [25, 26], modulators to key enzymes involved in epigenetic and metabolic pathways have the potential to inhibit tumor cell growth and proliferation, and to restore the normal functions of immune cells [27, 28]. Based on these tumor modulating and therapeutic properties, it is logical to explore the potential of combining these agents with OV as rational approaches to cancer therapy.
We have previously published three review articles highlighting various aspects of oncolytic immunotherapy [29,30,31]. In these reviews, we focused on two themes. The first was on the mode of cell death induced by OVs, mostly in the form of ICD, and the significance of ICD in eliciting potent antitumor immunity and its functionality as in situ therapeutic cancer vaccines [29, 30]. The second theme was on the development of vaccinia virus for cancer vaccines and as OV [31]. In the current review, we focus on combinatorial strategies integrating OVs with modulators of key signaling pathways in cancer and/or immune cells for optimized therapeutic efficacy. As small molecule modulators have shown promising results in preclinical and clinical studies, with some gaining approval for cancer therapy [17, 18, 32, 33], combining OV with small molecule modulators as rational therapeutic combinations will be the focus of discussion.
Single agent cancer therapies often produce limited efficacy [17, 34,35,36], and this includes immunotherapy [35]. One of many potential causes for lack of sufficient therapeutic response is tumor heterogeneity, which leads to an incomplete response to a particular monotherapy [36]. Rational combination regimens are badly needed to overcome the heterogeneous responses currently observed to these potentially curative therapies, as drug candidates could work additively or synergistically to produce enhanced therapeutic effects [34]. Surprisingly, a recent study showed that patient-to-patient variability and independent drug action are sufficient to explain the superiority of many FDA-approved drug combinations in the absence of drug synergy or additivity [37], suggesting that it may not be necessary for drugs to have additive or synergistic effects in order demonstrate therapeutic benefit for patients. This insight represents an unusual way to design combination therapies. In summary, combination therapy such as OVs with modulators of cellular signaling pathways are highly desired, although the clear rational for combining such approaches may not be immediately obvious and may require further mechanistic insights in determining potentially impactful therapeutic strategies.
Oncolytic viruses and immunotherapy: overview
Basic studies of OV-mediated immunotherapy
OVs preferentially infects and kills cancer cells without causing collateral harm to normal cells and tissues. As the infected cancer cells and/or tumor-associated stromal cells are destroyed by oncolysis, they release new infectious virus particles or virions to infect and destroy the remaining tumor cells/stromal cells. The tumor selectivity of OVs have been well studied in many cases [38,39,40]. Some viruses possess natural tumor cell tropism while others gain this property through genetic engineering [40]. A particular OV may work through one or more mechanisms. First, cellular entry via virus-specific, receptor-mediated mechanisms restricts the virus to cancer cells and cancer-associated cells. Second, rapid cell division in tumor cells with high metabolic and replicative activity may support increased viral replication compared with quiescent normal cells. Third, tumor-driver mutations specifically increase the selectivity of virus replication in tumor cells (Fig. 1). Reovirus and vaccinia virus naturally possess the ability to specially target cancer cells driven by the activated Ras pathway. Reovirus preferentially replicates in Ras-activated cells [41]. Vaccinia virus (VV) targets cancer cells that overexpress EGFR as it requires EGFR-Ras signaling to replicate [42]. The genetically engineered VV, Pexa-Vec (JX-594), targets cancer cells via multiple mechanisms, whereby virus replication is activated by EGFR/KRAS pathway signaling, cellular thymidine kinase (TK) levels, and resistance to type I interferons (IFNs) in cancer cells [42]. Finally, some OVs target cancer cells and/or tumor-associated stromal cells. Notably, multiple distinct mechanisms or OV features can underpin this process. For example, some OVs can selectively infect and replicate in stromal cells. Oncolytic vesicular stomatitis virus (VSV) infects and destroys tumor vasculature in vivo but leaves normal vasculature intact [43]. OVs expressing certain T-cell engagers simultaneously targets cancer and immunosuppressive stromal cells [44,45,46]. Importantly, it has been shown that stromal destruction is required for the eradication of established solid tumors by adaptive immunity involving T cells under certain conditions [47]. Therefore, those OVs that effectively target both cancer cells and stromal cells are likely to be advantageous.

Viral proteins and small molecule compounds may inhibit signaling in synergy to promote viral replication, and improve elicited inflammation, ICD and antitumor immune responses. We use RAS, IFN and dsRNA-dependent protein kinase (PKR) pathways as an example. Most viruses replicate poorly in cells that produce active PKR. In response to viral infection, PKR activates the transcription factor NF-κB by inducing degradation of IκBβ. NF-κB activates transcription of proinflammatory genes that induce an immune response against viruses. RAS activation by EGFR, v-Erb2, or platelet-derived growth-factor receptor (PDGFR) signaling inhibits PKR activity. RAS activation therefore allows viral oncolytic activity in cancer cells. PKR activity is also activated by interferon (IFN)-α/β signaling through the IFN receptor (IFNR). Tumor cells with defects in this signaling pathway allow a higher degree of viral replication than normal cells. Several viral proteins and RNAs (adenoviral VA RNA128, VV’s E3L and K3L, the hepatitis CE2 protein and influenza virus NS1 protein) inhibit PKR. Interfering with the different steps of signaling pathways using different classes of compounds have resulted in increased viral replication and subsequent efficacy. PKR shuts off protein synthesis by phosphorylating eIF2α. The protein phosphatase-1α is activated by the protein ICP34.5, which is expressed by HSV-1. ICP34.5 dephosphorylates eIF2α to allow protein synthesis to continue. An HSV1 strain that expresses a mutant form of ICP34.5 can therefore only replicate in cells with inactive PKR. Targeting this process by means of different small molecule inhibitors increased OV spread and efficacy. Note: Viral genes are shown in red, small molecule inhibitors are shown in green, whereas cellular proteins are shown in blue
OVs destroy cancer cells and/or inhibit tumor progression through four distinct mechanisms: oncolysis, vascular collapse, antitumor immunity, and expression of therapeutic transgene(s) [48]. Antitumor immune responses are potentiated through immunogenic cell death (ICD) of cancer cells and subsequent presentation of danger signals to dendritic cells (DCs), and release/presentation of cellular debris, viral and tumor antigens (including neoantigens) to the local and systemic immune cells [30, 49]. In fact, as early as 1999, Rabkin, Martuza, Toda and others have observed that an oncolytic HSV (oHSV) could function as an in-situ cancer vaccine and stimulate anti-tumor immunity [50]. In that study, OV delivery and tumor cell killing resulted in generation of CD8+ CTL responses against the dominant “tumor-specific” major histocompatibility complex (MHC) class I-restricted epitope (AH1) from the Gp70 antigen expressed by CT26 colon cancer cells [50]. The fact that tumor antigen-specific adaptive immunity plays a key role in OV-mediated cancer therapeutics has been verified subsequently in numerous studies. Based on these data, we and others believe that OVs function as therapeutic cancer vaccines [29, 51,52,53], or a specific type of immunotherapy [54].
OVs themselves can modulate the TME and turn cold tumor hot [55, 56]. The evidence comes by examining the release of DAMPs and PAMPs [30, 49, 57], the cytokine/chemokines produced [30, 49, 58], infiltration of immune cells [59, 60], induction of ICD [61, 62], activities of infiltrated immune cells and elicited antitumor immunity in tumor models [30, 49, 63, 64], among other properties after treating the tumors with OV. In order to further improve its immunostimulatory functions, several strategies have been exploited. One is simply to integrate immunostimulatory genes such as Th1-cytokines into the viral vectors [65]. This was done with GM-CSF and IL-2 in the early iterations of this approach [66, 67]. IL-10, originally considered to be a Th2 cytokine, but later shown to be a Th1 cytokine in certain environments, has been incorporated into OVs and demonstrated improved antitumor immunity and enhanced efficacy [68, 69]. IL-24, a member of the IL-10 family, has also been shown to be potent antitumor factor when expressed from an OV [70, 71]. We and others have engineered oncolytic vaccinia viruses (VVs) expressing recombinant IL-2, IL-12, IL-15, IL-21, IL-23, and IL-36γ for improved efficacy and safety in multiple tumor models [64, 72,73,74,75,76,77]. In some cases, two cytokines may complement each other and synergize to activate antitumor immunity and lead to complete tumor regression in non-immunogenic tumor models. This is the case for IL-7 and IL-12 expressed by OV [78, 79]. The second approach is to engineer co-stimulatory ligands such as ICOS ligand (ICOS-L) into OV to enhance co-stimulation of immune cells within the TME. As OV delivery often leads to upregulation of T-cell co-stimulatory receptors, with the inducible co-stimulator (ICOS) being most notable [80], this is a rational approach to further augmenting anti-tumor immunity using OV. Third, it is also possible to use the reverse approach, whereby OV can be engineered to express inhibitors or antagonists targeting co-inhibitory molecules. For example, recent studies showed that engineered OV expressing ICIs could activate anti-tumor responses [81,82,83]. Fourth, there are a few “don’t eat me” signals (CD24 and CD47) that that cancers seem to use to evade detection and destruction by the immune system. Thus, OVs could be engineered with an antibody against CD47 or CD24 and improved innate immunity, in addition to its known functions of oncolysis and modulation of immune cells [83, 84]. A fifth strategy is to engineer an OV with a tumor antigen, helping such armed OV elicit robust tumor antigen-specific CD8+ T cell responses, leading to improved antitumor therapy [85]. The sixth is to disrupt the signaling pathways that facilitate interactions between cancer cells and their environment. For example, CXCR4 is one of the key stimuli involved in signaling interactions. Studies found that targeting CXCL12/CXCR4 signaling by an OV expressing CXCR4 antagonist effectively disrupted the tumor vasculature, induced ICD of cancer cells, reversed immunosuppressive TME and improved antitumor immunity, including inhibition of cancer metastasis [86, 87]. Lastly, engineering OV to express a T-cell engager can engage naïve T cells and cancer cells, which activates these T cells to kill cancer cells, bypassing the dependence of MHC antigen on cancer cells [88,89,90]. In addition, OVs armed with bispecific engager targeting both T cells (CD3) and fibroblast (e.g, fibroblast activation protein) can target both cancer proper and associated stroma [89, 90].
In summary, as novel class of antitumor agents, one unique property of OVs is that they replicate selectively in cancer cells, yet express other therapeutic proteins locally to amplify its antitumor effects and modulate the TME to turn cold tumor hot. As they elicit both innate and adaptive antitumor immunity, they target tumor locally yet act systemically to inhibit/eliminate not only primary tumor, but also micro-metastases at a distance.
Clinical studies of OV immunotherapy
Three OVs have been approved for treatment of human cancers: H101 (adenovirus, AdV), T-VEC (herpes simplex virus, HSV), and Delytact (G47∆; HSV) were approved in China (in 2005), USA (in 2015), and Japan (in 2021) [91], respectively.
As summarized in a recent review, 97 clinical trials with OVs enrolling a total of 3233 cancer patients have been completed, resulting in 119 published reports [15]. Among these studies, objective clinical responses were reported in only 9% of patients and disease control was achieved in only 21% of patients, suggesting a clear need to enhance therapeutic responses to OVs. Three GM-CSF-armed OVs highlighted clinical studies and progression of the field. They are genetically engineered from HSV-1 (T-VEC), human adenovirus (CG0070), or vaccinia virus (Pexa-Vec), respectively. For the approved T-VEC, local and distant antitumor immunity was induced by intralesional vaccination with the virus in patients with stage IIIc and IV melanoma [92, 93]. This antitumor immunity was associated with overall objective clinical response that led to approval of T-VEC by the FDA for patients with advanced melanoma [94]. A recent study in melanoma patients of stage IIIB-IVM1a with injectable, unresectable metastatic lesions demonstrated that treatment of a “real-life” cohort of patients with T-VEC resulted in high overall response rate (64%) and a large fraction of durable complete responses (43%) [95]. However, as observed in the randomized, double-blind phase III trial (NCT02263508), combining T-VEC with pembrolizumab did not lead to a survival benefit compared to pembrolizumab alone for patients with advanced melanoma [96]. The second OV, CG0070, has undergone phase II testing in patients with BCG-unresponsive non-muscle-invasive bladder cancer. In this patient cohort, intravesical CG0070 yielded an overall 47% complete response (CR) rate at 6 months for all patients and 50% for patients with carcinoma-in-situ, with an acceptable level of toxicity [97]. This OV is currently being evaluated in the phase III BOND-003 trial (NCT04452591), and in phase II trial in combination with pembrolizumab. The preliminary report of the phase II trial showed a CR rate of 88% (14/16) at the 3 month assessment interim timepoint [98]. Finally, Pexa-Vec demonstrated oncolytic and immune-mediated mechanisms of action, tumor responses and dose-related survival in individuals with hepatocellular carcinoma (HCC) in a phase 2 trial [99]. In the phase III PHOCUS trial (NCT02562755), unfortunately Pexa-Vec/Nexavar combination therapy failed to meet the intended criteria (mainly improved clinical benefit when compared to Nexavar alone). However, 10 additional clinical studies in phase I/II are evaluating Pexa-Vec in other solid cancer indications and may reveal additional therapeutic opportunities for this OV. For example, a phase I/II study of Pexa-Vec in combination with immune checkpoint inhibitor (ICI) in refractory colorectal cancer (NCT03206073) may demonstrate that this combination is efficacious [100]. In summary, OV-elicited antitumor immunity contributes significantly to, or is essential for the overall therapeutic efficacy mediated by an OV. However, other mechanisms of action, such as cytotoxicity and anti-angiogenesis, also contribute to the overall therapeutic effects and represented additional opportunities to enhance the effectiveness of OVs. As monotherapy, OVs exert limited efficacy and thus rational combination strategies are desperately needed to further improve the efficacy of this novel class of immunotherapy.
In regard to OV toxicity, multiple studies have investigated this potential therapeutic challenge using animal models. For example, Lun et al. have studied the toxicity of Pexa-Vec (JX-594) [101]. A supratherapeutic dose of JX-594 demonstrated GM-CSF-dependent inflammation and necrosis in non-tumor-bearing rodents. In another study, Tang et al. found that some brain tumor-bearing mice died soon after treatment with vvDD-IL15-Rα, and close examination revealed that viral infection of ependymal cells, subventricular cells, and meninges was widespread, leading to death [102]. VSV exhibits natural neurotropism, but genetic engineering can abrogate neurotoxicity [103]. So far clinical trials with OVs have not shown significant toxicity or safety issues. Influenza-like symptoms (such as chills and fever) have been noted for both local and systemic administration of OVs but are mild [104, 105].
Biomarkers for OV immunotherapy
As only a fraction of patients treated with OV go on to derive treatment benefit, it stands to reason that identifying clinical biomarkers that can successfully predict patients who will respond favorably will improve outcomes to OV therapy [106]. These may include both predictive and response biomarkers, and they may guide OV therapy [106].
Cancer cells possess multiple signaling or cellular hallmarks [107] that can be exploited for OV infection, replication and oncolysis (Fig. 1). It has been shown that defects in interferon pathways potentiate the sensitivity of cancer cells to various OVs, including VSV [108]. Many OVs depend on oncogenic signaling pathway that are constitutively activated in cancer cells for their selective viral replication and oncolysis. For example, a number OVs depend on activated Ras pathway for their replication, including reovirus [109], influenza A virus delINS1 [110], poxvirus Pexa-Vec [42], coxsackievirus Type B3 [111], and alphavirus M1 [112]. For alphavirus M1, there have been four biomarkers identified thus far that correspond to increased OV activity: zinc-finger antiviral protein (ZAP), inositol-requiring kinase 1α, Ras homolog family member Q, and mutated and activated KRAS [113].
Until now, a limited number of response biomarkers for OV-mediated therapy have been identified [6]. The first one may be immunoglobulin-like transcript 2 (ILT2) for oncolytic VV [114]. The Kaufman lab observed an inverse association between ILT2 expression in the tumor and clinical response. They further identified ILT2 as a marker of regulatory CD4+ and suppressor CD8+ T cell responses, and ILT2 down-regulation was predictive of therapeutic responses in patients treated with oncolytic VV-mediated immunotherapy. Serum HMGB1 may be a predictive and prognostic biomarker for immunotherapy with oncolytic adenovirus [115]. Additionally, Nguyen and colleagues showed that defects in IFN-JAK-STAT pathway as response biomarkers to virotherapy mediated by oncolytic VSV and HSV-1 [116]. Another recent study identified the receptor of oncolytic alphavirus M1 as a therapeutic predictor for multiple solid tumors [117]. In summary, we believe that some of these biomarkers are tumor type-specific and/or OV-specific. As summarized correctly by Kaufman [106], each OV is unique and contains a different set of viral genes, other genetic components, and arming transgenes. These genetic factors and/or strategies to modify the OV may influence their biologic interactions in different tumors, and impact different gene expression status within infected cells, including cell death pathways. As such, we need to define how changes in specific innate sensing and antiviral machinery elements influence the ability of specific OVs to infect and replicate in individual tumor cells and how these changes impact antiviral and antitumor immune responses by the host. In addition to validating these identified biomarkers, uncovering additional biomarkers that can reliably predict therapeutic response will be of tremendous value.
In the following section, we will discuss biologically relevant and targetable signaling pathways and the development status of the current and emerging inhibitors/modulators. We then focus on rational combinations, including those that produce mixed responses when combined with OV, where OVs combined with inhibitors/activators of signal transduction pathways have been evaluated in preclinical studies or advanced to clinical testing.
Signaling pathways, modulators and combinations with OVs for improved therapy
Cancer development is driven by genetic and epigenetic alterations that allow cells to proliferate abnormally and escape mechanisms that normally control their survival and migration. Many of these alterations can be attributed to signaling pathways that control cell growth and signaling networks that fuel cancer progression [107, 118]. In addition to the well-known original hallmarks of cancer [107], it is intriguing to note that cancer cells display cancer-associated metabolic changes, or hallmarks of cancer metabolism [119, 120]. Importantly, it is not only cancer cells, but also immune cells in the TME that undergo metabolic reprogramming some of which may lead to immune tolerance [121]. Thus, these key-signaling molecules including metabolic enzymes in cancer cells, immune cells, and likely stromal cells are all potential therapeutic targets.
Many investigators have focused on the discovery and exploration of small molecules to modulate key signaling pathways as cancer treatments. Many small molecules can effectively modulate immune responses and the immunosuppressive TME [122]. Overall, data thus far has demonstrated a series of successful targeting, future therapeutic opportunities, as well as potential challenges [32, 33]. Some investigators consider the development of these small molecule drugs to be the next generation of immunotherapy for cancer [123]. Readers are referred to recent reviews related to small molecule modulators of signaling pathways and their expanding role in immuno-oncology [18, 28, 122, 123].
In this section, we will explore key signaling pathways as well as how these pathways could be explored as targets for cancer therapy (Fig. 2). As the body of work in this area is extensive and includes a large list of both targetable pathways and candidate therapeutics, it is not possible to comprehensively review all potential approaches currently under development. As such, we focus specifically on those small molecules targeting signaling pathways that may involve antitumor immunity. Then in the second part of each sub-section, we discuss the combination strategies and studies of OV with modulators of that signaling pathway and relevant findings in preclinical studies.

The Effects of small-molecule drugs on the TME. Targeted therapies have been shown to alter the TME in multiple ways, either directly or indirectly. Some agents could reverse the immunosuppressive environment by inhibiting the infiltration and function of MDSCs and Tregs. Numerous therapies increase the expression and presentation of tumor antigens, increasing cross-priming of DCs for enhanced T cell activation. Targeted therapies also can increase the expression of NKG2D ligands on NK cells, which serve as co-stimulatory molecules for CTL,as well as activators of NK cells, which also increase the NK cytotoxicity
The rationale for combining OVs with small molecule modulators
Many cell types and molecules play roles in shaping the immunosuppressive TME [124]. Those cell types and molecules discussed earlier may have fundamental roles in the TME. As stated earlier, OVs themselves can modulate the TME and turn cold tumor hot [55, 56]. However, the strength of immunogenic ‘hot’ property and antitumor immunity elicited by OVs may not be strong enough to eliminate the primary tumor and secondary metastasis, and thus combination with other antitumor agents deem necessary in most cases, especially for reversing the immunosuppressive nature of the TME. Many small molecules can effectively modulate immune responses and the immunosuppressive nature of the TME [122, 125, 126]. Some investigators consider the development of these small molecules to be the next generation of immunotherapy for cancer [18, 123]. Due to their unique mechanisms of action and anti-tumor properties, these agents may act in synergy with OVs, leading to enhanced antitumor immunity and therapeutic efficacy (Figs. 2 and 3).

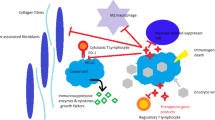
Rationale for the combination of OV with immuno-and targeted therapies in the TME for cancer. The TME is composed of diverse cell types, secreted factors, and extracellular matrix that provide targets for combination of OV therapies. We could arbitrarily divide these targets and mechanisms of action into steps A-G. A. OV replicate selectively in tumor and have the capacity for direct oncolysis. B. OV-mediated increases in the release of DAMPs, PAMPs and cytokines promote the accumulation of CTLs at tumor beds and retention of their killing capability. C. OV induce IFN pathways followed by elicitation of immune responses, thus mediating a broader range of long-lasting antitumor effects. D. OV infection leads to increased expression of immune checkpoint molecules such as PD-L1 and CTLA-4 from cancer and stromal cells, that sensitizes infected tumors to ICI. E. Cytotoxic chemotherapy destroys tumor cells by induction of cell death, often via ICD, or targeted therapies interrupt aberrant signaling pathways and potentially death of cancer cells. This may induce weak or moderate immune responses against tumor. F. Relevant cells such as TAMs, DCs, CAFs, and MDSCs secrete ECM components, growth factors, and cytokines, which can contribute to the regulation of tumor progression and therapeutic response in unique ways, such as CAFs suppress T and NK cells via cytokines and growth factors including PGE2 and TGF-β. Some OVs are designed to target not only cancer cells, but also stromal cells (e.g., CAFs). G. OV shape the TME for immunotherapy by shifting the tumor status from ‘cold’ to ‘hot’, thus, improving immune cell recruitment and effector function
EGFR/KRAS/MAPK signaling pathway
The epidermal growth factor receptor (EGFR) family is among the most investigated receptor tyrosine kinase (RTK) groups owing to its general role in signal transduction and oncogenesis. A large fraction of human cancers displays elevated EGFR and kinase activity through overexpression and/or mutations [127]. EGFR is one of the most frequently altered oncogenes in solid cancers. Therefore, efforts have been taken to develop small-molecule inhibitors as well as large molecules or biologics (such as antibodies) to inhibit this survival and activation signaling pathway for cancer cells. For a comprehensive review of small molecule inhibitors targeting the EGFR/ErbB family of RTKs, please refer to a recently published review article [128]. Regarding toxicities related to treatment with TKIs, it is important to note that treatment was almost uniformly associated with considerable toxicities [129], with the most frequent AEs including hand–foot syndrome, diarrhea, and nausea/vomiting. Therefore, careful consideration needs to be taken when considering combination therapy using these TKIs and other inhibitors.
Since 2004, several small molecules functioning as EGFR tyrosine kinase inhibitors (TKIs) have been approved by the FDA and other authorities as cancer therapies, particularly for the treatment of non-small cell lung cancer (NSCLC) (Table 1). For example, Erlotinib was initially approved to treat NSCLC in 2004, then approved to treat pancreatic cancer in 2005.
Two small molecule drugs, sunitinib and sorafenib, inhibits cellular signaling by targeting multiple RTKs (Table 1). Sorafenib is an inhibitor with activity against many protein kinases, including VEGFR, PDGFR and RAF kinases [130]. Both sunitinib and sorafenib were approved for treating patients with advanced renal cell carcinoma (RCC). Sorafenib has also been approved for advanced primary liver cancer, FLT3-ITD positive AML and radioactive iodine resistant advanced thyroid carcinoma.
KRAS was considered to be undruggable until recently. Rigosertib (RGS) is a non-ATP-competitive small molecule RAS mimetic. It has the potential to block RAS-RAF-MEK-ERK and PI3K-AKT-mTOR signaling pathways and to interfere with CRAF interaction with polo-like kinase 1 (PLK1). Mechanistically, the RAS-mimetic disrupts RAS association with effector proteins to block signaling. In one study, the authors showed that RGS inhibits tumor growth in models of implanted colorectal and lung cancer, and it blocks tumor growth of a transgenic model of KRASG12D-induced pancreatic cancer in mice [131]. Yan et al. have recently shown that RGS treatment led to induction of CD40 as a result of RAS/RAF/PI3K pathway disruption, followed with cancer cell death through ICD, and augmented cancer response to checkpoint blockade [132].
The progress towards developing small molecule inhibitors for mutant KRAS, particularly KRASG12C, has been even more striking [133]. Sotorasib (AMG510; Lumakras™) and Adagrasib (MRTX849) have demonstrated potent clinical efficacy and high selectivity in human patients with KRASG12C-driven cancers. This has been an unprecedented breakthrough. Sotorasib has been rapidly progressed from preclinical to clinical studies, to accelerated approval by FDA. Scientists at Amgen Inc. discovered and assessed the activity of this covalent inhibitor of KRASG12C [134]. In immune-competent mice, Sotorasib treatment initiated the conversion to a pro-inflammatory TME and generated durable cures alone or in combination with ICIs. In a preclinical study, Sotorasib treatment led to the regression of KRASG12C tumors and improved the efficacy of chemotherapy and targeted agents in mice [135]. A phase 1 trial of sotorasib in 129 human patients with advanced solid tumors harboring the KRASG12C mutation was published in 2020 [136]. The study showed 32% objective response and 88% disease control in the subgroup of patients with NSCLC. In a phase II trial, sotorasib therapy resulted in durable clinical benefit without new safety signals in patients with previously treated KRASG12C-mutated NSCLC [137]. In 2021, the FDA approved Sotorasib for use in patients with KRASG12C-mutated NSCLC (Table 1). Adagrasib has also proceeded through phase I-II clinical trials and showed clinical efficacy without new safety signals in patients with advanced KRASG12C solid tumors [138, 139]. Based on these promising clinical outcomes, two phase III trials have been planned [138].
Combination of OVs with inhibitor of EGFR/KRAS/MAPK signaling pathway
Investigators have tested OV combined with a small molecule inhibitor of one of the RTKs as rational combination therapy. Malignant peripheral nerve sheath tumors (MPNSTs), driven in part by hyperactive Ras and EGFR signaling, are often incurable. Cripe and his team developed a xenograft model of human MPNST and evaluated the combined antitumor effects of oHSV and the EGFR inhibitor, erlotinib. oHSV injection exhibited more dramatic antitumor activity than erlotinib. Combination therapies showed a trend toward an increased antiproliferative effect [140]. In another study with a peritoneally disseminated model of human xenograft pancreatic cancer, Canerpaturev (C-REV) combined with erlotinib had no beneficial effect on survival. However, in the subcutaneous tumor model, this combination resulted in the inhibition of tumor growth to a greater extent than using either agent on its own [141]. At present, the combination of an OV with mAb against EGFR have generated greatly improved therapeutic results in multiple studies [142, 143]. In our opinion, more definitive studies will be needed using the currently greatly improved small molecule inhibitors [128], to determine the future potential of this approach in the context of small molecule inhibitors.
Combining inhibitors of other TKRs with OVs has also been explored. For example, VEGFR TKI axitinib and oHSV have been evaluated in vitro and then assessed in two orthotopic glioblastoma (GBM) models derived from GBM stem-like cells [144]. The results showed that systemic TKI (Axitinib) beneficially combined with G47Δ-mIL12 to enhance antitumor efficacy. In another study, the authors showed that OV and SCF-1R inhibition (with PLX3397) reprogrammed the TME, enhancing the infiltration of CD8+ T cells and improving the impact of anti-PD-1 immunotherapy [145].
As mentioned, sunitinib is an inhibitor for multiple RTKs and its potential for use in combination with OVs has been explored by two groups. In the first study, sunitinib improved VSV-mediated virotherapy through inhibition of antiviral innate immunity [146]. In the second study, the authors explored its use together with an oncolytic VV [147]. The oncolytic VV mpJX-594 targets tumor blood vessels, spreads secondarily to tumor cells, and produces widespread CD8+ T-cell-dependent tumor cell killing in primary tumors and metastases. Importantly, these effects can be amplified by coadministration of sunitinib. Importantly, this study revealed multiple unrecognized features of the antitumor properties of oncolytic VV, all of which can be amplified by the multitargeted kinase inhibitor sunitinib [147].
Many cancers are driven by oncoproteins and some OVs selectively replicate in and destroy cancer cells overexpressing oncoproteins, suggesting a potentially exploitable therapeutic opportunity. It has been known for over 2 decades that human reovirus requires an activated Ras signaling pathway for infection of cultured cells, creating a clear opportunity to treat Ras activated cancers with this OV [148]. Further studies indicated that Ras-transformation affects multiple steps of the virus life cycle, including viral uncoating and disassembly, releasing PKR-induced translational inhibition, production of viral progeny, release of progeny, and viral spread following reovirus-induced cancer cell death occurring through necrotic, apoptotic, and autophagic pathways [149]. Another study suggested that reovirus induced cell death is immunogenic [150]. In this context, it is exciting to note that tumors driven by RAS signaling display a natural vulnerability to oncolytic alphavirus M1 [112]. Inhibition of the RAS/RAF/MEK signaling axis suppresses M1 infection and the subsequent cytopathic effects [112]. As such it would not be logical to combine these OVs with RAS inhibitors, highlighting that a clear understanding of the mechanistic interplay between OV and small molecule inhibitors is necessary in designing effective and potentially synergistic combination therapies.
Melanomas often have mutations in BRAF and RAS, and investigators have explored the use of BRAF/MEK inhibitors to treat melanoma in preclinical and clinical studies [151]. In one study, Roulstone et al. explored the combination of an oncolytic reovirus (TR3D) with BRAF- and MEK-targeted inhibitors in vitro and in tumor models [152]. Combined this OV with a BRAF inhibitor (PLX4720) led to significantly increased antitumor activity in BRAF mutant tumors in both immune-deficient and immune-competent models.
T-VEC has been approved for advanced melanoma, yet therapeutic responses to this OV are limited. Trametinib is a MEK inhibitor also approved for treatment of melanoma and in a study by Bommareddy et al., the combination of T-VEC and trametinib resulted in enhanced melanoma cell death in vitro [153]. The combination treatment resulted in delayed tumor growth and improved survival in tumor models. Mechanistically, regression of treated tumors was dependent on activated CD8+ T cells and Batf3+ DCs. Interestingly, authors also observed antigen spreading and induction of an inflammatory gene signature, including PD-L1. Adding anti-PD-1 antibody to T-VEC + MEK inhibition further augmented responses through enhanced tumor antigen-specific T cell responses. Interestingly, MEK inhibitor (trametinib) also enhances oncolytic VV viral replication in doxorubicin-resistant ovarian cancer cells and the combined approach attenuated A2780-R ovarian cancer growth [154]. The preclinical studies by Bommareddy et al. [153] and others, strongly support clinical evaluation of this triple combination as a rational approach in melanoma and other cancers [155].
PI3K/AKT/mTOR pathway
The PI3K/Akt/mTOR pathway regulates cell proliferation, growth, cell size, metabolism, and motility [156]. In many cancers such as breast, lung ovarian and prostate, this pathway is often activated aberrantly [156,157,158]. The enhanced activity of this crucial intracellular signaling pathway is often associated with tumor progression, and cancer’s resistance to therapies, and thus targeting this signaling pathway is a rational approach for cancer therapy [159]. There are 4 different isoforms of PI3K: alpha, beta, delta, and gamma. Small molecule inhibitors against a specific isoform of the enzyme, or as pan inhibitor, have been developed and evaluated in preclinical models with some progressing to advanced phase clinical trials [156,157,158].
Macrophage PI3Kγ drives progression of pancreatic ductal adenocarcinoma, and possibly other cancers [160]. Importantly, while the Syk-PI3Kγ axis in macrophages has been reported to inhibit antitumor immunity, SRX3207, a novel dual Syk-PI3K inhibitor, has been shown to block these inhibitory effects, thereby relieving tumor immunosuppression [161]. In another study, authors’ demonstrated that targeting PI3K-γ with a selective inhibitor (IPI-549) reshapes the immune milieu of the TME and promote CTL-mediated tumor regression without targeting cancer cells directly [162]. IPI-549 can inhibit PI3Kγ in MDSCs, resulting in downregulation of arginase 1 (Arg-1) and ROS to promote MDSCs apoptosis and reduce their immunosuppressive activity to CD8+ T cells [163]. This inhibitor is currently being evaluated in human cancer patients in multiple clinical trials.
Three different PI3K inhibitors have been approved by the FDA for the treatment of follicular lymphoma [164]. Following a successfully phase III clinical study [165], the FDA approved the first PI3K inhibitor, Piqray (alpelisib), for breast cancer patients with advanced disease and where their tumors have the PIK3CA mutation and are hormone receptor (HR) positive and HER2 negative in 2019. However, the overall landscape and therapeutic potential, as summarized by Mishra et al., is that “very few PI3K inhibitors are approved by the FDA as PI3K inhibitors suffer from many adverse effects, and poor solubility and permeability” [158].
AKT, as a key component of the PI3K/AKT/mTOR signaling pathway, exerts a pivotal role in cell growth, proliferation, survival, and metabolism [166, 167]. Small molecule inhibitors for AKT have been synthesized and evaluated in both preclinical and clinical trials [167, 168]. Capivasertib is a potent selective oral agent and inhibits all three isoforms of the AKT kinase. In the phase II FAKTION trial with postmenopausal women who have inoperable breast cancers that are aromatase inhibitor–resistant and estrogen receptor (ER)-positive/HER2-negative, Jones et al. found that the addition of capivasertib to endocrine therapy with fulvestrant prolonged progression-free survival in these patients [169]. At this time, only a few AKT inhibitors have been approved for cancer treatment [168].
Several mTOR inhibitors have been approved to treat human cancer [170]. The FDA-approved mTOR inhibitors include Sirolimus for treating patients with lymphangioleiomyomatosis with gene mutations of the tuberous sclerosis complex 2 gene in RCC, and Everolimus for RCC, pancreatic, and breast cancers. Currently, additional mTOR inhibitors are being evaluated in clinical trials. In general, it appears that mTOR inhibitors have mixed efficacy in patients across tumor indications and among patients with the same type of cancer. While mTOR inhibition alone has clear efficacy in some types of cancer, preclinical studies demonstrate strong rationale for combining mTOR inhibitors with other drugs, including OVs. While therapeutic efficacy has been demonstrated, small molecule inhibitors for the PI3K/AKT/mTOR pathway can exert certain toxicities, and the mechanisms behind these effects have been described [171].
Combination of OVs with inhibitors of PI3K/AKT/mTOR pathway
GBM is a lethal primary brain cancer with a median survival of less than 2 years. Rabkin, Martuza, and their teams showed that oHSVs could synergize with PI3K/AKT pathway inhibitors to target glioblastoma (GBM) stem cells [172], and prostate cancer stem-like cells [173]. Similarly, these small molecule inhibitors have also been incorporated into therapeutic regimens using other OVs, such as adenovirus [174], and Newcastle disease virus (NDV) [175]. However, not all the combinations would generate a synergy. For example, temozolomide has been used as a standard care for GBM, however it can only extend overall survival to a few months. Thus, investigators have tried to combine it with other antitumor agents for improved efficacy. A recent study found that temozolomide induces activation of Wnt/β-catenin signaling in glioma cells via PI3K/Akt pathway [176]. Yet temozolomide antagonize oncolytic immunotherapy in GBM using G47Δ-IL12 [177].
ICI is ineffective in GBM with PTEN deficiency [178]. Xing et al. have explored the impact of PTEN deficiency on OV therapy. They found that OV and PI3K inhibition work synergistically on the TME and restore immune checkpoint therapy response in PTEN-deficient GBM [179].
Wang and his team demonstrated improved systemic delivery of an oncolytic VV by using an inhibitor of PI3Kδ [180]. Transient inhibition of PI3Kδ with PI3Kδ-selective inhibitor IC87114 or the clinically approved idelalisib, enhanced the delivery and therapeutic effects of intravenously delivered oncolytic VV. This occurred by inhibiting attachment of the virus to, but not internalization by, systemic macrophages through perturbation of signaling pathways involving RhoA/ROCK, AKT, and Rac. They also applied this approach to increase the potential for intertumoral and intratumoral spread of oncolytic VV, effectively treating pancreatic cancer [181].
Rapamycin, or Sirolimus, is a macrolide compound and has immunosuppressant functions in humans and is especially useful in preventing the rejection of kidney transplants. The mammalian target of rapamycin, or mTOR, play a central role in Akt-mediated cell proliferation, differentiation, maturation and survival [182].
In 2005, Iggo and the team found that RAD001 (Everolimus), an mTOR inhibitor, improves the efficacy of oncolytic adenoviruses that target colon cancer [183]. They believed that RAD001 has three useful properties: inhibiting tumor cell growth directly, blocking angiogenesis, and suppressing the immune response. However, how RAD001 enhanced treatment efficacy was not completely understood. In 2007, McFadden, Forsyth and colleagues found that rapamycin increased myxoma virus tropism for human cancer cells and thus enhanced oncolytic virotherapy [184, 185]. Later, this combination was also applied to VV [186], HSV [187, 188], vesicular stomatitis virus (VSVΔM51) [189], and AdV [190]. As the PI3K/AKT/mTOR signaling pathway is multifunctional, it is not surprising that rapamycin could promote oncolytic virotherapy through multiple mechanisms. First, in the case of HSV and myxoma viruses, rapamycin functions to enhance the permissiveness of cancer cells to OV by reconfiguring the internal cell signaling environment to one that is optimal for productive virus replication [184, 187]. Second, rapamycin increases viral replication by impairing mTORC1-dependent type I IFN production and a reduction of intra-tumoral infiltration of macrophages [189, 191]. Third, active-site dual mTORC1 and mTORC2 inhibitors (but not rapamycin) augment HSV1-dICP0 infection in cancer cells via the eIF4E/4E-BP axis [188].
JAK-STAT3 pathway
Many cytokines function as crucial drivers of cancer as well as autoimmune conditions. They bind to receptors and trigger signaling cascades through Janus kinase (JAK) and signal transducer and activator of transcription (STAT) pathways. IL-6/JAK/STAT3 signaling acts to drive cancer cell proliferation, survival, invasiveness, and metastasis, while strongly suppressing antitumor immunity [192]. Thus, targeting JAKs and STATs could be an efficacious strategy [192, 193].
Currently, only four JAK inhibitors (ruxolitinib, tofacitinib, baricitinib and fedratinib) are approved for the treatment of myeloproliferative neoplasms and other disorders [194]. Many innovative studies and approaches to developing small molecule inhibitors/activators of this signaling pathways have been performed. In 2008, Xiong et al. demonstrated that inhibition of JAK1/2 signaling with AG490 (and inhibition of STAT3 as well) induced apoptosis, cell cycle arrest, and reduced tumor cell invasion in colorectal cancer cells [195]. However, despite some promising preclinical results, to date no clinical studies have shown efficacy in solid tumors treated with JAK inhibitors.
Therapeutically exploiting STAT3 activity in cancer appears to be more promising therapeutic approach [196, 197]. In 2012, Zhang et al. reported on an orally bioavailable small-molecule inhibitor of transcription factor STAT3, BP-1-102, that led to regression of human breast and lung cancer xenografts [198]. In a recent study, the authors showed that a potent and selective small-molecule degrader of STAT3 could produce complete tumor regression [199]. The inhibitor pyrimethamine displays anti-cancer activity and immune stimulatory effects in mouse breast cancer models [200]. Additionally, some FDA-approved compounds, such as pyrimethamine and celecoxib, have also been identified as STAT3 inhibitors [197]. Interestingly, another FDA-approved drug atovaquone was identified as a STAT3 inhibitor and anticancer agent using a gene expression-based discovery platform [201]. Currently, there are 18 clinical trials testing STAT3 inhibitors as cancer treatments listed in the database (clinicaltrials.gov), with 8 of these trials now completed, three recruiting, and one not yet recruiting.
As a note of caution when considering combination approaches, the JAK-inhibitor ruxolitinib has been shown to impair DC cell function both in vitro and in vivo [202]. To overcome this issue, the same team has identified a different JAK inhibitor, pacritinib, that can effectively conserve DC function when compared to ruxolitinib [203].
Combination of OVs with modulators of IFNγ-JAK-STAT pathway
The effects of STAT inhibitors on OV are complex and appear to depend on both selected OV and type of tumor. One of the major mechanisms of resistance to VSV infection is the type I IFN response, leading to the development of IFNβ-armed VSV. This VSV-IFNβ virus leads to resistance of viral replication in normal cells with intact IFN signaling but allows viral replication in cancer cells with defective IFNβ signaling. However, some cancer cells have intact or partially intact IFN signaling pathways and can resist VSV-mediated therapy. To overcome this issue, the authors utilized the JAK/STAT inhibitor, ruxolitinib, in combination with VSV-IFNβ to see if inhibition of the signaling could enhance VSV-IFNβ therapy for lung cancer [204]. Combination of ruxolitinib and VSV-IFNβ therapy resulted in a trend toward improved survival of mice, suggesting that further evaluation is needed. In another study, Nguyen and colleagues found that mutations in the IFNγ-JAK-STAT pathway cause resistance to ICI immunotherapy can increase sensitivity to OV infection and treatment [116]. This study also supports JAK inhibitor-OV combination for treatment-naïve melanomas without IFN signaling defects.
p53 pathway and its roles in cancer, immune function, and cancer immunity
p53 is one of the most well-studied tumor suppressors. TP53 is mutated in approximately 50% of all human cancers. Interestingly, in the other ~ 50% of cancer carrying wild type p53, the signaling pathway is often disrupted at other interaction points [205]. The activity of p53 can be inhibited through the function of the negative regulators MDM2 and MDMX [206]. As the functional domains of the p53, mutation hot spots, and loss/gain of function mutations are out of the scope of this article, readers are referred to two excellent reviews for these details [207, 208].
As the p53 pathway is one of the most crucial signaling pathways in cells, it has been widely investigated as a target for developing anti-cancer drugs [205, 206, 209]. A series of small molecules targeting MDM2 and MDMX have been developed and studied in preclinical models and in clinical studies [209, 210]. Lane and colleagues have found that an old antibiotic, actinomycin D (ActD), when used at low dose, could mimic nutlin-3 and impart highly specific activation of p53 dependent transcription, and induce a reversible protective growth arrest in normal cells and enhance the activity of chemotherapy-induced killing of p53-positive human tumor cells [211]. At low cytostatic concentrations, ActD promotes ribosomal stress, which decreases MDM2 activity, resulting in p53 stabilization and activation. Chen et al. showed that ActD-induced p53 expression is mediated by AKT [212]. This property of ActD led to a series of clinical studies, including 6 phase III clinical trials using ActD in combination with other anti-cancer agents in a number of tumor indications [209]. Most of these trials are ongoing and final reports have not yet been published.
p53 is also functionally important in normal immune processes and antitumor immunity [213]. The key conclusion from recent studies is that wildtype p53 has fundamental roles in cancer immunity, however, mutations in p53 not only cripple wildtype p53 immune functions but also subvert the immune functions through its gain-of-functions [213]. The mutant p53 is associated with inflammation and immune dysfunction, indicating that it modulates immunity associated with cancer. In one key study, the authors identified a role for cancer-cell-intrinsic p53 as a key regulator of pro-metastatic neutrophils by using a panel of 16 distinct genetically engineered murine models of breast cancer. Mechanistic studies revealed that loss of p53 in cancer cells induced the secretion of WNT ligands that could stimulate tumor-associated macrophages to produce IL-1β, thus driving neutrophil infiltration and systemic inflammation [214]. In another study, the authors discovered a novel mechanism of targeting p53 for cancer immunotherapy. Activation of p53 in immature myeloid precursor cells was observed to dictate their differentiation into Ly6c+CD103+ monocytic antigen-presenting cells in tumors [215]. Increasing p53 expression using a p53-agonist drug elicits a sustained increase in Ly6c+CD103+ cells in tumors during immunotherapy, leading to markedly enhanced efficacy and duration of response [215].
Combination of OV with small molecules targeting p53-pathways
At this time, no combination of OV and a small molecule modulator of p53 has been assessed experimentally, based on our extensive literature search. However, Tagawa et al. studied how an MDM2 inhibitor could interact with an OV to produce synergistic cytotoxicity of cancer cells. Specifically, they found that an MDM2 inhibitor achieves synergized effect with an oncolytic AdV (Ad-delE1B) lacking E1B-55 kDa gene to target mesothelioma with wildtype p53 through augmenting NFI expression [216].
Epigenetic reprogramming in cancer cells and immune cells
DNA methylation, histone modifications including acetylation, methylation and phosphorylation, are major forms of epigenetic modifications that play vital roles in gene regulation, key biological processes and in cancer biology [217]. Under the influence of the TME and during activation, immune cells undergo epigenetic reprogramming to adapt to their environments and which directly impact the resulting cell differentiation and functionality [218, 219]. Therefore, small molecule inhibitors of enzymes involved in DNA methylation and histone modifications, can be explored to potentially reverse the suppressed functions of immune cells in the TME [28, 220, 221]. For example, epigenetic silencing of CXCL9/10 expression by promoter DNA methylation and H3K27me3 in tumor cells limits the infiltration of cytotoxic T cells, preventing tumor attack and promoting tumor growth [222, 223]. To reverse these effects, inhibitors of epigenetic enzymes DNMT or EZH2, alone or in combination, restore CD8+ T cell infiltration and improve sensitivity to ICI [223, 224]. We and others have showed that DNA demethylating agents can enhance cancer immunogenicity and induce de novo expression of cancer-testis antigens that improve responses to adoptive T cell therapy [225, 226]. These and other studies strongly suggested that epigenetic modulation may turn cold tumors hot [227, 228], thereby increasing the responsiveness of tumors to immunotherapy.
Another subtype of epigenetic modification is on SUMOylation. The covalent conjugation of small ubiquitin-like modifier (SUMO) proteins to protein substrates may lead to suppress type I interferon responses. Lightcap and colleagues recently studied the effects of TAK-981, a small-molecule SUMOylation inhibitor, on human and mouse immune cells. They found that the compound promoted the activation of dendritic cells and T cells. Further, they demonstrated that this compound activates antitumor immune responses and potentiates immune therapies in preclinical tumor models [229].
So far 10 small-molecule epigenetic drugs have been approved to treat several types of cancer (Table 2). Many other small molecule inhibitors targeting key enzymes involved in epigenetic pathways have been discovered and are at preclinical and clinical development as cancer therapies [230,231,232]. These emerging agents have the potential to be also explored in the context of immunotherapy, as has been recently reviewed [27, 28, 233].
Combination of OVs with inhibitors of epigenetic enzymes
One major hurdle in cancer therapy is heterogeneity of cancer, which results in variations in the ability of tumors to support productive infection by OVs and to induce adaptive anti-tumor immunity. Mounting evidence suggests tumor epigenetics may play a key role in this heterogeneity. In the last decade, therapeutic strategies aiming to exploit the epigenetic identity of tumors have been developed [28, 234, 235]. Some recent studies combining OV with epigenetic drugs are shown in Table 3.
Histone deacetylases (HDACs) are a large family of enzymes that have crucial roles in numerous biological processes, largely through their repressive influence on transcription [245]. HDACi have been shown to inhibit the IFN response [246]. HDACs may modulate epigenetic modifications of histones and chromatin, and other cellular regulatory proteins (such as p53, E2F1–3), leading to diminished cellular antiviral response. In one study, two HDACi markedly enhance the infection and spread of VSV and OVV in primary human tumor tissue explants and multiple tumor models. In another study, two HDACi (Scriptaid and LBH589) combined with OV Delta24-RGD led to enhanced efficacy in patient-derived glioblastoma cells [247]. HDAC6 inhibition enhances oHSV replication in glioma [248]. This was in line with previous studies demonstrating that HDAC6 controls innate immune and autophagic responses to TLR-mediated signaling by intracellular bacteria [249], and thus likely viruses too.
Reduced cellular IFN responses and enhanced virus-induced apoptosis may explain the increased viral replication and oncolytic activity in some cases [250]. In one study, an unexpected property of HDACi on adaptive immunity was found [251]. Entinostat (MS-275), which is a class I-specific HDACi, induced lymphopenia, leading to selective depletion of bystander lymphocytes and Treg cells but allowed expansion of antigen-specific secondary responses. Intriguingly, during the boosting phase, coadministration of an oVSV with entinostat biased the immune response towards anti-tumor immunity by suppressing the primary anti-viral immune response and enhancing the boost response against tumor antigens. Overall, this HDACi enhanced OV-mediated therapeutic efficacy, suppressed autoimmunity and thus improved the therapeutic index [251].
In a recent study, Muscolini et al reported that the NAD+-dependent histone deacetylase SIRT1 plays a key role in the permissivity of PC-3 prostate cancer cells to VSVΔM51 viral replication and oncolysis. Enhanced VSVΔM51 infection and cancer cell killing were mediated by HDACs and directly correlated with a decrease of SIRT1 expression [239]. In addition, pharmacological inhibition sensitized prostate cancer cells to VSVΔM51 infection, resulting in augmentation of virus replication and spread. Mechanistically, HDACi upregulated the microRNA miR-34a that down-regulated the level of SIRT1 [239].
Immune checkpoint pathways
The discovery of key immune checkpoint molecules (CTLA-4 and PD-1) and their blockade as novel approaches in cancer immunotherapy led to the Nobel Prize in Medicine in 2018 [252]. Monoclonal antibodies against PD-1 or PD-L1 have been approved to treat various types of cancer with proven efficacy [253]. However, therapeutic antibodies face functional and practical limitations, including inadequate tissue accessibility, pharmacokinetic challenges, impaired interactions with the immune system, as well as high production costs [254]. The toxicities observed following delivery of anti-PD-1/PD-L1 antibodies are well documented [255]. The general adverse events (AEs) may include fatigue, pyrexia, infusion reactions; while organ-specific AEs may include dermatologic toxicities, diarrhea/colitis, endocrine toxicities, hepatic toxicities, and pneumonitis.
Small molecule inhibitors targeting PD-1/PD-L1 signaling pathway may provide an attractive alternative approach. The rational design strategies of these small molecules are based on three approaches as summarized in an excellent review paper by Wu et al. [256]: 1). Blocking direct interaction of PD-1 and PD-L1 proteins; 2). Inhibiting the expression (transcription and translation) of PD-L1 protein; 3). Promoting the degradation of the PD-L1 protein. Bristol-Myers Squibb (BMS) and Aurigene Discovery Technologies each disclosed several promising PD-1/PD-L1 inhibitors, including small molecules and peptides, although only a fraction of experimental data have been disclosed to the public [257, 258]. Following on this approach, other inhibitors have been discovered by both industry and academic groups. One recent study detailed a series of novel inhibitors for PD-1/PD-L1 interactions, with A9 as a standout [259]. We have listed some representative inhibitors in Table 4.
Basic and preclinical studies have been performed to assess the functional properties and potency of small molecules or peptides that interact with the PD-1/PD-L1 axis and how these agents impact antitumor immunity. In one study, the authors conducted systematic in vitro characterization on BMS-103, BMS-142 and others [272]. These compounds are strongly active in biochemical assays, yet their acute cytotoxicity greatly compromised their immunological activity. In contrast, Wang et al. have found that a new small molecule inhibitor, called APBC, could effectively interrupt the PD-1/PD-L1 by directly binding to PD-L1, presenting the KD and IC50 values at low-micromolar levels. Better yet, it could elevate cytokine secretions of the primary T-lymphocytes that are cocultured with cancer cells in a dose-dependent manner. It displayed superior antitumor efficacy in B16F10 melanoma in hPD-L1 knock-in mouse model without the induction of observable liver toxicity [260]. Koniecny and the team have developed di-bromo-based small molecule inhibitors for PD-1/PD-L1 interaction and showed its activity in vitro [273]. Fang et al. discovered 1,3,4-oxadiazole derivatives as inhibitors for PD-1/PD-L1 interaction and found compound II-14 as the most potent one in biochemical activity and antitumor activity [261].
It is important to note that small molecules have been developed for targeting additional immune checkpoints. Cbl-b is expressed in all leukocyte subsets and regulates multiple signaling pathways in T cells, NK cells, B cells, and some types of myeloid cells. Cbl-b negatively regulates activation signals through antigen or pattern recognition receptors and co-stimulatory molecules. Several studies showed that Cbl-b-gene knockout mice reject tumors. Thus, targeting Cbl-b may be an promising strategy to enhance antitumor immunity [274]. Src homology-2-containing protein tyrosine phosphatase 2 (SHP2) is a major phosphatase involved in several cellular processes. Recent studies showed that this enzyme plays vital roles not only in T lymphocytes and macrophages, but also cancer cells. Thus, exploring the use of SHP2 inhibitors has potential promise for cancer immunotherapy [275]. Hematopoietic progenitor kinase 1 (HPK1/MAP4K1) is a hematopoietic cell-restricted member of the serine/threonine Ste20-related protein kinases. It is a negative regulator of the T cell receptor, B cell receptor, and DCs. It is probable that blocking the HPK1 kinase activity with a small molecule inhibitor may elicit superior anti-tumor activity of both T and B cells, resulting in a synergistic amplification of anti-tumor potential [269, 276]. Small molecule inhibitors for these enzymes have been developed and some of them are in clinical studies for cancer immunotherapy (Table 4).
Combination of OV with immune checkpoint blockade
As we have noticed, the discovery of small molecule modulators for PD-1/PD-L1 (Table 3) are relatively recent events, thus no studies combining them with OVs have been published. However, many studies with OVs in combination with immune checkpoint blockade using PD-1, PD-L1, CTLA-4 antibodies have studied. Many reviews on this topic have been published and we refer to two recent great reviews [7, 277]. Worth specific mentioning are two clinical studies of T-VEC with either anti-PD-1 or anti-CTLA-4 antibodies, achieving dramatically improved clinical responses with up to ~ 67% objective response rate in patients with advanced melanoma [55, 105].
Other pathways of particular importance to innate immunity
The cGAS–STING signaling pathway is a key mediator of inflammation and innate immunity in settings of infection, cellular stress, and cancer [278]. A network of pattern recognition receptors (PRRs) can detect invading viruses and trigger the host antiviral response. Stimulator of interferon genes (STING) functions as a node and integrator of the signal network—now called cGAS-STING-TBK1 pathway [279]. At this time, the cGAS-STING-TBK1 axis is considered the major signaling pathway in innate immune response across different species. Indeed, cGAS-STING is an important pathway in cancer immunotherapy [280]. In addition to their role in antitumor immunity, STING agonists have recently been shown to be effective in promoting tumor vascular normalization and formation of tertiary lymphoid structures within the therapeutic TME [281].
Small molecules targeting this innate immune signaling pathway have been developed and some are now undergoing clinical testing [282]. It is noteworthy that high potency STING agonists can engage unique myeloid pathways to reverse pancreatic cancer immune privilege [283]. In this study, potent synthetic STING agonists (e.g., IACS-8803) reprogrammed suppressive myeloid populations, both human and murine origins, in part through inhibition of Myc signaling, metabolic modulation, and antagonism of the cell cycle. Inhibitors of upstream molecules may also lead to the activation of STING pathway. CX-5461 is one of the most promising inhibitors for RNA polymerase I, and previous reports have shown that CX-5461 treatment induces DNA damage response through ATM/ATR kinase. In a recent study, the authors reported that CX-5461 could induce a rapid accumulation of cytosolic DNA. This accumulation led to transcriptional upregulation of STING, phosphorylation of IRF3, and activation of type I interferon response. Thus, CX-5461 therapy-induced immune activation may be exploited as novel drug combinations with the potential to increase immunotherapy efficacy [284]. In another study, inhibition of Ataxia telangiectasia mutated (ATM) enhanced cancer immunotherapy by promoting mitochondrial DNA leakage and cGAS/STING activation [285]. The authors suggested that that ATM may serve as both a therapeutic target and a biomarker to enable ICI therapy.
Combination of OVs with small molecule modulators of other pathways
The STING axis may be silenced during malignant transformation, allowing cancers to escape immune surveillance, which may in turn allow OV to efficiently replicate and exert therapeutic benefit in these cancers [286]. A recent study demonstrated that STING restricts oHSV replication and spread in resistant malignant peripheral nerve sheath tumors [287]. However, while STING knockout tumors could support increased lytic potential by HER2-retargeted oHSV-1, these tumors also showed molecular signatures of an immunosuppressive TME. These signatures were correspondingly associated with ineffectiveness of the combination therapy in a tumor model. Accordingly, these authors proposed that antiviral, tumor-resident Sting provides a fundamental contribution to immunotherapeutic efficacy of OV [288]. Kaufman, Rabkin and associates conducted a study that suggests T-VEC induces ICD and promotes tumor immunity, and it can induce therapeutic responses in anti-PD-1-refractory, low STING-expressing melanoma [155]. To summarize, these data that OV immunotherapy induces ICD and overcomes STING deficiency in melanoma.
Some studies have explored inhibitors targeting the transforming growth factor beta receptor 1 (TGF-βR1). Cripe and associates combined OV HSV1716 with an TGF-βR1 inhibitor (A8301) to treat syngeneic models of murine rhabdomyosarcoma [289]. They observed enhanced efficacy appears to depend on an improved anti-tumor T cell response. In another study with patient-derived recurrent GBM models, dual therapy with an oHSV and inhibitors of TGF-β receptor kinase also led to enhanced efficacy [290]. These studies revealed a novel synergy of virotherapy and blockade of TGF-β signaling and warrant further preclinical investigation to support clinical translation of this combination strategy. In addition, these two agents, either alone or in combination, can increase the susceptibility of immune-silent tumors to immune checkpoint therapy [291].
Aurora-A kinase is also a promising therapeutic target in cancer [292]. Two studies showed that, the kinase inhibitors alone or in combination with OVs, can possess additive/synergistic killing effects on cancer cells [293,294,295]. Interestingly, in one study, the authors found that aurora A kinase inhibition enhances oHSV virotherapy through cytotoxic synergy and innate cellular immune modulation, noting that alisertib inhibited virus-induced accumulation of intra-tumoral MDSCs [295].
Small molecule experimental drugs can target cancers with intrinsic or acquired resistance to the infection of various OVs. Xiao et al. have recently demonstrated that DNA-dependent protein kinase (DNA-PK) inhibition sensitizes cancer cells to alphavirus M1 and improves therapeutic effects in refractory cancer models and in patient tumor samples. It turned out that DNA-PK inhibition synergizes with OV M1 by inhibiting antiviral response and potentiating DNA damage [296].
One mechanism of OV therapeutic failure is tumor cell resistance to OV infection [297]. Dimethyl fumarate (DMF) is a common treatment for psoriasis and multiple sclerosis, and it also exhibits anticancer properties. Selman et al. observed that DMF and various fumaric and maleic acid esters (FMAEs) could enhance viral infection of cancer cells as well as human tumor biopsies by OVs, including VSV, AdV and HSV. Together, these combination therapies improved therapeutic outcome in OV-resistant syngeneic tumors and xenograft models in mice [298]. This study demonstrates that unconventional application of FDA–approved drugs and biological agents can result in improved anticancer therapy.
ICD inducers are useful in induction of cancer cell death and eliciting antitumor immunity [299]. Most OVs themselves are ICD inducers [30]. However, small molecules can also function as ICD inducers, further potentiating the induction of antitumor immunity when combined with OVs. For example, CDK4/6 inhibitor and VSVΔ51 synergistically induced ICD and boosted antitumor immunity, leading to enhanced efficacy in refractory glioblastoma [300]. Mechanistically, CDK4/6 inhibition led to autophagic degradation of MAV, resulting in dampened antiviral responses and thus enhanced tumor-selective replication of the OV. This CDK4/6 inhibition cooperated with OV infection to induce severe DNA damage stress and amplify ICD, leading to increased numbers of activated CD8+ cells [300]. Another study tested the combination of a ferroptosis enhancer with an OV. Erastin itself is a typical activator of ferroptosis, considered to be one type of ICD. We showed that Erastin alone had a limited effect on systemic immunity or localized intratumoral immunity in tumor-bearing mice. When combined with an OV, however, erastin enhanced the number of activated DCs and the activity of tumor-infiltrating T lymphocytes (based on increased IFN-γ+CD8+ and PD-1+CD8+ T cells), leading to improved therapy in tumor models in mice [301].
Efforts have been undertaken to identify novel small molecules that can work with OVs for improved antitumor effects. Diallo et al. uncovered a novel small molecule to target the complex cellular defense mechanisms that permit effective viral infection and replication. They have designed a high-throughput pharmacoviral approach and this identified novel chemical sensitizers (e.g., VSe1) to allow effective infection and replication of OVs such as VSVΔ51 [302]. Through this screen, they identified compound #1 that can sensitize resistant cancer cells to infection with VSVΔ51 by damping the activation of antiviral responses in cancer cells. However, compound 1 is rapidly degraded and thus derivatives with improved stability and activity would be desirable. To achieve this aim, Dornan et al endeavored to develop lead compounds (pyrrole-derivatives) that increased OV growth up to 2000-fold in vitro and demonstrated remarkable tumor selectivity both ex vivo and in vivo [303]. This expands the scope of OVs to include the originally resistant tumors, further potentiating this promising therapy. In another study, investigators found that vanadium compounds can potentiate oncolytic immunotherapy though multiple mechanisms [304]. The compounds work by subverting the antiviral type I IFN response toward a death-inducing and pro-inflammatory type II IFN response, leading to improved OV spread, increased bystander killing of cancer cells, and enhanced antitumor immunity [304].
Combinations of OV with small-molecule modulator-containing cocktails is a highly effective strategy in modulating the TME and enhancing therapeutic efficacy. For example, Kalinski and colleagues demonstrated that IFN-α and polyinosinic: polycytidylic acid (p-I:C) synergize with the ‘classical’ type-1-polarizing cytokine cocktail (TNFα/IL-1β), allowing for serum-free generation of fully mature type-1-polarized DCs [305]. Further studies indicated that such polarized DCs produce much higher levels of IL-12p70 and induces up to a 40-fold increase in long-lived CTLs specific for melanoma-associated antigens. Later, the slightly modified triple cocktail (IFN-α, poly I:C, and a COX-2 inhibitor), termed chemokine modulating cocktail (CKM), was used for a series of preclinical and clinical studies [306]. The sequential treatment with an OV followed by CKM resulted in the upregulation of Th1-attracting cytokines (CKs) and reduction of Treg-attracting CKs (CCL22 and CXCL12), concurrent with enhanced trafficking of tumor-specific CD8+ T cells and NK cells into the TME. As a result, we observed highly significant antitumor activity and long-term survival of tumor-bearing mice [58]. Additionally, rapamycin and celecoxib have been evaluated in combination with OV and adoptive T cell transfer (ACT) [307]. The authors hypothesized that the combination of local tumor-specific T cells activation delivered alongside an anti-immunosuppressant would improve therapy for gliomas. They utilized an IL15Rα-IL15-encoding OV as the T-cell activating stimulus and a prostaglandin synthesis inhibitor as the anti-immunosuppressant, together with ACT of tumor-specific T cells. IL15Rα-IL15-armed OVs, in conjunction with ACT, rapamycin, and celecoxib provide potent antitumor effects against brain tumors, suggesting that complex but rationally designed therapeutic combinations can produce robust anti-tumor effects.
OV in combination with other regimens
Conventional approaches to cancer therapy, such as chemotherapy, radiotherapy, and other types of immunotherapy have been well studied and combining OV with these classes of antitumor agents have been widely investigated. As these are outside the scope of this review, we will only briefly discuss these strategies. Readers are referred to recent reviews that have included more intensive discussion [6, 7, 9, 308].
Some traditional chemotherapeutic and targeted agents are now known to function, at least in part, through immunologic mechanisms [309]. If a particular chemo- agent and a particular OV work to target different immune cells and/or mechanisms that are favorable for antitumor immunity, it is likely that they may act synergistically to potentiate antitumor immunity and therapeutic efficacy.
In addition to surgery, radiotherapy remains the preferred treatment for locoregional tumors. Radiation therapy and chemotherapy are designed to target cancer cells by compromising cellular integrity during cell division. However, these agents can also induce immune modulation that can either impede or augment overall therapeutic efficacy. The impact of radiotherapy, as well as chemotherapy, on the immune system depends highly on context, making it challenging but imperative to understand how each cytotoxic therapy may compromise immune function [310].
It is interesting to note that radiotherapy has been used as a tool to elicit changes in clinically actionable signaling pathways in cancer [311] which can serve as targets for small molecule inhibitors in combination therapy. In this regard, radiotherapy and OV combinations have been evaluated, with some important preclinical observations that may support further study. Importantly, some armed OVs can replicate and express genes of interest selectively in tumor cells, thus improving noninvasive precision molecular imaging and radiotherapy [312]. Oncolytic VVs in combination with radiation have been extensively studied in tumor models [313,314,315,316]. Stereotactic body radiation combined with oncolytic VV induces potent anti-tumor effects by triggering tumor cell necroptosis and DAMPs, markers of ICD [317].
In another study, Delta-24-RGD has been combined with radiotherapy in diffuse intrinsic pontine glioma and pediatric high grade glioma models [318]. The combination led to synergistic anti-glioma effects. Interestingly, OV treatment led to the downregulation of relevant DNA damage repair proteins, further sensitizing tumors cells to radiotherapy. This is a rational combination and serves as a guide for clinical studies. Considering the clinical context and potential optimism for this combination approach, a recent clinical trial with oncolytic DNX-2401 followed by radiotherapy in pediatric patients with diffuse intrinsic pontine glioma [319] produced early promising outcomes. A total of 12 patients received up to 1 × 1010 or 5 × 1010 VP of DNX-2401 and 11 received subsequent radiotherapy. Intra-tumoral infusion of OV followed by radiotherapy resulted in changes in T-cell activity and a reduction in or stabilization of tumor size in some patients but was associated with adverse events.
Clinical studies of combination strategy
We have conducted an extensive survey of the literature and ongoing clinical trials (clinicaltrials.gov). Only a few current clinical trials meet the specified criteria and involve delivery of OV with small molecules targeting key signaling pathways. These trials are listed in Table 5. From these analyses, five types of combinations were identified: OV combined with RTKi (sorafenib or apatinib), non-receptor TKI (Ruxolitinib, an inhibitor to Janus kinase, or JAK), irinotecan (topoisomerase inhibitor), or cyclophosphamide (immunomodulatory agent), or traditional chemotherapeutic agents (Carfilzomib; Gemcitabine; Nab-paclitaxel).
It is worth emphasizing that ICI has now become standard of care for a variety of cancers. Many preclinical studies evaluating the combination of OVs with anti-PD-1/PD-L1 or anti-CTLA4 monoclonal antibodies have been performed with impressive therapeutic efficacy. Two clinical studies on melanoma patients, using T-VEC and either anti-PD-1 or anti-CTLA-4 antibody, have demonstrated striking efficacy for this combinatorial approach [55, 105]. However, as previously noted a phase III trial with anti-PD-1 and T-VEC failed to achieve better clinical response than anti-PD-1 alone in melanoma [96]. So far, small molecule inhibitors for checkpoint molecules PD-L1, MAP4K1, CBL-B and SHP2 are being investigated in clinical studies (Table 3). As such, the combination of an OV with small molecule ICI should be initiated within next few years.
While clinical trials combining OV with small molecule inhibitors are currently ongoing, the definitive data on their toxicity still await publication. Previous trials using OV or small molecule inhibitors as single agents generally indicate limited OV toxicity while small molecule inhibitors tend to induce more toxicities. For this reason, we expect toxicities resulting from these combination regimens to be mostly driven by the small molecule component, although some toxicities may expect to be potentiated by OV-driven effects. Therefore, successful combination therapies may require careful consideration and selection of small molecule inhibitor(s) with known activity/efficacy and limited toxicity as important criteria.
Conclusions and perspectives
The clinical success of anti-CTLA4 and anti-PD-1/PD-L1 ICIs has led to vast expansion of the immuno-oncology field and the combination approaches to cancer therapy currently under investigation. Among them, the use of OV is an exciting and emerging branch, with three OVs showing clear efficacy in three cancer indications that led to subsequent approval in three countries. OVs work through multiple mechanisms, and act locally but function systemically via adaptive antitumor immunity. On the other hand, the development of small molecules targeting key signaling pathways continues to play an expanding role in immuno-oncology, however in most cases their efficacy as monotherapies has been limited. Logically, these two classes of antitumor agents can be combined, likely by using mechanistic insights to guide rational approaches to further improve treatment efficacy. Preclinical studies have demonstrated that combining OVs with small molecule modulators of key signaling pathways in cancer- and/or immune cells can have enhanced therapeutic efficacy (Figs. 2 and 3). Ongoing and upcoming clinical studies of these rational combinations will ultimately determine their relevance for treating human cancer.
Multiple factors are likely to determine the efficacy of these combinations. First, oncogenic signaling pathway have emerged as key targets for small molecules. These signaling pathways (such as AKT-PI3K-mTOR, KRAS-ERK/MAPK) play important functional roles not only in tumor development and progression, but also essential roles in non-transformed healthy cells, including immune cells. In this regard, the potential interactions between small molecule inhibitors, cancer cells, and immune cells may be complex and, in some cases, result in opposing effects. Importantly, the inhibitor may not only inhibit or kill cancer cells, but also suppress functions of immune cells, resulting in antagonistic, rather than synergistic or additive action. For example, mTOR, a vital sensor of signals within the immune microenvironment, is a central regulator of T cell biology [320]. Thus, mTOR inhibition may lead to disastrous effects and abrogate antitumor immunity. Extreme caution needs to be exercised when employing such inhibitors as part of a combination regimen, with careful understanding of the mechanistic impacts considered. Second, as many of the targetable signaling pathways are essential in normal cells, the potential toxicity induced by these signaling inhibitors will need to be carefully evaluated, monitored, and/or mitigated. Third, how the activity of small molecules intersect with the biological effects of OV need to be carefully evaluated. Specifically, if a small molecule induces cancer cell death prior to productive infection/replication by OV that is sufficient to produce robust oncolysis, then these agents may again functionally antagonize and limit overall therapeutic efficacy. Fourth, inhibitors specific for a mutated signaling molecule in cancer cells, such as KRASG12C, are ideal ones to be explored in combination regimens, as they may provide much higher specificity for tumor targeting and thus would be anticipated to limit toxicity. The recently approved small molecule drug, Sotorasib, a highly specific inhibitor for KRASG12C protein, is such an example [137]. Our unpublished study showed that the combination of a small molecule inhibitor of KRASG12C with an OV elicited potent antitumor immunity and led to regression of tumors with KRASG12C-mutant protein (Zhu Z et al., submitted for publication). Lastly, depending on the mechanism of action driving potential synergy between agents, the timing and order of the treatments could have significant effects on treatment outcome [4] and may directly determine whether the combined effects are synergistic/additive, or potentially reverse any beneficial effects. For example, it has been shown that sequential administration with OV Pexa-Vec (JX-594), followed by Sorafenib, results in better efficacy in HCC [321]. In that study, a potential problem with this combination emerged where if given simultaneously, sorafenib could block Pexa-Vec replication through RAF inhibition. The study’s authors explored simultaneous and sequential combinations in vitro and in animal tumor models, finding that the regimen of Pexa-Vec followed by sorafenib was statistically superior to reverse approach. Importantly, additional well designed mechanistic studies are needed to identify optimal combinations, as the combination of two classes of agents may produce novel and/or unexpected mechanisms of action at the molecular, cellular, and/or host level.
Novel experimental systems and cutting-edge technologies that can be used to dissect complex mechanism(s) of action help to facilitate the development of novel combination regimens, inform clinical studies and, eventually, can help establish new standards of treatment or therapeutic paradigms These developments include but are not limited to: 1) Three-dimensional organoids that represent a promising, near-physiological model for human cancers and tremendously support diverse potential applications in cancer research including immunotherapy and OV combinations [322], and 2) human tumor tissue explant cultures provide patient-derived models for short term studies (data available within days, rather months) and involve all components of the TME, including cancer, stroma, inflammatory lymphoid and myeloid infiltrate, as well as vasculature (although without active blood flow) [306]. The results from these simplified in vitro systems may guide in-depth in vivo studies, saving time and costs. In addition, novel tools to analyze the immune landscape of tumor tissues before and after combination therapy may provide useful data to further inform combination strategies, with single cell RNA sequencing (scRNA Seq) and proteomics providing two powerful examples. In fact, scRNA Seq has been used to analyze the immune profile in OV-immunotherapy [323]. Finally, CRISPR/Cas9 knockout of selected candidate genes provides a breakthrough technology to study gene functions in cancer in vivo [324]. More importantly, in vivo CRISPR screens have identified important regulators of antitumor immunity and candidate targets for cancer immunotherapy [325, 326]. We envision that these powerful technologies and experimental systems will strongly accelerate the identification and translation of novel combination regimens into highly effective immunotherapies for cancer patients.
At this time, while clinical trials evaluating these approaches have been limited and have generally focused on OV in combination with RTKi and other TKi, topoisomerase inhibitor, or cyclophosphamide, new opportunities are rapidly emerging. As preclinical studies have shown that inhibitors of EGFR/KRAS/MAPK, HDACi and certain metabolic enzymes effectively synergize with OVs, these approaches are likely to translate into new therapies for clinical testing. In worst cases, even if some combinations may not deem to be rational, they may still work better than either monotherapy as one study explained the superiority of many combinations of approved drugs in the absence of drug synergy or additivity [37].
With increased understanding of cancer biology, and improved small molecule inhibitor/modulators available, it will be necessary to explore additional combinations of OV with modulators of signaling pathways in future preclinical studies to identify optimal approaches. As a branch of precision personalized medicine, one of the biggest challenges is to manage the most efficient ways to identify which combination to use, when to use them, and which patients are most likely to benefit from these approaches.
Availability of data and materials
The materials supporting the conclusions of this review has been included within the article.
Abbreviations
- AdV:
-
Adenovirus
- ATM:
-
Ataxia telangiectasia mutated
- BCG:
-
Bacillus Calmette-Guérin vaccine
- CAF:
-
Carcinoma-associated fibroblasts
- CK:
-
Chemokine
- CKM:
-
Chemokine modulating cocktail
- CTL:
-
Cytotoxic T lymphocytes
- DAMP:
-
Damage-associated molecular pattern
- DC:
-
Dendritic cells
- EGFR:
-
Epidermal growth factor receptor
- 4E-BP1:
-
Eukaryotic translation initiation factor 4E-binding protein 1
- GBM:
-
Glioblastoma
- HCC:
-
Hepatocellular carcinoma
- HDAC:
-
Histone deacetylase
- HSV:
-
Herpes simplex virus
- ICD:
-
Immunogenic cell death
- ICI:
-
Immune checkpoint inhibitor
- JTKi:
-
Janus (tyrosine) kinase inhibitor
- MV:
-
Measles virus
- MDSC:
-
Myeloid-derived suppressor cells
- mTOR:
-
The mammalian target of rapamycin
- Myx:
-
GTP-binding protein MX
- NDV:
-
Newcastle disease virus
- NSCLC:
-
Non-small cell lung cancer
- OAS:
-
Oligoadenylate synthetase
- oHSV:
-
Oncolytic HSV
- OV:
-
Oncolytic virus
- PAMP:
-
Pathogen-associated molecular pattern
- Pexa-Vec:
-
Pexastimogene devacirepvec
- PD1:
-
Programmed cell death 1
- PD-L1:
-
Programmed cell death ligand-1
- PGE2:
-
Prostaglandin E2
- PI3K:
-
Phosphoinositide 3-kinases
- PKR:
-
Protein kinase R
- RCC:
-
Renal cell carcinoma
- ROS:
-
Reactive oxygen species
- RTK:
-
Receptor tyrosine kinase
- STING:
-
Stimulator of interferon genes
- TAM:
-
tumor-associated macrophages
- TGF-β:
-
Transforming growth factor-β
- TME:
-
Tumor microenvironment
- TKI:
-
Tyrosine kinase inhibitor
- T-VEC:
-
Talimogene laherparepvec
- VSV:
-
Vesicular stomatitis virus
- VV:
-
Vaccinia virus
References
Tortorella D, Gewurz BE, Furman MH, Schust DJ, Ploegh HL. Viral subversion of the immune system. Annu Rev Immunol. 2000;18:861–926.
Duggal NK, Emerman M. Evolutionary conflicts between viruses and restriction factors shape immunity. Nat Rev Immunol. 2012;12:687–95.
Kelly E, Russell SJ. History of oncolytic viruses: genesis to genetic engineering. Mol Ther. 2007;15:651–9.
Ottolino-Perry K, Diallo JS, Lichty BD, Bell JC, McCart JA. Intelligent design: combination therapy with oncolytic viruses. Mol Ther. 2010;18:251–63.
Guo ZS, Bartlett DL. Oncolytic viruses as platform for multimodal cancer therapeutics: a promising land. Cancer Gene Ther. 2014;21:261–3.
Bommareddy PK, Shettigar M, Kaufman HL. Integrating oncolytic viruses in combination cancer immunotherapy. Nat Rev Immunol. 2018;18:498–513.
Twumasi-Boateng K, Pettigrew JL, Kwok YYE, Bell JC, Nelson BH. Oncolytic viruses as engineering platforms for combination immunotherapy. Nat Rev Cancer. 2018;18:419–32.
Martin NT, Bell JC. Oncolytic virus combination therapy: killing one bird with two stones. Mol Ther. 2018;26:1414–22.
Zhang B, Cheng P. Improving antitumor efficacy via combinatorial regimens of oncolytic virotherapy. Mol Cancer. 2020;19:158.
Achard C, Surendran A, Wedge ME, Ungerechts G, Bell J, Ilkow CS. Lighting a fire in the tumor microenvironment using oncolytic immunotherapy. EBioMedicine. 2018;31:17–24.
Wang L, Chard Dunmall LS, Cheng Z, Wang Y. Remodeling the tumor microenvironment by oncolytic viruses: beyond oncolysis of tumor cells for cancer treatment. J Immunother Cancer. 2022;10:e004167.
Harrington K, Freeman DJ, Kelly B, Harper J, Soria JC. Optimizing oncolytic virotherapy in cancer treatment. Nat Rev Drug Discov. 2019;18:689–706.
Lawler SE, Speranza MC, Cho CF, Chiocca EA. Oncolytic viruses in cancer treatment: a review. JAMA Oncol. 2017;3:841–9.
Hemminki O, Dos Santos JM, Hemminki A. Oncolytic viruses for cancer immunotherapy. J Hematol Oncol. 2020;13:84.
Macedo N, Miller DM, Haq R, Kaufman HL. Clinical landscape of oncolytic virus research in 2020. J Immunother Cancer. 2020;8:e001486.
Bedard PL, Hyman DM, Davids MS, Siu LL. Small molecules, big impact: 20 years of targeted therapy in oncology. Lancet. 2020;395:1078–88.
Zhong L, Li Y, Xiong L, Wang W, Wu M, Yuan T, et al. Small molecules in targeted cancer therapy: advances, challenges, and future perspectives. Signal Transduct Target Ther. 2021;6:201.
Offringa R, Kotzner L, Huck B, Urbahns K. The expanding role for small molecules in immuno-oncology. Nat Rev Drug Discov. 2022. https://doi.org/10.1038/s41573-022-00538-9.
Hossain DMS, Javaid S, Cai M, Zhang C, Sawant A, Hinton M, et al. Dinaciclib induces immunogenic cell death and enhances anti-PD1-mediated tumor suppression. J Clin Invest. 2018;128:644–54.
Liu P, Zhao L, Pol J, Levesque S, Petrazzuolo A, Pfirschke C, et al. Crizotinib-induced immunogenic cell death in non-small cell lung cancer. Nat Commun. 2019;10:1486.
Huang KC, Chiang SF, Yang PC, Ke TW, Chen TW, Hu CH, et al. Immunogenic cell death by the novel topoisomerase I inhibitor TLC388 enhances the therapeutic efficacy of radiotherapy. Cancers (Basel). 2021;13:1218.
San Jose-Eneriz E, Agirre X, Rabal O, Vilas-Zornoza A, Sanchez-Arias JA, Miranda E, et al. Discovery of first-in-class reversible dual small molecule inhibitors against G9a and DNMTs in hematological malignancies. Nat Commun. 2017;8:15424.
Gross S, Rahal R, Stransky N, Lengauer C, Hoeflich KP. Targeting cancer with kinase inhibitors. J Clin Invest. 2015;125:1780–9.
Schaer DA, Beckmann RP, Dempsey JA, Huber L, Forest A, Amaladas N, et al. The CDK4/6 inhibitor abemaciclib induces a T cell inflamed tumor microenvironment and enhances the efficacy of PD-L1 checkpoint blockade. Cell Rep. 2018;22:2978–94.
Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science. 2020;368:eaaw5473.
Suva ML, Riggi N, Bernstein BE. Epigenetic reprogramming in cancer. Science. 2013;339:1567–70.
Topper MJ, Vaz M, Marrone KA, Brahmer JR, Baylin SB. The emerging role of epigenetic therapeutics in immuno-oncology. Nat Rev Clin Oncol. 2020;17:75–90.
Dai E, Zhu Z, Wahed S, Qu Z, Storkus WJ, Guo ZS. Epigenetic modulation of antitumor immunity for improved cancer immunotherapy. Mol Cancer. 2021;20:171.
Bartlett DL, Liu Z, Sathaiah M, Ravindranathan R, Guo Z, He Y, et al. Oncolytic viruses as therapeutic cancer vaccines. Mol Cancer. 2013;12:103.
Guo ZS, Liu Z, Bartlett DL. Oncolytic immunotherapy: dying the right way is a key to eliciting potent antitumor immunity. Front Oncol. 2014;4:74.
Guo ZS, Lu B, Guo Z, Giehl E, Feist M, Dai E, et al. Vaccinia virus-mediated cancer immunotherapy: cancer vaccines and oncolytics. J Immunother Cancer. 2019;7:6.
Hoelder S, Clarke PA, Workman P. Discovery of small molecule cancer drugs: successes, challenges and opportunities. Mol Oncol. 2012;6:155–76.
Falzone L, Salomone S, Libra M. Evolution of cancer pharmacological treatments at the turn of the third millennium. Front Pharmacol. 2018;9:1300.
Bayat Mokhtari R, Homayouni TS, Baluch N, Morgatskaya E, Kumar S, Das B, et al. Combination therapy in combating cancer. Oncotarget. 2017;8:38022–43.
Murciano-Goroff YR, Warner AB, Wolchok JD. The future of cancer immunotherapy: microenvironment-targeting combinations. Cell Res. 2020;30:507–19.
Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. 2018;15:81–94.
Palmer AC, Sorger PK. Combination cancer therapy can confer benefit via patient-to-patient variability without drug additivity or synergy. Cell. 2017;171:1678–1691 e13.
Guo ZS, Thorne SH, Bartlett DL. Oncolytic virotherapy: molecular targets in tumor-selective replication and carrier cell-mediated delivery of oncolytic viruses. Biochim Biophys Acta. 2008;1785:217–31.
Howells A, Marelli G, Lemoine NR, Wang Y. Oncolytic viruses-interaction of virus and tumor cells in the battle to eliminate cancer. Front Oncol. 2017;7:195.
Jhawar SR, Thandoni A, Bommareddy PK, Hassan S, Kohlhapp FJ, Goyal S, et al. Oncolytic viruses-natural and genetically engineered cancer immunotherapies. Front Oncol. 2017;7:202.
Strong JE, Coffey MC, Tang D, Sabinin P, Lee PW. The molecular basis of viral oncolysis: usurpation of the Ras signaling pathway by reovirus. EMBO J. 1998;17:3351–62.
Parato KA, Breitbach CJ, Le Boeuf F, Wang J, Storbeck C, Ilkow C, et al. The oncolytic poxvirus JX-594 selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers. Mol Ther. 2012;20:749–58.
Breitbach CJ, De Silva NS, Falls TJ, Aladl U, Evgin L, Paterson J, et al. Targeting tumor vasculature with an oncolytic virus. Mol Ther. 2011;19:886–94.
Yu F, Hong B, Song XT. A T-cell engager-armed oncolytic vaccinia virus to target the tumor stroma. Cancer Translat Med. 2017;3:122.
Freedman JD, Duffy MR, Lei-Rossmann J, Muntzer A, Scott EM, Hagel J, et al. An oncolytic virus expressing a T-cell engager simultaneously targets cancer and immunosuppressive stromal cells. Cancer Res. 2018;78:6852–65.
de Sostoa J, Fajardo CA, Moreno R, Ramos MD, Farrera-Sal M, Alemany R. Targeting the tumor stroma with an oncolytic adenovirus secreting a fibroblast activation protein-targeted bispecific T-cell engager. J Immunother Cancer. 2019;7:19.
Zhang B. Targeting the stroma by T cells to limit tumor growth. Cancer Res. 2008;68:9570–3.
Gujar S, Bell J, Diallo JS. SnapShot: cancer immunotherapy with oncolytic viruses. Cell. 2019;176:1240–1240 e1.
Workenhe ST, Mossman KL. Oncolytic virotherapy and immunogenic cancer cell death: sharpening the sword for improved cancer treatment strategies. Mol Ther. 2014;22:251–6.
Toda M, Rabkin SD, Kojima H, Martuza RL. Herpes simplex virus as an in situ cancer vaccine for the induction of specific anti-tumor immunity. Hum Gene Ther. 1999;10:385–93.
Li QX, Liu G, Wong-Staal F. Oncolytic virotherapy as a personalized cancer vaccine. Int J Cancer. 2008;123:493–9.
Aitken AS, Roy DG, Bourgeois-Daigneault MC. Taking a stab at cancer; oncolytic virus-mediated anti-cancer vaccination strategies. Biomedicines. 2017;5:3.
Russell SJ, Barber GN. Oncolytic viruses as antigen-agnostic cancer vaccines. Cancer Cell. 2018;33:599–605.
Melcher A, Harrington K, Vile R. Oncolytic virotherapy as immunotherapy. Science. 2021;374:1325–6.
Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, et al. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-PD-1 immunotherapy. Cell. 2017;170:1109–1119 e10.
Gujar S, Pol JG, Kroemer G. Heating it up: oncolytic viruses make tumors ‘hot’ and suitable for checkpoint blockade immunotherapies. Oncoimmunology. 2018;7:e1442169.
Guo ZS, Naik A, O'Malley ME, Popovic P, Demarco R, Hu Y, et al. The enhanced tumor selectivity of an oncolytic vaccinia lacking the host range and antiapoptosis genes SPI-1 and SPI-2. Cancer Res. 2005;65:9991–8.
Francis L, Guo ZS, Liu Z, Ravindranathan R, Urban JA, Sathaiah M, et al. Modulation of chemokines in the tumor microenvironment enhances oncolytic virotherapy for colorectal cancer. Oncotarget. 2016;7:22174–85.
Liu Z, Ravindranathan R, Kalinski P, Guo ZS, Bartlett DL. Rational combination of oncolytic vaccinia virus and PD-L1 blockade works synergistically to enhance therapeutic efficacy. Nat Commun. 2017;8:14754.
Feist M, Zhu Z, Dai E, Ma C, Liu Z, Giehl E, et al. Oncolytic virus promotes tumor-reactive infiltrating lymphocytes for adoptive cell therapy. Cancer Gene Ther. 2021;28:98–111.
Donnelly OG, Errington-Mais F, Steele L, Hadac E, Jennings V, Scott K, et al. Measles virus causes immunogenic cell death in human melanoma. Gene Ther. 2013;20:7–15.
Ma J, Ramachandran M, Jin C, Quijano-Rubio C, Martikainen M, Yu D, et al. Characterization of virus-mediated immunogenic cancer cell death and the consequences for oncolytic virus-based immunotherapy of cancer. Cell Death Dis. 2020;11:48.
Nishio N, Diaconu I, Liu H, Cerullo V, Caruana I, Hoyos V, et al. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res. 2014;74:5195–205.
Ge Y, Wang H, Ren J, Liu W, Chen L, Chen H, et al. Oncolytic vaccinia virus delivering tethered IL-12 enhances antitumor effects with improved safety. J Immunother Cancer. 2020;8:e0007.
Pearl TM, Markert JM, Cassady KA, Ghonime MG. Oncolytic virus-based cytokine expression to improve immune activity in brain and solid tumors. Mol Ther Oncolytics. 2019;13:14–21.
Mastrangelo MJ, Maguire HC Jr, Eisenlohr LC, Laughlin CE, Monken CE, McCue PA, et al. Intratumoral recombinant GM-CSF-encoding virus as gene therapy in patients with cutaneous melanoma. Cancer Gene Ther. 1999;6:409–22.
Carew JF, Kooby DA, Halterman MW, Kim SH, Federoff HJ, Fong Y. A novel approach to cancer therapy using an oncolytic herpes virus to package amplicons containing cytokine genes. Mol Ther. 2001;4:250–6.
Chard LS, Maniati E, Wang P, Zhang Z, Gao D, Wang J, et al. A vaccinia virus armed with interleukin-10 is a promising therapeutic agent for treatment of murine pancreatic cancer. Clin Cancer Res. 2015;21:405–16.
Chen D, Huang L, Zhou H, Zhang Y. Combining IL-10 and oncolytic adenovirus demonstrates enhanced antitumor efficacy through CD8(+) T cells. Front Immunol. 2021;12:615089.
Deng L, Yang X, Fan J, Ding Y, Peng Y, Xu D, et al. IL-24-armed oncolytic vaccinia virus exerts potent antitumor effects via multiple pathways in colorectal cancer. Oncol Res. 2021;28:579–90.
Hu HJ, Liang X, Li HL, Wang HY, Gu JF, Sun LY, et al. Enhanced anti-melanoma efficacy through a combination of the armed oncolytic adenovirus ZD55-IL-24 and immune checkpoint blockade in B16-bearing immunocompetent mouse model. Cancer Immunol Immunother. 2021;70:3541–55.
Liu Z, Ge Y, Wang H, Ma C, Feist M, Ju S, et al. Modifying the cancer-immune set point using vaccinia virus expressing re-designed interleukin-2. Nat Commun. 2018;9:4682.
Kowalsky SJ, Liu Z, Feist M, Berkey SE, Ma C, Ravindranathan R, et al. Superagonist IL-15-armed oncolytic virus elicits potent antitumor immunity and therapy that are enhanced with PD-1 blockade. Mol Ther. 2018;26:2476–86.
Yang M, Giehl E, Feng C, Feist M, Chen H, Dai E, et al. IL-36gamma-armed oncolytic virus exerts superior efficacy through induction of potent adaptive antitumor immunity. Cancer Immunol Immunother. 2021;70:2467–81.
Chen L, Chen H, Ye J, Ge Y, Wang H, Dai E, et al. Intratumoral expression of interleukin 23 variants using oncolytic vaccinia virus elicit potent antitumor effects on multiple tumor models via tumor microenvironment modulation. Theranostics. 2021;11:6668–81.
Chen T, Ding X, Liao Q, Gao N, Chen Y, Zhao C, et al. IL-21 arming potentiates the anti-tumor activity of an oncolytic vaccinia virus in monotherapy and combination therapy. J Immunother Cancer. 2021;9:e0001647.
Wang N, Wang J, Zhang Z, Cao H, Yan W, Chu Y, et al. A novel vaccinia virus enhances anti-tumor efficacy and promotes a long-term anti-tumor response in a murine model of colorectal cancer. Mol Ther Oncolytics. 2021;20:71–81.
Nakao S, Arai Y, Tasaki M, Yamashita M, Murakami R, Kawase T, et al. Intratumoral expression of IL-7 and IL-12 using an oncolytic virus increases systemic sensitivity to immune checkpoint blockade. Sci Transl Med. 2020;12:eaax7992.
Tasaki M, Yamashita M, Arai Y, Nakamura T, Nakao S. IL-7 coupled with IL-12 increases intratumoral T cell clonality, leading to complete regression of non-immunogenic tumors. Cancer Immunol Immunother. 2021;70:3557–71.
Zamarin D, Holmgaard RB, Ricca J, Plitt T, Palese P, Sharma P, et al. Intratumoral modulation of the inducible co-stimulator ICOS by recombinant oncolytic virus promotes systemic anti-tumour immunity. Nat Commun. 2017;8:14340.
Lin C, Ren W, Luo Y, Li S, Chang Y, Li L, et al. Intratumoral delivery of a PD-1-blocking scFv encoded in oncolytic HSV-1 promotes antitumor immunity and synergizes with TIGIT blockade. Cancer Immunol Res. 2020;8:632–47.
Wang G, Kang X, Chen KS, Jehng T, Jones L, Chen J, et al. An engineered oncolytic virus expressing PD-L1 inhibitors activates tumor neoantigen-specific T cell responses. Nat Commun. 2020;11:1395.
Xu B, Tian L, Chen J, Wang J, Ma R, Dong W, et al. An oncolytic virus expressing a full-length antibody enhances antitumor innate immune response to glioblastoma. Nat Commun. 2021;12:5908.
Tian L, Xu B, Teng KY, Song M, Zhu Z, Chen Y, et al. Targeting fc receptor-mediated effects and the “Don‘t eat me” signal with an oncolytic virus expressing an anti-CD47 antibody to treat metastatic ovarian cancer. Clin Cancer Res. 2022;28:201–14.
McGray AJR, Huang RY, Battaglia S, Eppolito C, Miliotto A, Stephenson KB, et al. Oncolytic Maraba virus armed with tumor antigen boosts vaccine priming and reveals diverse therapeutic response patterns when combined with checkpoint blockade in ovarian cancer. J Immunother Cancer. 2019;7:189.
Gil M, Seshadri M, Komorowski MP, Abrams SI, Kozbor D. Targeting CXCL12/CXCR4 signaling with oncolytic virotherapy disrupts tumor vasculature and inhibits breast cancer metastases. Proc Natl Acad Sci U S A. 2013;110:E1291–300.
Komorowski MP, McGray AR, Kolakowska A, Eng K, Gil M, Opyrchal M, et al. Reprogramming antitumor immunity against chemoresistant ovarian cancer by a CXCR4 antagonist-armed viral oncotherapy. Mol Ther Oncolytics. 2016;3:16034.
Yu F, Wang X, Guo ZS, Bartlett DL, Gottschalk SM, Song XT. T-cell engager-armed oncolytic vaccinia virus significantly enhances antitumor therapy. Mol Ther. 2014;22:102–11.
Guo ZS, Lotze MT, Zhu Z, Storkus WJ, Song XT. Bi- and tri-specific T cell engager-armed oncolytic viruses: next-generation cancer immunotherapy. Biomedicines. 2020;8:204.
Heidbuechel JPW, Engeland CE. Oncolytic viruses encoding bispecific T cell engagers: a blueprint for emerging immunovirotherapies. J Hematol Oncol. 2021;14:63.
Frampton JE. Teserpaturev/G47Delta: First Approval. BioDrugs. 2022;36:667–72.
Kaufman HL, Kim DW, DeRaffele G, Mitcham J, Coffin RS, Kim-Schulze S. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann Surg Oncol. 2010;17:718–30.
Kohlhapp FJ, Kaufman HL. Molecular pathways: mechanism of action for Talimogene Laherparepvec, a new oncolytic virus immunotherapy. Clin Cancer Res. 2016;22:1048–54.
Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33:2780–8.
Ressler JM, Karasek M, Koch L, Silmbrod R, Mangana J, Latifyan S, et al. Real-life use of talimogene laherparepvec (T-VEC) in melanoma patients in centers in Austria, Switzerland and Germany. J Immunother Cancer. 2021;9:e001701.
Chesney JA, Ribas A, Long GV, Kirkwood JM, Dummer R, Puzanov I, et al. Randomized, double-blind, placebo-controlled, global phase III trial of Talimogene Laherparepvec combined with Pembrolizumab for advanced melanoma. J Clin Oncol. 2022;JCO2200343. https://doi.org/10.1200/JCO.22.00343.
Packiam VT, Lamm DL, Barocas DA, Trainer A, Fand B, Davis RL 3rd, et al. An open label, single-arm, phase II multicenter study of the safety and efficacy of CG0070 oncolytic vector regimen in patients with BCG-unresponsive non-muscle-invasive bladder cancer: interim results. Urol Oncol. 2018;36:440–7.
Li R, Steinberg GD, Uchio EM, Lamm DL, Shah P, Kamat AM, et al. CORE1: phase 2, single-arm study of CG0070 combined with pembrolizumab in patients with nonmuscle-invasive bladder cancer (NMIBC) unresponsive to bacillus Calmette-Guerin (BCG). J Clin Oncol. 2022;40:4597.
Heo J, Reid T, Ruo L, Breitbach CJ, Rose S, Bloomston M, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med. 2013;19:329–36.
Monge BMC, Xie C, Steinberg SM, Fioraventi S, Walker M, Mabry-Hrones D, et al. A phase I/II study of Pexa-Vec oncolytic virus in combination with immune checkpoint inhibition in refractory colorectal cancer. J Clin Oncol. 2020;38(4_suppl):117.
Lun X, Chan J, Zhou H, Sun B, Kelly JJ, Stechishin OO, et al. Efficacy and safety/toxicity study of recombinant vaccinia virus JX-594 in two immunocompetent animal models of glioma. Mol Ther. 2010;18:1927–36.
Tang B, Guo ZS, Bartlett DL, Liu J, McFadden G, Shisler JL, et al. A cautionary note on the selectivity of oncolytic poxviruses. Oncolytic Virother. 2019;8:3–8.
Muik A, Stubbert LJ, Jahedi RZ, Geibeta Y, Kimpel J, Dold C, et al. Re-engineering vesicular stomatitis virus to abrogate neurotoxicity, circumvent humoral immunity, and enhance oncolytic potency. Cancer Res. 2014;74:3567–78.
Corrigan PA, Beaulieu C, Patel RB, Lowe DK. Talimogene Laherparepvec: an oncolytic virus therapy for melanoma. Ann Pharmacother. 2017;51:675–81.
Chesney J, Puzanov I, Collichio F, Singh P, Milhem MM, Glaspy J, et al. Randomized, open-label phase II study evaluating the efficacy and safety of Talimogene Laherparepvec in combination with Ipilimumab versus Ipilimumab alone in patients with advanced, unresectable melanoma. J Clin Oncol. 2018;36:1658–67.
Kaufman HL. Can biomarkers guide oncolytic virus immunotherapy? Clin Cancer Res. 2021;27:3278–9.
Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. 2022;12:31–46.
Matveeva OV, Chumakov PM. Defects in interferon pathways as potential biomarkers of sensitivity to oncolytic viruses. Rev Med Virol. 2018;28:e2008.
Shmulevitz M, Marcato P, Lee PW. Unshackling the links between reovirus oncolysis, Ras signaling, translational control and cancer. Oncogene. 2005;24:7720–8.
Bergmann M, Romirer I, Sachet M, Fleischhacker R, Garcia-Sastre A, Palese P, et al. A genetically engineered influenza A virus with ras-dependent oncolytic properties. Cancer Res. 2001;61:8188–93.
Deng H, Liu H, de Silva T, Xue Y, Mohamud Y, Ng CS, et al. Coxsackievirus type B3 is a potent oncolytic virus against KRAS-mutant lung adenocarcinoma. Mol Ther Oncolytics. 2019;14:266–78.
Cai J, Lin K, Cai W, Lin Y, Liu X, Guo L, et al. Tumors driven by RAS signaling harbor a natural vulnerability to oncolytic virus M1. Mol Oncol. 2020;14:3153–68.
Cai J, Yan G. The identification and development of a novel oncolytic virus: alphavirus M1. Hum Gene Ther. 2021;32:138–49.
Zloza A, Kim DW, Kim-Schulze S, Jagoda MC, Monsurro V, Marincola FM, et al. Immunoglobulin-like transcript 2 (ILT2) is a biomarker of therapeutic response to oncolytic immunotherapy with vaccinia viruses. J Immunother Cancer. 2014;2:1.
Liikanen I, Koski A, Merisalo-Soikkeli M, Hemminki O, Oksanen M, Kairemo K, et al. Serum HMGB1 is a predictive and prognostic biomarker for oncolytic immunotherapy. Oncoimmunology. 2015;4:e989771.
Nguyen TT, Ramsay L, Ahanfeshar-Adams M, Lajoie M, Schadendorf D, Alain T, et al. Mutations in the IFNgamma-JAK-STAT pathway causing resistance to immune checkpoint inhibitors in melanoma increase sensitivity to oncolytic virus treatment. Clin Cancer Res. 2021;27:3432–42.
Song D, Jia X, Liu X, Hu L, Lin K, Xiao T, et al. Identification of the receptor of oncolytic virus M1 as a therapeutic predictor for multiple solid tumors. Signal Transduct Target Ther. 2022;7:100.
Sever R, Brugge JS. Signal transduction in cancer. Cold Spring Harb Perspect Med. 2015;5:a006098.
Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23:27–47.
Martinez-Reyes I, Chandel NS. Cancer metabolism: looking forward. Nat Rev Cancer. 2021;21:669–80.
Biswas SK. Metabolic reprogramming of immune cells in cancer progression. Immunity. 2015;43:435–49.
Osipov A, Saung MT, Zheng L, Murphy AG. Small molecule immunomodulation: the tumor microenvironment and overcoming immune escape. J Immunother Cancer. 2019;7:224.
Kerr WG, Chisholm JD. The next generation of immunotherapy for cancer: small molecules could make big waves. J Immunol. 2019;202:11–9.
Labani-Motlagh A, Ashja-Mahdavi M, Loskog A. The tumor microenvironment: a milieu hindering and obstructing antitumor immune responses. Front Immunol. 2020;11:940.
Sun G, Rong D, Li Z, Sun G, Wu F, Li X, et al. Role of small molecule targeted compounds in cancer: progress, opportunities, and challenges. Front Cell Dev Biol. 2021;9:694363.
Spiesschaert B, Angerer K, Park J, Wollmann G. Combining oncolytic viruses and small molecule therapeutics: mutual benefits. Cancers (Basel). 2021;13:3386.
Nicholson RI, Gee JM, Harper ME. EGFR and cancer prognosis. Eur J Cancer. 2001;37(Suppl 4):S9–15.
Roskoski R Jr. Small molecule inhibitors targeting the EGFR/ErbB family of protein-tyrosine kinases in human cancers. Pharmacol Res. 2019;139:395–411.
Klein Hesselink EN, Steenvoorden D, Kapiteijn E, Corssmit EP, van der Horst-Schrivers AN, Lefrandt JD, et al. Therapy of endocrine disease: response and toxicity of small-molecule tyrosine kinase inhibitors in patients with thyroid carcinoma: a systematic review and meta-analysis. Eur J Endocrinol. 2015;172:R215–25.
Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D, et al. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006;66:11851–8.
Athuluri-Divakar SK, Vasquez-Del Carpio R, Dutta K, Baker SJ, Cosenza SC, Basu I, et al. A small molecule RAS-mimetic disrupts RAS Association with effector proteins to block signaling. Cell. 2016;165:643–55.
Yan C, Saleh N, Yang J, Nebhan CA, Vilgelm AE, Reddy EP, et al. Novel induction of CD40 expression by tumor cells with RAS/RAF/PI3K pathway inhibition augments response to checkpoint blockade. Mol Cancer. 2021;20:85.
Huang L, Guo Z, Wang F, Fu L. KRAS mutation: from undruggable to druggable in cancer. Signal Transduct Target Ther. 2021;6:386.
Lanman BA, Allen JR, Allen JG, Amegadzie AK, Ashton KS, Booker SK, et al. Discovery of a covalent inhibitor of KRAS(G12C) (AMG 510) for the treatment of solid tumors. J Med Chem. 2020;63:52–65.
Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575:217–23.
Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRAS(G12C) inhibition with Sotorasib in advanced solid tumors. N Engl J Med. 2020;383:1207–17.
Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, et al. Sotorasib for lung cancers with KRAS p.G12C mutation. N Engl J Med. 2021;384:2371–81.
Ou SI, Janne PA, Leal TA, Rybkin II, Sabari JK, Barve MA, et al. First-in-human phase I/IB dose-finding study of Adagrasib (MRTX849) in patients with advanced KRAS(G12C) solid tumors (KRYSTAL-1). J Clin Oncol. 2022;40:2530–8.
Janne PA, Riely GJ, Gadgeel SM, Heist RS, Ou SI, Pacheco JM, et al. Adagrasib in non-small-cell lung cancer harboring a KRAS(G12C) mutation. N Engl J Med. 2022;387:120–31.
Mahller YY, Vaikunth SS, Currier MA, Miller SJ, Ripberger MC, Hsu YH, et al. Oncolytic HSV and erlotinib inhibit tumor growth and angiogenesis in a novel malignant peripheral nerve sheath tumor xenograft model. Mol Ther. 2007;15:279–86.
Yamamura K, Kasuya H, Sahin TT, Tan G, Hotta Y, Tsurumaru N, et al. Combination treatment of human pancreatic cancer xenograft models with the epidermal growth factor receptor tyrosine kinase inhibitor erlotinib and oncolytic herpes simplex virus HF10. Ann Surg Oncol. 2014;21:691–8.
Menotti L, Nicoletti G, Gatta V, Croci S, Landuzzi L, De Giovanni C, et al. Inhibition of human tumor growth in mice by an oncolytic herpes simplex virus designed to target solely HER-2-positive cells. Proc Natl Acad Sci U S A. 2009;106:9039–44.
Wu Z, Ichinose T, Naoe Y, Matsumura S, Villalobos IB, Eissa IR, et al. Combination of Cetuximab and oncolytic virus canerpaturev synergistically inhibits human colorectal cancer growth. Mol Ther Oncolytics. 2019;13:107–15.
Saha D, Wakimoto H, Peters CW, Antoszczyk SJ, Rabkin SD, Martuza RL. Combinatorial effects of VEGFR kinase inhibitor Axitinib and oncolytic virotherapy in mouse and human glioblastoma stem-like cell models. Clin Cancer Res. 2018;24:3409–22.
Shi G, Yang Q, Zhang Y, Jiang Q, Lin Y, Yang S, et al. Modulating the tumor microenvironment via oncolytic viruses and CSF-1R inhibition synergistically enhances anti-PD-1 immunotherapy. Mol Ther. 2019;27:244–60.
Jha BK, Dong B, Nguyen CT, Polyakova I, Silverman RH. Suppression of antiviral innate immunity by sunitinib enhances oncolytic virotherapy. Mol Ther. 2013;21:1749–57.
Kim M, Nitschke M, Sennino B, Murer P, Schriver BJ, Bell A, et al. Amplification of oncolytic vaccinia virus widespread tumor cell killing by Sunitinib through multiple mechanisms. Cancer Res. 2018;78:922–37.
Coffey MC, Strong JE, Forsyth PA, Lee PW. Reovirus therapy of tumors with activated Ras pathway. Science. 1998;282:1332–4.
Gong J, Mita MM. Activated ras signaling pathways and reovirus oncolysis: an update on the mechanism of preferential reovirus replication in cancer cells. Front Oncol. 2014;4:167.
Errington F, White CL, Twigger KR, Rose A, Scott K, Steele L, et al. Inflammatory tumour cell killing by oncolytic reovirus for the treatment of melanoma. Gene Ther. 2008;15:1257–70.
Subbiah V, Baik C, Kirkwood JM. Clinical development of BRAF plus MEK inhibitor combinations. Trends Cancer. 2020;6:797–810.
Roulstone V, Pedersen M, Kyula J, Mansfield D, Khan AA, McEntee G, et al. BRAF- and MEK-targeted small molecule inhibitors exert enhanced Antimelanoma effects in combination with oncolytic Reovirus through ER stress. Mol Ther. 2015;23:931–42.
Bommareddy PK, Aspromonte S, Zloza A, Rabkin SD, Kaufman HL. MEK inhibition enhances oncolytic virus immunotherapy through increased tumor cell killing and T cell activation. Sci Transl Med. 2018;10:eaau0417.
Lee S, Yang W, Kim DK, Kim H, Shin M, Choi KU, et al. Inhibition of MEK-ERK pathway enhances oncolytic vaccinia virus replication in doxorubicin-resistant ovarian cancer. Mol Ther Oncolytics. 2022;25:211–24.
Bommareddy PK, Zloza A, Rabkin SD, Kaufman HL. Oncolytic virus immunotherapy induces immunogenic cell death and overcomes STING deficiency in melanoma. Oncoimmunology. 2019;8:1591875.
Alzahrani AS. PI3K/Akt/mTOR inhibitors in cancer: at the bench and bedside. Semin Cancer Biol. 2019;59:125–32.
De Santis MC, Gulluni F, Campa CC, Martini M, Hirsch E. Targeting PI3K signaling in cancer: challenges and advances. Biochim Biophys Acta Rev Cancer. 2019;1871:361–6.
Mishra R, Patel H, Alanazi S, Kilroy MK, Garrett JT. PI3K inhibitors in cancer: clinical implications and adverse effects. Int J Mol Sci. 2021;22:3464.
He Y, Sun MM, Zhang GG, Yang J, Chen KS, Xu WW, et al. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct Target Ther. 2021;6:425.
Kaneda MM, Cappello P, Nguyen AV, Ralainirina N, Hardamon CR, Foubert P, et al. Macrophage PI3Kgamma drives pancreatic ductal adenocarcinoma progression. Cancer Discov. 2016;6:870–85.
Joshi S, Liu KX, Zulcic M, Singh AR, Skola D, Glass CK, et al. Macrophage Syk-PI3Kgamma inhibits antitumor immunity: SRX3207, a novel dual Syk-PI3K inhibitory chemotype relieves tumor immunosuppression. Mol Cancer Ther. 2020;19:755–64.
De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells. Nature. 2016;539:443–7.
Ding D, Zhong H, Liang R, Lan T, Zhu X, Huang S, et al. Multifunctional nanodrug mediates synergistic photodynamic therapy and MDSCs-targeting immunotherapy of colon cancer. Adv Sci (Weinh). 2021;8:e2100712.
Hanel W, Epperla N. Evolving therapeutic landscape in follicular lymphoma: a look at emerging and investigational therapies. J Hematol Oncol. 2021;14:104.
Andre F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS, et al. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N Engl J Med. 2019;380:1929–40.
Song M, Bode AM, Dong Z, Lee MH. AKT as a therapeutic target for cancer. Cancer Res. 2019;79:1019–31.
Yang J, Nie J, Ma X, Wei Y, Peng Y, Wei X. Targeting PI3K in cancer: mechanisms and advances in clinical trials. Mol Cancer. 2019;18:26.
Martorana F, Motta G, Pavone G, Motta L, Stella S, Vitale SR, et al. AKT inhibitors: new weapons in the fight against breast cancer? Front Pharmacol. 2021;12:662232.
Jones RH, Casbard A, Carucci M, Cox C, Butler R, Alchami F, et al. Fulvestrant plus capivasertib versus placebo after relapse or progression on an aromatase inhibitor in metastatic, oestrogen receptor-positive breast cancer (FAKTION): a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 2020;21:345–57.
Hua H, Kong Q, Zhang H, Wang J, Luo T, Jiang Y. Targeting mTOR for cancer therapy. J Hematol Oncol. 2019;12:71.
Zhang Y, Yan H, Xu Z, Yang B, Luo P, He Q. Molecular basis for class side effects associated with PI3K/AKT/mTOR pathway inhibitors. Expert Opin Drug Metab Toxicol. 2019;15:767–74.
Kanai R, Wakimoto H, Martuza RL, Rabkin SD. A novel oncolytic herpes simplex virus that synergizes with phosphoinositide 3-kinase/Akt pathway inhibitors to target glioblastoma stem cells. Clin Cancer Res. 2011;17:3686–96.
Wang L, Ning J, Wakimoto H, Wu S, Wu CL, Humphrey MR, et al. Oncolytic herpes simplex virus and PI3K inhibitor BKM120 synergize to promote killing of prostate cancer stem-like cells. Mol Ther Oncolytics. 2019;13:58–66.
Tong Y, Zhu W, Huang X, You L, Han X, Yang C, et al. PI3K inhibitor LY294002 inhibits activation of the Akt/mTOR pathway induced by an oncolytic adenovirus expressing TRAIL and sensitizes multiple myeloma cells to the oncolytic virus. Oncol Rep. 2014;31:1581–8.
Jiang K, Li Y, Zhu Q, Xu J, Wang Y, Deng W, et al. Pharmacological modulation of autophagy enhances Newcastle disease virus-mediated oncolysis in drug-resistant lung cancer cells. BMC Cancer. 2014;14:551.
Tomar VS, Patil V, Somasundaram K. Temozolomide induces activation of Wnt/beta-catenin signaling in glioma cells via PI3K/Akt pathway: implications in glioma therapy. Cell Biol Toxicol. 2020;36:273–8.
Saha D, Rabkin SD, Martuza RL. Temozolomide antagonizes oncolytic immunovirotherapy in glioblastoma. J Immunother Cancer. 2020;8:e000345.
Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med. 2019;25:462–9.
Xing F, Xiao J, Wu J, Liang J, Lu X, Guo L, et al. Modulating the tumor microenvironment via oncolytic virus and PI3K inhibition synergistically restores immune checkpoint therapy response in PTEN-deficient glioblastoma. Signal Transduct Target Ther. 2021;6:275.
Ferguson MS, Chard Dunmall LS, Gangeswaran R, Marelli G, Tysome JR, Burns E, et al. Transient inhibition of PI3Kdelta enhances the therapeutic effect of intravenous delivery of oncolytic vaccinia virus. Mol Ther. 2020;28:1263–75.
Marelli G, Chard Dunmall LS, Yuan M, Di Gioia C, Miao J, Cheng Z, et al. A systemically deliverable vaccinia virus with increased capacity for intertumoral and intratumoral spread effectively treats pancreatic cancer. J Immunother Cancer. 2021;9:e001624.
Populo H, Lopes JM, Soares P. The mTOR signalling pathway in human cancer. Int J Mol Sci. 2012;13:1886–918.
Homicsko K, Lukashev A, Iggo RD. RAD001 (everolimus) improves the efficacy of replicating adenoviruses that target colon cancer. Cancer Res. 2005;65:6882–90.
Stanford MM, Barrett JW, Nazarian SH, Werden S, McFadden G. Oncolytic virotherapy synergism with signaling inhibitors: rapamycin increases myxoma virus tropism for human tumor cells. J Virol. 2007;81:1251–60.
Lun XQ, Zhou H, Alain T, Sun B, Wang L, Barrett JW, et al. Targeting human medulloblastoma: oncolytic virotherapy with myxoma virus is enhanced by rapamycin. Cancer Res. 2007;67:8818–27.
Lun XQ, Jang JH, Tang N, Deng H, Head R, Bell JC, et al. Efficacy of systemically administered oncolytic vaccinia virotherapy for malignant gliomas is enhanced by combination therapy with rapamycin or cyclophosphamide. Clin Cancer Res. 2009;15:2777–88.
Fu X, Tao L, Rivera A, Zhang X. Rapamycin enhances the activity of oncolytic herpes simplex virus against tumor cells that are resistant to virus replication. Int J Cancer. 2011;129:1503–10.
Zakaria C, Sean P, Hoang HD, Leroux LP, Watson M, Workenhe ST, et al. Active-site mTOR inhibitors augment HSV1-dICP0 infection in cancer cells via dysregulated eIF4E/4E-BP axis. PLoS Pathog. 2018;14:e1007264.
Alain T, Lun X, Martineau Y, Sean P, Pulendran B, Petroulakis E, et al. Vesicular stomatitis virus oncolysis is potentiated by impairing mTORC1-dependent type I IFN production. Proc Natl Acad Sci U S A. 2010;107:1576–81.
Kim SY, Kang S, Song JJ, Kim JH. The effectiveness of the oncolytic activity induced by Ad5/F35 adenoviral vector is dependent on the cumulative cellular conditions of survival and autophagy. Int J Oncol. 2013;42:1337–48.
Lun X, Alain T, Zemp FJ, Zhou H, Rahman MM, Hamilton MG, et al. Myxoma virus virotherapy for glioma in immunocompetent animal models: optimizing administration routes and synergy with rapamycin. Cancer Res. 2010;70:598–608.
Johnson DE, O'Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol. 2018;15:234–48.
Hosseini A, Gharibi T, Marofi F, Javadian M, Babaloo Z, Baradaran B. Janus kinase inhibitors: a therapeutic strategy for cancer and autoimmune diseases. J Cell Physiol. 2020;235:5903–24.
Talpaz M, Kiladjian JJ. Fedratinib, a newly approved treatment for patients with myeloproliferative neoplasm-associated myelofibrosis. Leukemia. 2021;35:1–17.
Xiong H, Zhang ZG, Tian XQ, Sun DF, Liang QC, Zhang YJ, et al. Inhibition of JAK1, 2/STAT3 signaling induces apoptosis, cell cycle arrest, and reduces tumor cell invasion in colorectal cancer cells. Neoplasia. 2008;10:287–97.
Huynh J, Chand A, Gough D, Ernst M. Therapeutically exploiting STAT3 activity in cancer - using tissue repair as a road map. Nat Rev Cancer. 2019;19:82–96.
Zou S, Tong Q, Liu B, Huang W, Tian Y, Fu X. Targeting STAT3 in cancer immunotherapy. Mol Cancer. 2020;19:145.
Zhang X, Yue P, Page BD, Li T, Zhao W, Namanja AT, et al. Orally bioavailable small-molecule inhibitor of transcription factor Stat3 regresses human breast and lung cancer xenografts. Proc Natl Acad Sci U S A. 2012;109:9623–8.
Bai L, Zhou H, Xu R, Zhao Y, Chinnaswamy K, McEachern D, et al. A potent and selective small-molecule degrader of STAT3 achieves complete tumor regression in vivo. Cancer Cell. 2019;36:498–511 e17.
Khan MW, Saadalla A, Ewida AH, Al-Katranji K, Al-Saoudi G, Giaccone ZT, et al. The STAT3 inhibitor pyrimethamine displays anti-cancer and immune stimulatory effects in murine models of breast cancer. Cancer Immunol Immunother. 2018;67:13–23.
Xiang M, Kim H, Ho VT, Walker SR, Bar-Natan M, Anahtar M, et al. Gene expression-based discovery of atovaquone as a STAT3 inhibitor and anticancer agent. Blood. 2016;128:1845–53.
Heine A, Held SA, Daecke SN, Wallner S, Yajnanarayana SP, Kurts C, et al. The JAK-inhibitor ruxolitinib impairs dendritic cell function in vitro and in vivo. Blood. 2013;122:1192–202.
Heine A, Wolf AM, Schlaweck S, Daecke SN, Brossart P, Wolf D. Pacritinib protects dendritic cells more efficiently than ruxolitinib. Exp Hematol. 2021;100:37–40.
Patel MR, Dash A, Jacobson BA, Ji Y, Baumann D, Ismail K, et al. JAK/STAT inhibition with ruxolitinib enhances oncolytic virotherapy in non-small cell lung cancer models. Cancer Gene Ther. 2019;26:411–8.
Huang J. Current developments of targeting the p53 signaling pathway for cancer treatment. Pharmacol Ther. 2021;220:107720.
Karni-Schmidt O, Lokshin M, Prives C. The roles of MDM2 and MDMX in cancer. Annu Rev Pathol. 2016;11:617–44.
Oren M, Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol. 2010;2:a001107.
Mantovani F, Collavin L, Del Sal G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019;26:199–212.
Sanz G, Singh M, Peuget S, Selivanova G. Inhibition of p53 inhibitors: progress, challenges and perspectives. J Mol Cell Biol. 2019;11:586–99.
Fang Y, Liao G, Yu B. Small-molecule MDM2/X inhibitors and PROTAC degraders for cancer therapy: advances and perspectives. Acta Pharm Sin B. 2020;10:1253–78.
Choong ML, Yang H, Lee MA, Lane DP. Specific activation of the p53 pathway by low dose actinomycin D: a new route to p53 based cyclotherapy. Cell Cycle. 2009;8:2810–8.
Chen CS, Ho DR, Chen FY, Chen CR, Ke YD, Su JG. AKT mediates actinomycin D-induced p53 expression. Oncotarget. 2014;5:693–703.
Agupitan AD, Neeson P, Williams S, Howitt J, Haupt S, Haupt Y. P53: a Guardian of immunity becomes its saboteur through mutation. Int J Mol Sci. 2020;21:3452.
Wellenstein MD, Coffelt SB, Duits DEM, van Miltenburg MH, Slagter M, de Rink I, et al. Loss of p53 triggers WNT-dependent systemic inflammation to drive breast cancer metastasis. Nature. 2019;572:538–42.
Sharma MD, Rodriguez PC, Koehn BH, Baban B, Cui Y, Guo G, et al. Activation of p53 in immature myeloid precursor cells controls differentiation into Ly6c(+)CD103(+) monocytic antigen-presenting cells in tumors. Immunity. 2018;48:91–106 e6.
Nguyen TTT, Shingyoji M, Hanazono M, Zhong B, Morinaga T, Tada Y, et al. An MDM2 inhibitor achieves synergistic cytotoxic effects with adenoviruses lacking E1B55kDa gene on mesothelioma with the wild-type p53 through augmenting NFI expression. Cell Death Dis. 2021;12:663.
Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27.
Alvarez-Errico D, Vento-Tormo R, Sieweke M, Ballestar E. Epigenetic control of myeloid cell differentiation, identity and function. Nat Rev Immunol. 2015;15:7–17.
Henning AN, Roychoudhuri R, Restifo NP. Epigenetic control of CD8(+) T cell differentiation. Nat Rev Immunol. 2018;18:340–56.
Jones PA, Ohtani H, Chakravarthy A, De Carvalho DD. Epigenetic therapy in immune-oncology. Nat Rev Cancer. 2019;19:151–61.
Murphy SA, Mapes NJ Jr, Dua D, Kaur B. Histone modifiers at the crossroads of oncolytic and oncogenic viruses. Mol Ther. 2022;30:2153–62.
Nagarsheth N, Peng D, Kryczek I, Wu K, Li W, Zhao E, et al. PRC2 epigenetically silences Th1-type chemokines to suppress effector T-cell trafficking in colon cancer. Cancer Res. 2016;76:275–82.
Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature. 2015;527:249–53.
Li J, Wang W, Zhang Y, Cieslik M, Guo J, Tan M, et al. Epigenetic driver mutations in ARID1A shape cancer immune phenotype and immunotherapy. J Clin Invest. 2020;130:2712–26.
Guo ZS, Hong JA, Irvine KR, Chen GA, Spiess PJ, Liu Y, et al. De novo induction of a cancer/testis antigen by 5-aza-2′-deoxycytidine augments adoptive immunotherapy in a murine tumor model. Cancer Res. 2006;66:1105–13.
Villanueva L, Alvarez-Errico D, Esteller M. The contribution of epigenetics to cancer immunotherapy. Trends Immunol. 2020;41:676–91.
Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. 2015;162:974–86.
Liu YT, Sun ZJ. Turning cold tumors into hot tumors by improving T-cell infiltration. Theranostics. 2021;11:5365–86.
Lightcap ES, Yu P, Grossman S, Song K, Khattar M, Xega K, et al. A small-molecule SUMOylation inhibitor activates antitumor immune responses and potentiates immune therapies in preclinical models. Sci Transl Med. 2021;13:eaba7791.
Christman JK. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene. 2002;21:5483–95.
Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26:5541–52.
Duan YC, Zhang SJ, Shi XJ, Jin LF, Yu T, Song Y, et al. Research progress of dual inhibitors targeting crosstalk between histone epigenetic modulators for cancer therapy. Eur J Med Chem. 2021;222:113588.
Hogg SJ, Beavis PA, Dawson MA, Johnstone RW. Targeting the epigenetic regulation of antitumour immunity. Nat Rev Drug Discov. 2020;19:776–800.
Forbes NE, Abdelbary H, Lupien M, Bell JC, Diallo JS. Exploiting tumor epigenetics to improve oncolytic virotherapy. Front Genet. 2013;4:184.
Chianese A, Santella B, Ambrosino A, Stelitano D, Rinaldi L, Galdiero M, et al. Oncolytic viruses in combination therapeutic approaches with epigenetic modulators: past, present, and future perspectives. Cancers (Basel). 2021;13:2761.
Ma J, Li N, Zhao J, Lu J, Ma Y, Zhu Q, et al. Histone deacetylase inhibitor trichostatin A enhances the antitumor effect of the oncolytic adenovirus H101 on esophageal squamous cell carcinoma in vitro and in vivo. Oncol Lett. 2017;13:4868–74.
Jennings VA, Scott GB, Rose AMS, Scott KJ, Migneco G, Keller B, et al. Potentiating oncolytic virus-induced immune-mediated tumor cell killing using histone deacetylase inhibition. Mol Ther. 2019;27:1139–52.
Qiu W, Ding X, Li S, He Y, Zhu L. Oncolytic bovine herpesvirus 1 inhibits human lung adenocarcinoma A549 cell proliferation and tumor growth by inducing DNA damage. Int J Mol Sci. 2021;22:8582.
Muscolini M, Castiello L, Palermo E, Zevini A, Ferrari M, Olagnier D, et al. SIRT1 modulates the sensitivity of prostate cancer cells to vesicular stomatitis virus oncolysis. J Virol. 2019;93:e00626–19.
Jaime-Ramirez AC, Yu JG, Caserta E, Yoo JY, Zhang J, Lee TJ, et al. Reolysin and histone deacetylase inhibition in the treatment of Head and neck squamous cell carcinoma. Mol Ther Oncolytics. 2017;5:87–96.
Stiff A, Caserta E, Sborov DW, Nuovo GJ, Mo X, Schlotter SY, et al. Histone deacetylase inhibitors enhance the therapeutic potential of reovirus in multiple myeloma. Mol Cancer Ther. 2016;15:830–41.
Islam S, Espitia CM, Persky DO, Carew JS, Nawrocki ST. Resistance to histone deacetylase inhibitors confers hypersensitivity to oncolytic reovirus therapy. Blood Adv. 2020;4:5297–310.
Ellerhoff TP, Berchtold S, Venturelli S, Burkard M, Smirnow I, Wulff T, et al. Novel epi-virotherapeutic treatment of pancreatic cancer combining the oral histone deacetylase inhibitor resminostat with oncolytic measles vaccine virus. Int J Oncol. 2016;49:1931–44.
Fox CR, Parks GD. Histone deacetylase inhibitors enhance cell killing and block interferon-beta synthesis elicited by infection with an oncolytic parainfluenza virus. Viruses. 2019;11:431.
Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42.
West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. 2014;124:30–9.
Berghauser Pont LM, Kleijn A, Kloezeman JJ, van den Bossche W, Kaufmann JK, de Vrij J, et al. The HDAC inhibitors Scriptaid and LBH589 combined with the oncolytic virus Delta24-RGD exert enhanced anti-tumor efficacy in patient-derived glioblastoma cells. PLoS One. 2015;10:e0127058.
Nakashima H, Kaufmann JK, Wang PY, Nguyen T, Speranza MC, Kasai K, et al. Histone deacetylase 6 inhibition enhances oncolytic viral replication in glioma. J Clin Invest. 2015;125:4269–80.
Moreno-Gonzalo O, Ramirez-Huesca M, Blas-Rus N, Cibrian D, Saiz ML, Jorge I, et al. HDAC6 controls innate immune and autophagy responses to TLR-mediated signalling by the intracellular bacteria listeria monocytogenes. PLoS Pathog. 2017;13:e1006799.
Nguyen TL, Abdelbary H, Arguello M, Breitbach C, Leveille S, Diallo JS, et al. Chemical targeting of the innate antiviral response by histone deacetylase inhibitors renders refractory cancers sensitive to viral oncolysis. Proc Natl Acad Sci U S A. 2008;105:14981–6.
Bridle BW, Chen L, Lemay CG, Diallo JS, Pol J, Nguyen A, et al. HDAC inhibition suppresses primary immune responses, enhances secondary immune responses, and abrogates autoimmunity during tumor immunotherapy. Mol Ther. 2013;21:887–94.
Guo ZS. The 2018 Nobel prize in medicine goes to cancer immunotherapy (editorial for BMC cancer). BMC Cancer. 2018;18:1086.
Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8:1069–86.
Chames P, Van Regenmortel M, Weiss E, Baty D. Therapeutic antibodies: successes, limitations and hopes for the future. Br J Pharmacol. 2009;157:220–33.
Naidoo J, Page DB, Li BT, Connell LC, Schindler K, Lacouture ME, et al. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann Oncol. 2015;26:2375–91.
Wu Q, Jiang L, Li SC, He QJ, Yang B, Cao J. Small molecule inhibitors targeting the PD-1/PD-L1 signaling pathway. Acta Pharmacol Sin. 2021;42:1–9.
Ashizawa T, Iizuka A, Tanaka E, Kondou R, Miyata H, Maeda C, et al. Antitumor activity of the PD-1/PD-L1 binding inhibitor BMS-202 in the humanized MHC-double knockout NOG mouse. Biomed Res. 2019;40:243–50.
Sasikumar PG, Ramachandra RK, Adurthi S, Dhudashiya AA, Vadlamani S, Vemula K, et al. A rationally designed peptide antagonist of the PD-1 signaling pathway as an immunomodulatory agent for cancer therapy. Mol Cancer Ther. 2019;18:1081–91.
Zhang H, Xia Y, Yu C, Du H, Liu J, Li H, et al. Discovery of novel small-molecule inhibitors of PD-1/PD-L1 interaction via structural simplification strategy. Molecules. 2021;26:3347.
Wang F, Ye W, Wang S, He Y, Zhong H, Wang Y, et al. Discovery of a new inhibitor targeting PD-L1 for cancer immunotherapy. Neoplasia. 2021;23:281–93.
Fang L, Tian J, Zhang K, Zhang X, Liu Y, Cheng Z, et al. Discovery of 1,3,4-oxadiazole derivatives as potential antitumor agents inhibiting the programmed cell death-1/programmed cell death-ligand 1 interaction. Bioorg Med Chem. 2021;46:116370.
Chang HN, Liu BY, Qi YK, Zhou Y, Chen YP, Pan KM, et al. Blocking of the PD-1/PD-L1 interaction by a D-peptide antagonist for cancer immunotherapy. Angew Chem Int Ed Engl. 2015;54:11760–4.
Zhu H, Bengsch F, Svoronos N, Rutkowski MR, Bitler BG, Allegrezza MJ, et al. BET Bromodomain inhibition promotes anti-tumor immunity by suppressing PD-L1 expression. Cell Rep. 2016;16:2829–37.
Xu Y, Poggio M, Jin HY, Shi Z, Forester CM, Wang Y, et al. Translation control of the immune checkpoint in cancer and its therapeutic targeting. Nat Med. 2019;25:301–11.
Wang H, Yao H, Li C, Shi H, Lan J, Li Z, et al. HIP1R targets PD-L1 to lysosomal degradation to alter T cell-mediated cytotoxicity. Nat Chem Biol. 2019;15:42–50.
Lim SO, Li CW, Xia W, Cha JH, Chan LC, Wu Y, et al. Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell. 2016;30:925–39.
Cha JH, Yang WH, Xia W, Wei Y, Chan LC, Lim SO, et al. Metformin promotes antitumor immunity via endoplasmic-reticulum-associated degradation of PD-L1. Mol Cell. 2018;71:606–620 e7.
Sasikumar PG, Sudarshan NS, Adurthi S, Ramachandra RK, Samiulla DS, Lakshminarasimhan A, et al. PD-1 derived CA-170 is an oral immune checkpoint inhibitor that exhibits preclinical anti-tumor efficacy. Commun Biol. 2021;4:699.
You D, Hillerman S, Locke G, Chaudhry C, Stromko C, Murtaza A, et al. Enhanced antitumor immunity by a novel small molecule HPK1 inhibitor. J Immunother Cancer. 2021;9:e001402.
Loeser S, Loser K, Bijker MS, Rangachari M, van der Burg SH, Wada T, et al. Spontaneous tumor rejection by cbl-b-deficient CD8+ T cells. J Exp Med. 2007;204:879–91.
LaMarche MJ, Acker M, Argintaru A, Bauer D, Boisclair J, Chan H, et al. Identification of TNO155, an allosteric SHP2 inhibitor for the treatment of cancer. J Med Chem. 2020;63:13578–94.
Ganesan A, Ahmed M, Okoye I, Arutyunova E, Babu D, Turnbull WL, et al. Comprehensive in vitro characterization of PD-L1 small molecule inhibitors. Sci Rep. 2019;9:12392.
Konieczny M, Musielak B, Kocik J, Skalniak L, Sala D, Czub M, et al. Di-bromo-based small-molecule inhibitors of the PD-1/PD-L1 immune checkpoint. J Med Chem. 2020;63:11271–85.
Lutz-Nicoladoni C, Wolf D, Sopper S. Modulation of immune cell functions by the E3 ligase Cbl-b. Front Oncol. 2015;5:58.
Liu Q, Qu J, Zhao M, Xu Q, Sun Y. Targeting SHP2 as a promising strategy for cancer immunotherapy. Pharmacol Res. 2020;152:104595.
Sawasdikosol S, Zha R, Yang B, Burakoff S. HPK1 as a novel target for cancer immunotherapy. Immunol Res. 2012;54:262–5.
Sivanandam V, LaRocca CJ, Chen NG, Fong Y, Warner SG. Oncolytic viruses and immune checkpoint inhibition: the best of both worlds. Mol Ther Oncolytics. 2019;13:93–106.
Zheng J, Mo J, Zhu T, Zhuo W, Yi Y, Hu S, et al. Comprehensive elaboration of the cGAS-STING signaling axis in cancer development and immunotherapy. Mol Cancer. 2020;19:133.
Motwani M, Pesiridis S, Fitzgerald KA. DNA sensing by the cGAS-STING pathway in health and disease. Nat Rev Genet. 2019;20:657–74.
Jiang M, Chen P, Wang L, Li W, Chen B, Liu Y, et al. cGAS-STING, an important pathway in cancer immunotherapy. J Hematol Oncol. 2020;13:81.
Filderman JN, Appleman M, Chelvanambi M, Taylor JL, Storkus WJ. STINGing the tumor microenvironment to promote therapeutic tertiary lymphoid structure development. Front Immunol. 2021;12:690105.
Ding C, Song Z, Shen A, Chen T, Zhang A. Small molecules targeting the innate immune cGAS-STING-TBK1 signaling pathway. Acta Pharm Sin B. 2020;10:2272–98.
Ager CR, Boda A, Rajapakshe K, Lea ST, Di Francesco ME, Jayaprakash P, et al. High potency STING agonists engage unique myeloid pathways to reverse pancreatic cancer immune privilege. J Immunother Cancer. 2021;9:e003246.
Cornelison R, Biswas K, Llaneza DC, Harris AR, Sosale NG, Lazzara MJ, et al. CX-5461 treatment leads to cytosolic DNA-mediated STING activation in ovarian cancer. Cancers (Basel). 2021;13:5056.
Hu M, Zhou M, Bao X, Pan D, Jiao M, Liu X, et al. ATM inhibition enhances cancer immunotherapy by promoting mtDNA leakage and cGAS/STING activation. J Clin Invest. 2021;131:e139333.
Lee J, Ghonime MG, Wang R, Cassady KA. The antiviral apparatus: STING and oncolytic virus restriction. Mol Ther Oncolytics. 2019;13:7–13.
Lee JM, Ghonime MG, Cassady KA. STING restricts oHSV replication and spread in resistant MPNSTs but is dispensable for basal IFN-stimulated gene upregulation. Mol Ther Oncolytics. 2019;15:91–100.
Froechlich G, Caiazza C, Gentile C, D'Alise AM, De Lucia M, Langone F, et al. Integrity of the antiviral STING-mediated DNA sensing in tumor cells is required to sustain the immunotherapeutic efficacy of herpes simplex oncolytic virus. Cancers (Basel). 2020;12:3407.
Hutzen B, Chen CY, Wang PY, Sprague L, Swain HM, Love J, et al. TGF-beta inhibition improves oncolytic herpes Viroimmunotherapy in murine models of rhabdomyosarcoma. Mol Ther Oncolytics. 2017;7:17–26.
Esaki S, Nigim F, Moon E, Luk S, Kiyokawa J, Curry W Jr, et al. Blockade of transforming growth factor-beta signaling enhances oncolytic herpes simplex virus efficacy in patient-derived recurrent glioblastoma models. Int J Cancer. 2017;141:2348–58.
Groeneveldt C, van Hall T, van der Burg SH, Ten Dijke P, van Montfoort N. Immunotherapeutic potential of TGF-beta inhibition and oncolytic viruses. Trends Immunol. 2020;41:406–20.
D'Assoro AB, Haddad T, Galanis E. Aurora-a kinase as a promising therapeutic target in cancer. Front Oncol. 2015;5:295.
Libertini S, Abagnale A, Passaro C, Botta G, Barbato S, Chieffi P, et al. AZD1152 negatively affects the growth of anaplastic thyroid carcinoma cells and enhances the effects of oncolytic virus dl922-947. Endocr Relat Cancer. 2011;18:129–41.
Iankov ID, Kurokawa CB, D'Assoro AB, Ingle JN, Domingo-Musibay E, Allen C, et al. Inhibition of the Aurora a kinase augments the anti-tumor efficacy of oncolytic measles virotherapy. Cancer Gene Ther. 2015;22:438–44.
Currier MA, Sprague L, Rizvi TA, Nartker B, Chen CY, Wang PY, et al. Aurora a kinase inhibition enhances oncolytic herpes virotherapy through cytotoxic synergy and innate cellular immune modulation. Oncotarget. 2017;8:17412–27.
Xiao X, Liang J, Huang C, Li K, Xing F, Zhu W, et al. DNA-PK inhibition synergizes with oncolytic virus M1 by inhibiting antiviral response and potentiating DNA damage. Nat Commun. 2018;9:4342.
Bhatt DK, Chammas R, Daemen T. Resistance mechanisms influencing oncolytic virotherapy, a systematic analysis. Vaccines (Basel). 2021;9:1166.
Selman M, Ou P, Rousso C, Bergeron A, Krishnan R, Pikor L, et al. Dimethyl fumarate potentiates oncolytic virotherapy through NF-kappaB inhibition. Sci Transl Med. 2018;10:eaao1613.
Zhou J, Wang G, Chen Y, Wang H, Hua Y, Cai Z. Immunogenic cell death in cancer therapy: present and emerging inducers. J Cell Mol Med. 2019;23:4854–65.
Guo D, Xiao J, Liang J, Fan J, Hou P, Li X, et al. CDK4/6 inhibition enhances oncolytic virus efficacy by potentiating tumor-selective cell killing and T cell activation in refractory glioblastoma. Cancer Res. 2022;82:33–39-3374.
Liu W, Chen H, Zhu Z, Liu Z, Ma C, Lee YJ, et al. Ferroptosis inducer improves the efficacy of oncolytic virus-mediated cancer immunotherapy. Biomedicines. 2022;10:1425.
Diallo JS, Le Boeuf F, Lai F, Cox J, Vaha-Koskela M, Abdelbary H, et al. A high-throughput pharmacoviral approach identifies novel oncolytic virus sensitizers. Mol Ther. 2010;18:1123–9.
Dornan MH, Krishnan R, Macklin AM, Selman M, El Sayes N, Son HH, et al. First-in-class small molecule potentiators of cancer virotherapy. Sci Rep. 2016;6:26786.
Selman M, Rousso C, Bergeron A, Son HH, Krishnan R, El-Sayes NA, et al. Multi-modal potentiation of oncolytic virotherapy by vanadium compounds. Mol Ther. 2018;26:56–69.
Mailliard RB, Wankowicz-Kalinska A, Cai Q, Wesa A, Hilkens CM, Kapsenberg ML, et al. Alpha-type-1 polarized dendritic cells: a novel immunization tool with optimized CTL-inducing activity. Cancer Res. 2004;64:5934–7.
Muthuswamy R, Wang L, Pitteroff J, Gingrich JR, Kalinski P. Combination of IFNalpha and poly-I:C reprograms bladder cancer microenvironment for enhanced CTL attraction. J Immunother Cancer. 2015;3:6.
Tang B, Guo ZS, Bartlett DL, Yan DZ, Schane CP, Thomas DL, et al. Synergistic combination of oncolytic virotherapy and immunotherapy for glioma. Clin Cancer Res. 2020;26:2216–30.
Yun C-O, Hong J, Yoon A-R. Current clinical landscape of oncolytic viruses as novel cancer immunotherapeutic and recent preclinical advancements. Front Immunol. 2022;13:953410.
Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell. 2015;28:690–714.
Hiam-Galvez KJ, Allen BM, Spitzer MH. Systemic immunity in cancer. Nat Rev Cancer. 2021;21:345–59.
Petroni G, Cantley LC, Santambrogio L, Formenti SC, Galluzzi L. Radiotherapy as a tool to elicit clinically actionable signalling pathways in cancer. Nat Rev Clin Oncol. 2022;19:114–31.
Wu ZJ, Tang FR, Ma ZW, Peng XC, Xiang Y, Zhang Y, et al. Oncolytic viruses for tumor precision imaging and radiotherapy. Hum Gene Ther. 2018;29:204–22.
Mansfield D, Pencavel T, Kyula JN, Zaidi S, Roulstone V, Thway K, et al. Oncolytic vaccinia virus and radiotherapy in head and neck cancer. Oral Oncol. 2013;49:108–18.
Buckel L, Advani SJ, Frentzen A, Zhang Q, Yu YA, Chen NG, et al. Combination of fractionated irradiation with anti-VEGF expressing vaccinia virus therapy enhances tumor control by simultaneous radiosensitization of tumor associated endothelium. Int J Cancer. 2013;133:2989–99.
Gholami S, Chen CH, Lou E, Belin LJ, Fujisawa S, Longo VA, et al. Vaccinia virus GLV-1h153 in combination with 131I shows increased efficiency in treating triple-negative breast cancer. FASEB J. 2014;28:676–82.
Wilkinson MJ, Smith HG, McEntee G, Kyula-Currie J, Pencavel TD, Mansfield DC, et al. Oncolytic vaccinia virus combined with radiotherapy induces apoptotic cell death in sarcoma cells by down-regulating the inhibitors of apoptosis. Oncotarget. 2016;7:81208–22.
Chen WY, Chen YL, Lin HW, Chang CF, Huang BS, Sun WZ, et al. Stereotactic body radiation combined with oncolytic vaccinia virus induces potent anti-tumor effect by triggering tumor cell necroptosis and DAMPs. Cancer Lett. 2021;523:149–61.
Martinez-Velez N, Marigil M, Garcia-Moure M, Gonzalez-Huarriz M, Aristu JJ, Ramos-Garcia LI, et al. Delta-24-RGD combined with radiotherapy exerts a potent antitumor effect in diffuse intrinsic pontine glioma and pediatric high grade glioma models. Acta Neuropathol Commun. 2019;7:64.
Gallego Perez-Larraya J, Garcia-Moure M, Labiano S, Patino-Garcia A, Dobbs J, Gonzalez-Huarriz M, et al. Oncolytic DNX-2401 virus for pediatric diffuse intrinsic pontine glioma. N Engl J Med. 2022;386:2471–81.
Chapman NM, Chi H. mTOR links environmental signals to T cell fate decisions. Front Immunol. 2014;5:686.
Heo J, Breitbach CJ, Moon A, Kim CW, Patt R, Kim MK, et al. Sequential therapy with JX-594, a targeted oncolytic poxvirus, followed by sorafenib in hepatocellular carcinoma: preclinical and clinical demonstration of combination efficacy. Mol Ther. 2011;19:1170–9.
Xu H, Lyu X, Yi M, Zhao W, Song Y, Wu K. Organoid technology and applications in cancer research. J Hematol Oncol. 2018;11:116.
Ramelyte E, Tastanova A, Balazs Z, Ignatova D, Turko P, Menzel U, et al. Oncolytic virotherapy-mediated anti-tumor response: a single-cell perspective. Cancer Cell. 2021;39:394–406 e4.
Katti A, Diaz BJ, Caragine CM, Sanjana NE, Dow LE. CRISPR in cancer biology and therapy. Nat Rev Cancer. 2022;22:259–79.
Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature. 2017;547:413–8.
Wei J, Long L, Zheng W, Dhungana Y, Lim SA, Guy C, et al. Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature. 2019;576:471–6.
Acknowledgements
We thank reviewers for constructive suggestions that led to improved quality of the article, Ms. Sarah Fricano for proof-reading the manuscript.
Funding
ZZ was supported by a fellowship from China Medical University for his study at the University of Pittsburgh during 2020–2021. Some original research papers from our own groups discussed in this review was supported by NIH grants R21CA205727-01 and 1R21CA216574-01A1 (to BL), PO1CA132714, P01CA234212, 1RO1CA82016 and 1RO1CA 095128 (to PK).
Author information
Authors and Affiliations
Contributions
ZSG conceived this study, did the literature search, designed and wrote the manuscript. ZZ contributed to writing and prepared all figures. AJRM made critical suggestions and polished the whole manuscript. PK made critical suggestions to improve the quality of the article. All other authors made suggestions to improve the quality of this article. The author(s) read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors have read the manuscript and consent the submission for publication.
Competing interests
ZZ, BL and ZSG filed patent applications related to oncolytic virus and its combination with small molecule inhibitor for enzymes for cancer therapy. Other authors declare no conflict of interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhu, Z., McGray, A.J.R., Jiang, W. et al. Improving cancer immunotherapy by rationally combining oncolytic virus with modulators targeting key signaling pathways. Mol Cancer 21, 196 (2022). https://doi.org/10.1186/s12943-022-01664-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-022-01664-z