
Abstract
Though Forkhead box P (FOXP) transcription factors comprising of FOXP1, FOXP2, FOXP3 and FOXP4 are involved in the embryonic development, immune disorders and cancer progression, the underlying function of FOXP3 targeting CD4 + CD25+ regulatory T (Treg) cells and the dual roles of FOXP proteins as an oncogene or a tumor suppressor are unclear and controversial in cancers to date. Thus, the present review highlighted research history, dual roles of FOXP proteins as a tumor suppressor or an oncogene, their molecular networks with other proteins and noncoding RNAs, cellular immunotherapy targeting FOXP3, and clinical implications in cancer progression.
Similar content being viewed by others

Background
Cancer still remains a major factor of human deaths worldwide to date [1]. It is well documented that epigenetic and genetic alterations including transcription factors, growth factors, cytokines, chemokines and proteases are critically involved in cancer progression under specific microenvironment [2]. As one of transcription factors Forkhead box P (FOXP) family consist of FOXP1 (3p14.1), FOXP2 (7q31), FOXP3 (Xp11.23) and FOXP4 (6p21.1) with similar 110 amino acid DNA-binding domain termed winged helix\forkhead domain [3], since 19 Fox gene subfamilies (A-S) were identified with 50 genes in humans so far [4]. FOXP proteins play important roles in the regulation of gene transcription in associated with immune function [4], carcinogenesis [5, 6], differentiation [7, 8] and angiogenesis [6, 9, 10].
Accumulating evidence reveals that FOXP1 regulates development of B cells [11], FOXP2 controls language development [12] and FOXP4 mediates development of T cell [13]. Furthermore, FOXP3, so called scurfin, is critically involved in differentiation and function of regulatory T cells or CD4+/CD25+ regulatory T (Treg) cells for cancer immunotherapy [14, 15]. It is well documented that FOXP3 modulates Treg development and functions [16] by immune evasion of tumor cells through imbalance of immunoediting and immunosurveillance in some cancers [17].
Additionally, it is well documented that FOXP proteins interact with other molecules and signaling pathways. FOXP1 has protein-protein interaction with NFAT1 to enhance breast cancer cell motility [18] and FOXP2 is essential in growth arrest of 143 osteosarcoma cells via p21 activation [19]. Also, FOXP3 is associated with IL-17 [20], RUNX1 [21], STAT3 [22], NF-κB [23], FOXO3 [24] and other cofactors such as EOS (Ikzf4) [25], interferon regulatory factor 4 (IRF4) [26], special AT-rich sequence-binding protein-1 (SATB1) [25, 27] and GATA1, while FOXP4 is closely associated with miR 4316 in breast cancer cells [28], miR 491–5p in osteosarcoma [29] and miR 338-3p in hepatocellular carcinoma [30].
Nevertheless, the underlying functions of FOXP proteins in cancer progression and immunology still remain unclear and confused to readers. Thus, the present review highlighted research history, dual functions of FOXP family as a tumor suppressor or an oncogene, their interaction with other proteins and noncoding RNAs, and cellular therapy targeting FOXP proteins in cancer progression, their clinical implications and finally suggested research perspectives.
Overview of FOXP family: structure and domains and research history
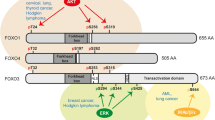
FOXP (Forkhead box P) family proteins share a highly conserved C2H2 zinc finger domain, leucine zipper (winged helix) domain (WHD) consisting of β-sheets, α-helices and wings or loops like a helix-turn helix-like motif [31], winged helix Forkhead DNA binding domain (FHD) and about 50 residues N terminal domain [32] (Fig. 1a). Of note, FOXP1 and FOXP2 contain C-terminal-binding protein 1 (CtBP1) binding domain different from FOXP3 and FOXP4 [33,34,35,36]. Also, tertiary structure of FOXP1 forkhead domain contains five α-helices (H1–5), three β-strands (S1, S2 and S3) and two wings (W1 and W2) [37, 38] and also FOXP2 and FOXP3 have similar crystal structure of FOXP1 with their specific dimers (Fig. 1b). Though the FHDs of FOXP1, FOXP2, FOXP3 and FOXP4 all have a C-terminal winged helix FHD to be dimerised, FOXP3 dimer is considered more stable than FOXP2 or FOXP1 dimer [36]. Notably, Mendoza et al. [39] identified homo- hetero dimers and an oligo composed of FOXP1/2/4 complex in HEK 293 cells and brain. Additionally, of two independent subdomains required for transcriptional repression activity, subdomain 1 with a highly conserved leucine zipper similar to that of N-Myc gives homo- and hetero-dimerisation to FOXP1/2/4 proteins. In contrast, subdomain 2 with a binding motif for the corepressor protein C-terminal binding protein 1 (CtBP-1) is found only in FOXP1 and FOXP2 but not in FOXP4,while FOXP3 binds to RUNX to repress target gene expression [13, 40].

Domains and crystal structures of FOXP family. a Domains of FOXP1, FOXP2, FOXP3 and FOXP4. FOXP members share a highly conserved C2H2 zinc finger domain, leucine zipper domain, Forkhead DNA binding domain and about 50 residues N terminal domain. Also, FOXP1 and FOXP2 contain CtBP1 binding domain different from FOXP3 and FOXP4. b Crystal structures of FOXP1 (PDB: 2KIU), FOXP2-DNA complex (PDB: 2A07) and FOXP3-DNA complex (PDB:3QRF) by using The PyMOL Molecular Graphics System Version 2.3.0
Looking back on research history of FOXP family, as first discovery of FOXP family, Godfrey et al. [41] suggested that T lymphocytes mediate scurfy lesions in abnormal thymic environment in 1991, since regulatory T cell-deficient scurfy mice induce severe autoimmune disorders, leading to death (Fig. 2). Thereafter, Brunkow et al. [42] first coined FOXP3 for scurfin essential for normal immune homeostasis, and human Treg cells were further characterized as CD4+CD25+ T cells by Taams and his colleagues [43] in 2001, since Sakaguchi et al. [44] reported that CD4+CD25+ cells enhance self-tolerance by immunosuppression in 1995. Then Shu and his colleagues [45] identified and characterized FOXP1 and FOXP2 in the lungs of mice. Also, Banham et al. [46] suggested FOXP1 as a novel tumor suppressor candidate localized to the chromosome 3p 14.1 region. However, FOXP1 was found as a tumor suppressor in breast cancer [47] and as an oncogene in MALT lymphoma [48]. Additionally, FOXP2 was recognized as an oncogene in several lymphomas including multiple myeloma, and as a tumor suppressor in gastric cancer [49] and hepatocellular carcinoma (HCC) [50] since Lai et al. [51] first demonstrated that FOXP2 is critically involved in a severe language and speech disorder in 2001.

Timeline for FOXP family research history. DLBCL, diffuse large B-cell lymphoma; FOXP1–4, forkhead box P1–4; NFAT, Nuclear factor of activated T-cells; Th17, T helper 17; Treg, T regulatory; nTreg cells, naïve T regulatory cells; NF-kappa B, Nuclear factor kappa B. The numbers in references indicate PMID of PubMed
Hori et al. [52] for the first time identified that FOXP3 is a key regulatory gene for the development of Treg cells in 2003 and Schubert et al. [53] reported that scurfin or FOXP3 represses NFAT transcription factor and cytokine production and proliferation by CD4+ T cell activation. Next, Teufel et al. [13] demonstrated FOXP4 as a tumor suppressor in patients with kidney tumors in 2003 and Wang et al. [30] reported FOXP4 as an oncogene in HCC in 2015. Consistently, FOXP4 depletion inhibits proliferation of HCC as a negative regulator of miR-338-3p [30]. Of note, the important role of Th17/Treg ratio has been a hot issue in cancers [54] and autoimmune diseases [55, 56], since Treg cells can be differentiated into Th17 cells [57] and then the significance of Th17/Treg from Th1/Th2 was revealed in immune response [58]. From this research chronicle, extensive research has been conducted targeting FOXP family.
Dual biologic functions of FOXP family proteins as a tumor suppressor or an oncogene
Despite accumulating evidence on dual functions of FOXP proteins as an oncogene or a tumor suppressor in specific cancer types [59, 60] and their related signaling pathways, it still remains unclear under what factors or circumstances the FOXP proteins act a tumor suppressive or oncogenic role. It is well documented that FOXP1 is overexpressed with poor prognosis in diffuse large B-cell lymphoma (DLBCL) [61,62,63,64], primary cutaneous large B-cell lymphomas (PCLBCL) [65, 66], follicular lymphoma [67] and gastric mucosa-associated lymphoid tissue lymphoma (MALT) [68] as an oncogene. Consistently, Wang et al. [69] reported that FOXP1 depletion reduced the proliferation of hepatocellular carcinoma via G1/S phase arrest and decreased phosphorylation of retinoblastoma protein (Rb). Notably, Brown et al. [70] indicated that the growth of DLBCL is mediated by suppression of MHC class II expression and immune response signatures and activation of Wnt/β-catenin signaling induced by FOXP1 [71]. Also, Bates et al. [47] reported that nuclear FOXP1 is significantly co-expressed with estrogen receptor beta or alpha in ER positive MCF-7 breast cancer patients following tamoxifen treatment, though they do not directly affect each other by siRNA transfection [72]. Also, FOXP1 works as an oncogene by activating chromosome translocations under the control of immunoglobulin heavy chain (IGH) enhancers [60, 73].
In contrast, FOXP1 is also known as a tumor suppressor, since FOXP1 gene maps to a tumor suppressor locus at 3p14.1 and so loss of FOXP1 expression is associated with a poor outcome in in breast cancer [74]. Furthermore, overexpression of FOXP1 inhibits proliferation and invasion in U251 glioma cells [75], while knockdown of FOXP1 promotes the development of lung carcinoma [76]. Similarly, FOXP1 represses AR-induced transcriptional activity or histone modification as a tumor suppressor [77, 78]. Interestingly, previous evidence reveals that FOXP1 and FOXP2 exert functional cooperativity during development. Indeed, FOXP2−/−FOXP1−/+ mice showed severe developmental defects and perinatal lethality compared to FOXP2−/−FOXP1+/+ mice [5]. Furthermore, FOXP1 is also known to interact with FOXP3 through NFAT-IL-2 promoter DNA complexes [74, 79].
Recently, critical roles of FOXP2 have been demonstrated in cancer progression as a tumor suppressor, though FOXP2 mutations are well known to cause language and speech development deficits. Also, FOXP2 was reported to suppress the transcriptional activity of target genes through the Zinc finger domain and also binds to domain for C-Terminal Binding Protein-1 (CtBP1) for suppressing E-cadherin and promoting invasion [59]. Furthermore, Cuiffo et al. reported that downregulation of FOXP2 enhances tumor initiation in breast cancers as a putative tumor/metastasis suppressor [80]. Also, FOXP2 was downregulated in hepatocellular carcinoma (HCC) tumor tissues with poor overall survival rate and its downregulation significantly promoted the invasiveness of HCC [50]. In addition, FOXP2 is essential for regulation of p21 in 143B osteosarcoma cell growth inhibition [19]. Of note, Morris et al. claimed that phosphorylation at Ser557 is identified as another means of regulating the transcriptional functions of FOXP2 [81]. Furthermore, FOXP2 is regarded as a SUMO target protein at cellular level, since FOXP2 is covalently modulated by both SUMO1 and SUMO3. SUMOylation of FOXP2 is significantly disturbed by a specific SUMO Specific Protease 2 (SENP2), since SUMOylation modulates transcriptional activity of FOXP2 in targeting downstream target genes (DISC1, SRPX2, and MiR200c) by reporter gene assay [82].
In contrast, mutations of transcription factor FOXP2 were shown in neoplastic plasma cells [83] and overexpression of FOXP2 is associated with high risk of early PSA recurrence in erythroblast transformation-specific-related gene (ERG) fusion-negative prostate cancers [84].
FOXP3 promotes the immune evasion as Treg cell marker suppressing immune response against cancer, while FOXP3 at the Xp11.23 revealed good prognosis in breast cancers as a tumor suppressor [85,86,87,88] by regulating HER-2/ErbB2 [88] or SKP2 [89, 90] oncogene. Furthermore, it is noteworthy that FOXP3 functions as dual roles through interaction with other transcription factors nuclear factor kappa-B (NF-κB), nuclear factor of activated T cells (NFAT) [91], and acute myeloid leukemia 1 (AML-1) [92] in the tumor microenvironment.
FOXP4 is closely associated with FOXP1 and FOXP2 with 54 and 60% identity, respectively since FOXP4 forms a large multidomain transcriptional repressors with FOXP1 and FOXP2 [40], while FOXP3 and FOXP4 protein sequences are merely 47% identical in the aligned sequence region [13]. FOXP4 was overexpressed in A549 and H1703 non-small cell lung cancer (NSCLC) cells and conversely FOXP4 depletion markedly reduced the growth and invasion of above two NSCLCs [93]. Furthermore, FOXP4 gene was closely associated with prostate cancer risk in Chinese men [94, 95] and also long non-coding RNA FOXP4-AS1 is suggested a poor prognostic factor in colorectal cancer [96] and osteosarcoma [97]. In contrast, FOXP4 was significantly downregulated in patients with kidney cancers [13]. Overall, despite accumulating evidence on dual functions of FOXPs, further study is required to verify the dual role mechanisms of FOXP proteins in association with their related molecules under specific microenvironment or phosphorylation condition in the near future.
Regulating tumor progression by FOXP3 in the tumor microenviroment
It is well documented that FOXP3 is a key transcription factor for development and function of Treg cells [98]. Treg cells are produced from the thymus, and the periphery, by constitutively expressing glucocorticoid-induced TNF receptor family-related gene (GITR), cytotoxic T lymphocyte associated antigen 4 (CTLA-4) and IL-2 receptor (IL-2R) α chain (CD25) [99, 100]. Treg cells induce immunosuppression by CTLA-4–mediated downregulation of costimulatory molecules or IL-2 deprivation on antigen-presenting cells (APCs), and by secretion of cytokines, such as IL-10 or TGF-β. Thus, Treg cells suppress tumor-specific CD8+ T cell cytotoxicity through TGF-β signaling [101] and some molecules including nuclear factor of activated T cells (NFAT) [15] and Runt-related transcription factor 1 (RUNX1) [92] are found to bind to the promoter regions of FOXP3-regulated genes for activation of Treg cells. FOXP3 overexpression of Tregs may promote tumor cell growth in non-small cell lung cancer (NSCLC) microenvironment [102].
FOXP3 regulates immune system as a specific marker for CD4+/ CD25+ or CD4+/CD25− Treg cell development and function [17, 103, 104]. CD4+/CD25+/FOXP3+ Treg contributes to immunosuppression and cancer progression by reducing the anticancer immunity of CD4+ or CD8+ effector T cells [17, 105]. Two major populations of Treg cells have been defined as peripherally induced Treg (iTreg) cells and thymically derived natural Treg (nTreg) cells. CD4+CD25+FOXP3+ nTreg cells derived from thymus are known to modulate immune disorders such as autoimmunity, allergy, and graft rejection by suppressing activation of naïve T cells, effector T cells and memory CD4+ and CD8+ T cells [106]. iTreg cells, so called as type 1 regulatory T cells (Tr1), are developed from naïve T cells in the periphery during an active immune response including antigens, IL-2, IL-10 and TGF-β [107]. Furthermore, human FOXP3 expressing nTreg cells can be subdivided into CD25+/CD45RA+/FOXP3lo (resting Treg cells; rTreg cells), CD25hi/CD45RA−/FOXP3hi (activated Treg cells; aTreg cells), and CD25+/CD45RA−/FOXP3lo (non-Treg cells) [108, 109]. Interestingly, Whiteside [110] suggested that iTreg cells should be depleted and nTreg cells are promoted in cancer patients, since iTreg cells produce immunosuppressive cytokines, notably TGF-β as well as prostaglandin E2 resistant to oncological therapy, while FOXP3+ nTreg cells are responsible for peripheral tolerance to avoid autoimmune disease [111].
In addition, CD4+CD25+ regulatory T cell deficiency due to loss-of-function mutations of FOXP3 gene induces the lethal autoimmune syndromes observed in FOXP3-null mice or FOXP3-mutant scurfy mice [98]. Consistently, the infiltration of effector Treg cells into tumor cells indicates poor prognosis of overall survival (OS) [87, 112] and FOXP3 is overexpressed in pancreatic [113], prostate [114] and gastric [115] cancers by suppressing antitumor immunity [116] and inducing effector CD4+ T cell death by activation of proapoptotic protein Bad and Bim [101].
The interplay between FOXP family members and other molecules
FOXP1 and interleukins (IL-7 and 21)
FOXP1 works as a negative regulator of tumor-specific CD4+ T helper 7 (Th7) cells for cancer immunotherapy, since mature naïve CD4+ T cells proliferate to exert antitumor effect programmed by IL-7 only in the absence of FOXP1 [117, 118]. Similarly, CD8+ T cells lacking the transcription factor FOXP1 show function and effector phenotype of IL-7, since FOXP1 represses expression of IL-7 receptor α-chain (IL-7Rα), phosphorylation of MEK and ERK [119]. Furthermore, FOXP1 suppresses the antitumor function of interleukin 21 (IL-21) to stimulate the secretion of IFNγ from CD4+ T or CD8+ T cells in estrogen positive MCF-7 breast cancers [70, 120]. In contrast, De Silva et al. indicated that FOXP1hi supernatant reduced lymphocyte migration by secretion of chemokines such as CXCL9, CXCL10, CXCL11, CXCL13, CX3CL, CCL20, IL-7, IL-21, and IFNγ compared to FOXP1lo supernatant [121]. Nevertheless, the mechanism by which FOXP1 represses IL-7 or IL-21 still remains unclear to date.
FOXPs and nuclear factor of activated T cell (NFAT)
Nuclear factor of activated T cells (NFAT) is an inducible nuclear factor binding to the antigen receptor response element-2 (ARRE-2) of IL-2 promoter in human T cells and also is involved in cell proliferation, survival, invasion, migration and angiogenesis [122]. NFAT family include five members such as four calcium-responsive isoforms named NFAT1 (NFATp or NFATc2), NFAT2 (NFATc or NFATc1), NFAT3 (NFATc4), NFAT4 (NFATx or NFATc3) and a tonicity-responsive enhancer-binding protein (NFAT5 or TonEBP) [122, 123]. The NFAT isoforms are constitutively activated in several cancer types [122, 124]. Interestingly, FOXP proteins form cooperative complexes with NFAT [74] and so the crystal structures of the FOXP2–NFAT2 DNA complex are also conserved with FOXP1 and FOXP3 [15, 123]. Also, NFAT1 depletion inhibited invasion and migration of human non-small cell lung cancer [125] and NFAT overexpression promoted invasion in breast cancers via upregulation of cyclooxygenase-2, α6β4 integrin and glypican-6 [126,127,128]. Likewise, Oskay et al showed that FOXP1 directly binds to NFAT1 on DNA and promotes migration in MDA-MB231 breast cancer cells [18]. However, FOXP1 binds poorly to the ARRE2 composite site in the absence of NFAT1 [15]. Overall, NFAT1 closely interacts with FOXP1 or FOXP3 in cancer progression.
FOXPs and p53/p21
It is well documented that p53 suppresses tumorigenesis by regulating apoptosis, metabolic networks, free radical and senescence [129]. Recently, Jung et al. demonstrated that p53 induction by genotoxic reagent upregulates FOXP3 expression and conversely FOXP3 is regulated in a p53-dependent manner by MDM2 inhibitor Nutlin-3 [130]. Of note, FOXP3 induced cellular senescence in MCF7 and HCT116 cells via activation of p53/p21 and reactive oxygen species(ROS) production [131]. Furthermore, FOXP1 known as a B cell oncogene is reduced by miR-34a via p53 networks [132], indicating .the cloe interaction between FOXP1/3 and p53 signaling.
Accumulating evidence demonstrate that the cyclin-dependent kinase inhibitor p21 WAF1/CIP1 is a widely-characterized p53 target gene during cell cycle arrest [133, 134]. Of note, Gascoyne et al. indicated that FOXP2 activation preceded up-regulation of p21WAF1/CIP1 in 143B osteosarcoma cells [19]. Likewise, FOXP2 overexpression upregulated the expression of p21 in hematopoietic stem cells (HSCs). However, though p21 is known a downstream effector of gp130/STAT3 activation [135], exogenous STAT3 promoter IL-6 could not rescue reduction of p21WAF1/CIP1 expression following FOXP2 depletion, implying that FOXP2-dependent regulation of p21 WAF1/CIP1 independent of IL-6 [19]. Also, it was known that FOXP2 regulates p21 independent of p53 status in cell lines (143B mutant, MG-63 null, U2OS wild-type, SAOS-2 null) different from FOXP1 [136, 137] proteins, which should be further investigated in specific cell lines and in vivo.
FOXP3 and Interleukin-17
Emerging evidence shows that Treg population is observed in tumor infiltrating lymphocytes of several cancers such as breast cancer [138], gastric cancer [139], pancreatic cancer [140] and, colorectal cancer [141] and lung cancer [142]. Interestingly, CD4 + CD25 + FOXP3 + (GFP+) T cells can differentiate into T helper 17 (Th17) cells in the presence of IL-6 [57] and Th17 cells, one of the CD4+ T cells, can produce IL-17 to protect cells against microbial infection [143]. In contrast, activation of Treg cells reduces antipathogenic or anticancer immunity, leading to cancer progression and infection [144].
Hence, the balance between FOXP3+ Treg cells and Th17 cells is considered an important factor for treatment of autoimmune diseases [145] and cancers [146]. Indeed, Maruyama et al. [54] reported that the infiltration of Th17 cells gradually decreased compared to increased Treg cells in gastric cancer progression. However, Hou et al. [20] claimed that Th17 cells and FOXP3-expressing T cells were significantly increased in uterine cervical cancer and cervical intraepithelial neoplasia while the ratio of Th17/FOXP3 Treg cells was decreased in tumor-infiltrating lymphocytes (TILs). Consistently, Beyer [147] suggested a novel strategy to suppress expansion and differentiation of naïve Treg cells induced by IL-2 therapy, since Treg cells induce immunosuppression in neoplastic patients. For reprogamming Treg cells into Th17 like cells, Sharma et al. [148] suggested that indoleamine 2,3-dioxygenase (IDO) inhibitor and antitumor vaccine converted Treg cells into Th17 phenotype cells in B16 melanoma mouse model. Similarly, Bahan et al. [149] reported that high dose of oligonucleotides (CpG) treatment directly reprogrammed splenic FOXP3 Treg cells to express IL-17 with efficiency of 6–7% in IDO-KO mice model.
In contrast, Xu et al. [57] suggested that activated FOXP3+ Treg cells have potential to stimulate CD4+CD25−FOXP3−T cells or can differentiate into Th17 phenotype cells in the presence of IL-6 regardless of exogenous TGF-β. Thereafter, the property of IL-17+FOXP3+ T cells has been recognized with low expression of Helios [150] and overexpression of ICOS [139] and RORγt for distinguishing peripherally induced and thymic-derived FOXP+ Treg cells. Thus, IL-17+FOXP3+ T cells were considered as an intermediate differentiation stage between Th17 cells and Treg cells, since Treg cells can be converted into IL-17+FOXP3+ T cells by stimulation of TGF-β and/or IL-6 [151]. Accumulating evidence reveals that IL-17 + FOXP3+ T cells are shown highly in colon [152] and esophageal [153] cancers, inflammatory bowel disease [154], periodontitis [155], and rheumatoid arthritis [156]. Nevertheless, further studies are required to explore the mechanisms and interplay with other molecules in the differentiation of IL-17+FOXP3+ T cells, and to assess clinical implications targeting the balance between FOXP3+ Treg cells and Th17 cells in the near future.
FOXP3 and RUNX
RUNX proteins such as RUNX1 (AML1) and RUNX3 induced by TGF-β play a critical role in embryonic development, hematopoiesis and T cell development by regulating CD4, CD8 and lymphokine genes [157, 158]. Emerging evidence indicates that the complex of core-binding factor subunit beta (CBFβ) and runt-related transcription factor 1 and 3 (RUNX1 and 3) is essential for Treg suppressive function [7, 159]. Recouvreux et al. suggested that RUNX1 plays a critical role in breast tumor progression, only depending on FOXP3 availability [21]. Also, RUNX1 is known as an important target for chromosomal translocations of leukemias with CD4+CD25highFOXP3+ Treg cells [92, 160], while RUNX3 methylation and silencing are observed in several epithelial cancers [161, 162]. Also, the role of RUNX1 or RUNX3 should be explored in the differentiation of I IL-17+FOXP3+ T cells in association with mTOR, CNOT2, HIF-1α and RIF-4 that are involved in tumor progression and immune tolerance.
FOXP3 and STAT3/5
There is accumulating evidence that STAT3 is involved in cancer initiation and development as an oncogenic transcription factor [163]. Regarding relationship between FOXP3 and STAT3, Hossain et al. revealed that FOXP3 silencing decreased the expression of STAT3-related genes such as IL-6, VEGFA, C-Myc, BCL2L1, and CCND1, but not TGF-β1 in tumor induced regulatory T cells by qRT-PCR analysis [22]. Conversely, STAT3 promoter IL-6 induced DNA-methyltransferase 1 (DNMT1) expression and promoted STAT3-dependent methylation of FOXP3 in Treg cells [164]. Also, Lam et al. demonstrated the important role of Cdk5 in phosphorylation of STAT3 (S727) to bind to FOXP3 gene in CD4+ T cells [165].
Additionally, activated STAT5 is associated with suppression of antitumor immunity, since STAT5 plays a critical role in the function and development of Treg cells to promote proliferation, invasion, and survival of tumor cells [166]. Furthermore, several studies demonstrated that STAT5 mediates the critical link between the IL-2/15 and FOXP3. Thus, in T-cell-specific STAT5-null mice, CD25- and FOXP3-expressing cells were reduced and also STAT5 was detected to bind to gamma interferon-activated sequence (GAS) sites of FOXP3 promoter [167, 168]. Similarly, Wang et al showed that activated STAT5 is associated with increased FOXP3 expression in melanoma cells and lymphocytes [169]. Hence, the pivotal role of STAT3 or STAT5 should be extensively explored in FOXP3+ Treg cells in vitro and in vivo.
FOXP1/3 and FOXO3a/1
Among FOXO subfamilies, FOXO3a is well known to act as a tumor suppressor by inducing apoptosis and cell cycle arrest [170]. Interestingly, FOXO3a phosphorylation as a downstream of E3 ubiquitin-protein ligase CBL-B increased FOXP3 expression in CBL-B deficient T cells and conversely FOXO3a depletion impaired TGF- β driven FOXP3 induction [171], since FOXO3a directly binds to the FOXP3 promoter in iTreg cells [172]. Furthermore, corporation of FOXO1 and STAT5 suppresses FOXP3 expression and production in response to the TCR signaling [173], since miR-182 downregulates FOXO1 [174] and PTPN2 dephosphorylates STAT5 [175], leading to suppression of FOXP3. Also, Du et al. reported that Mst1/Mst2 kinases enhanced FOXO3-mediated FOXP3 expression by maintaining the stability of FOXO1/3 proteins through phosphorylation of S212 and S207 and inhibited TCR induced Akt activation [176], implying the role of Mst1/Mst2 kinases in FOXO3-mediated FOXP3 expression. In addition, Bi et al. [118] reported that FOXO1 acts as a negative regulator and FOXP1 and a negative regulator of CD4+ T helper 9 (TH9) cell differentiation and antitumor activity. Likewise, van Boxtel et al. demonstrated that FOXO1/3 activation in FOXP1 depleted cells enhanced cell death, indicating the opposing roles between FOXO1/3 and FOXP1 [177]. Hence, the underlying mechanism of FOXO1 or FOXO3 mediated FOXP1/3 expression should be further explored in vitro and in vivo in tumor progression and chemoresistance.
FOXP3 and NF-κB
It is well documented that NF-κB is involved in inflammation, proliferation, cell adhesion and tumor progression [178]. Of note, Hao et al. revealed that FOXP3 suppresses cell migration by inhibition of NF-κB activity and COX-2 expression in gastric cancers [179]. However, the tumor suppressor role of FOXP3 was disturbed under inflammatory microenvironment in gastric cancers, since FOXP3 interacts with two key transcription factors such as nuclear factor of activated T cells (NFAT) and NF-κB [91, 180]. Consistently, Wang et al. suggested that silencing of FOXP3 promoted the proliferative, migratory and invasive properties of A549 cells by downregulation of ZO-1, upregulation of vimentin and phosphorylation of NF-κB at protein level. Likewise, FOXP3 downregulation attenuated the expression of LIM Domain Only 2 (LMO2) but increased the expression of an oncogenic transcription factor T-cell acute lymphocytic leukemia protein 1 (TAL1) at mRNA level in T-cell acute lymphoblastic leukemia (T-ALL) cells [181]. However, Chu et al. demonstrated that FOXP3 depletion downregulated cyclin D1 and NF-κB subunit p65, but upregulated caspase-3 levels in K1 and WRO thyroid cancer cells [182]. Also, Jia et al. indicated that blockade of toll-like receptor 4 (TLR4) signaling induced downregulation of FOXP3 after blocking NF-κB in A549 cells [183]. Here microenvironmental conditions where FOXP3 acts as a tumor suppressor or an oncogene should be clearly determined in specific cancers in association with TRL4, NFAT and NF-κB signaling in the future.
FOXPs and VEGF
It is well documented that angiogenesis related molecules including VEGF play pivotal role in carcinogenesis [184]. Le et al. reported that FOXP3 suppresses VEGF signaling to exert anti-angiogenic or anti-metastatic effect in MDA-MB-231 breast cancer cells [185, 186]. However, Tang and his colleagues demonstrated that FOXP3 is positively correlated with VEGF-C in lymphangiogenesis of cervical cancer [187]. Likewise, FOXP1 promoted proliferation, migration and tube formation in cultured endothelial cells [9]. Also, Wan et al. demonstrated the correlation between FOXP1 and VEGF in the patients with renal carcinoma [188]. Furthermore, He et al. suggested that FOXP1 and FOXP2 upregulates levels of angiogenic factor such as VEGF with G patch and FHA domains 1 (AGGF1) [189] and induces angiogenesis in glioma cells [190]. Overall, it is demonstrated that FOXP proteins are closely associated with VEGF signaling.
FOXPs and noncoding RNAs
Emerging evidence suggests that FOXP members modulate various noncoding RNAs during cancer development and progression, since noncoding RNAs (ncRNAs) are RNA molecules that are not translated into proteins, including transfer RNAs (tRNAs) and ribosomal RNAs (rRNAs), as well as small RNAs such as microRNAs (miRNAs), siRNAs, circular RNA and the long ncRNAs (lncRNAs) [191]. For instance, FOXP1 induction by inhibition of miR-9 promoted tumor growth, while FOXP1 knockdown suppressed the growth of epidermal growth factor receptor (EGFR) dependent cancers [192]. Also, downregulation of FOXP1 increased miR-34a level as a tumor suppressor in gastric diffuse large B-cell lymphoma (gDLBCL) cells [193]. Furthermore, elevation of miR-199a and repression of FOXP2 are prominent features of malignant breast cancer with poor survival rate [194]. Likewise, miR-181d-5p [195], miR-374b-5p [196], miR-122 [197], miR-150 [198] and miR-504 [199] act as a tumor suppressor by inhibition of FOXP1, while miR-376a [200] and miR-139 [201] work as a tumor suppressor via inhibition of FOXP2 in lymphoma and osteosarcoma. Additionally, miR-146 [23, 202], miR-7 and miR-155 [203] induced by FOXP3 act as a tumor suppressor in breast and prostate cancers. Notably, Liu et al. revealed that FOXP3-induced miR-146a/b suppressed tumor cell proliferation and enhanced apoptosis by inhibition of NF-κB activation through suppression of interleukin-1 receptor-associated kinase 1 (IRAK1) and TNF receptor associated factor-6 (TRAF-6) in MCF-7 breast cancer cells [202]. Likewise, McInnes et al. showed that FOXP3 induced miR-7 and miR-155 to target oncogenic SATB1 in BT549 breast cancer cells [203], whereas overexpression of miR-338-3p inhibited proliferation of hepatocellular carcinoma cells and induced cell cycle arrest partially through the downregulation of FOXP4 [30]. Of note, miR-338-3p [30], miR-491–5p [29] and miR-138 [93, 204] were downregulated in HCC, osteosarcoma and non-small lung cancer cells, targeting FOXP4. In addition, lncRNA MALAT1 [205] and SNHG12 [206] promoted proliferation of multiple myeloma and glioma targeting FOXP1, while lncRNA UFC1 [207] and 7SL [208] enhanced proliferation of cervical cancer and osteosarcoma, targeting FOXP3 and FOXP4, respectively. Notably, circular RNA SHKBP1 was upregulated in malignant glioma targeting FOXP1 or FOXP2 [190], while circular RNA ZNF609 [209] and MYO9B [28] were upregulated in renal cancer and breast cancer, respectively, targeting FOXP4. Taken together, the critical roles of these miRNAs, lncRNAs, circular RNAs should be further investigated during antitumor or oncogenic effects of FOXP families in vitro, in vivo and clinically in the future.
Clinical application and cellular cancer immunotherapy
Among clinical trials Dr. Sylvain Ladoire performed clinical trial (ClinicalTrials.gov Identifier: NCT01513408) at Centre Georges Francois Leclerc with 500 participants with a topic, “Prospective study of the relevance of T lymphocytes tumor infiltrates D8 and FOXP3 as a new immune prognostic biomarker in breast cancer treated by neoadjuvant chemotherapy to verify that accumulation of regulatory T-lymphocytes expressing FOXP3 is associated with poor prognosis in breast cancer patients. Also, another clinical trial (ClinicalTrials.gov Identifier: NCT03718923) on FOXP1 related neurodevelopmental disorders is underway at The Seaver Autism Center for Research and Treatment, New York. Also, pilot study (ClinicalTrials.gov Identifier: NCT01538485) was completed to assess the effects of Vitamin D supplementation on the number of regulatory FOXP3+ T cells in the gastrointestinal mucosa in healthy women and men in Austria in 2012. Besides, graft rejection studies in renal transplant recipients (ClinicalTrials.gov Identifier: NCT01446484) and liver transplant recipients (ClinicalTrials.gov Identifier: NCT01678937) were conducted targeting FOXP3. Here it was found that clinical trials have been conducted targeting FOXP1 or FOXP3. Emerging evidence indicates that FOXP3 regulates Treg development and functions [16] to induce the immune evasion of tumor cells through imbalance of immunoediting and immunosurveillance [17] and. FOXP1 acts as a transcriptional regulator for primary human CD4+ T cells [210]. Recently dendritic cells (DCs) based immunotherapy has been on the spotlight for cancer therapy, since DCs are considered the most powerful antigen-presenting cells (APCs) activating naïve and memory immune responses [211]. Through a lot of clinical trials with DCs via various routes (intradermal, intranodal, intravenous, subcutaneous, intratumoral) [212], cancer immunotherapy efficacy by DC vaccine was limited mainly due to inhibition of immune response by tumor-secreted TGF-β and FOXP3 related Treg cells and low quality of DC production [213]. Notably, CD4+CD25+FOXP3+ (GFP+) T cells can differentiate into Th17 cells in the presence of IL-6 [57] and T helper 17 (Th17) cells, one of the CD4+ T cells, can produce IL-17 to protect against microbial infection [214], while excessive activation of Treg cells suppresses antipathogenic or anticancer immunity, leading to chronic infection and tumor progression [144]. The balance between FOXP3+ Treg cells and Th17 cells is considered an important target for treatment of autoimmune diseases [145] and cancers [146]. Thus, next generation DC immunotherapy is required for more effective cancer therapy. Thus, Treg depletion and Th17 booster can be a potent strategy for DC cancer immunotherapy and DC vaccines were suggested as a next generation cancer immunotherapy [215], only if the standardization and quality of DC vaccines can be upgraded to enhance migrating activity to the lymph nodes, presenting antigen and costimulation to T cells, and surviving long enough for optimal T-cell activation. In the same line, combination therapy of DCs with checkpoint inhibitors including ipilimumab [216] or other immune cells or oncolytic virus [216] is considered attractive in cancer therapy [216, 217]. In this regard, we suggest the cocktail of specialized DC vaccines and Th17 cells by reprogramming Treg cells into Th17 cells [146] or ex vivo expansion of Th17 cells from human PBMCs [218] is suggested as a next generation cellular cancer immunotherapy, which should be further investigated in vivo and clinically.
Conclusions and perspectives
FOXP family consisting of FOXP1, FOXP2, FOXP3 and FOXP4 are involved in the embryonic development, immune disorders and cancer progression. Accumulating evidence reveals that FOXP family act as a tumor suppressor or an oncogene in several cancers. FOXP1 was overexpressed with poor prognosis in DLBCL, MALT, primary cutaneous large B-cell lymphomas and follicular lymphoma as an oncogene, while FOXP1 worked in breast, lung carcinoma and U251 glioma cells as a tumor suppressor. Also, FOXP2 was activated for up-regulation of p21 in 143B osteosarcoma cells, while FOXP4 was overexpressed in A549 and H1703 NSCLC cells along with prostate cancer risk. Likewise, CD4+/CD25+/FOXP3+Treg cells are overexpressed in pancreatic, prostate and gastric cancers through immunosuppression and cancer progression as an oncogene, while FOXP3 overexpression indicates good prognosis in patients with breast cancers as a tumor suppressor. Given that transcriptional activity of FOXP1, FOXP2, and FOXP4 is modulated by tissue-specific homo- and heterodimerisation via a zinc finger and a leucine zipper motif [33], their functional similarity is expected and so their more detailed protein-protein interactions and the molecular conditions for their dual roles as an oncogene or a tumor suppressor should be clarified in specific cancer types in the future, considering reports that dual functions of FOXP family may be closely associated with tumor microenvironmental factors such as dendritic cells (DCs), inflammatory cytokines especially in colon and esophageal cancers related to inflammation.
Regarding interplay of FOXP members with their related molecules, FOXP1 is closely associated with IL-7, IL-21, NFAT, while FOXP2 is more related to p21 and FOXP3 is critically associated with IL-17, RUNX, STAT3/5, FOXO3a/1 and NF-κB. Also, it is well documented that FOXP members are regulated by miRNAs, lncRNAs, circular RNAs (Table 1, Fig. 3). Nonetheless, their detailed interactions are not fully understood as a tumor suppressor or an oncogene, which indicates further mechanistic study in vitro and in transgenic mouse model. Also, the underlying mechanisms that FOXO1/3 suppresses FOXP1/3 should be further examined in vitro and KO mice model.

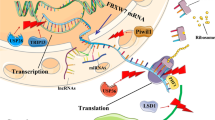
Interplay between FOXP family and their related molecules targeted by noncoding RNAs. FOXP family members consist of FOXP1, FOXP2, FOXP3 and FOXP4 that communicate with other molecules. Interleukin-6 (IL-6) activates the Janus tyrosine kinase (JAK) family members (JAK1, JAK2, and TYK2), leading to the activation of transcription factors of the signal transducer and activator of transcription (STAT) family including STAT3 and STAT5. Also, IL-6 induces DNA-methyltransferase 1 (DNMT1) expression and promotes STAT3-dependent methylation of FOXP3. FOXP2 overexpression upregulates the expression of p53/ p21, a downstream effector of gp130/STAT3. Transforming growth factor-β (TGF-β) activates phosphorylation of SMAD, which forms complex with CBFβ/RUNX1/3 for maintenance of FOXP3, but ThPOK blocks RUNX [219]. TNF-α stimulates protein phosphatase 1 (PP1) for dephosphorylation of FOXP3 (S418). FOXP3 interacts with two key transcription factors such as nuclear factor of activated T cells (NFAT) and NF-κB. FOXO3a phosphorylation increases FOXP3 and FOXO1 acts as a negative regulator and FOXP1. The receptor tyrosine kinases (RTKs) activate MEK-ERK signaling axis, which is repressed by FOXP1. Among noncoding RNAs, MALAT1, SNHG12 and CircRNA SHKBP1 activate FOXP1, while miR-9, miR-19a, miR-34a, miR-92a, miR-122, miR-150, miR-181–5p, miR-374-5p and miR-504 downregulate FOXP1. CircRNA SHKBP1 increases FOXP2, while miR-23a, miR-139, miR-190, miR-196b and miR-376a suppress FOXP2. UFC1 activates FOXP3 and miR-138, miR-338-3p and miR-491–5p downregulate FOXP4, while circR-SHKBP1, CircR-MYO9B and MFI2 upregulate FOXP4
Additionally, FOXP3 is known a key transcription factor for the development and function of CD4+CD25+ regulatory T (Treg) cells. Recently the balance between FOXP3+ Treg cells and Th17 cells provides a new insight into a potent cellular cancer therapy, since FOXP3+Treg cells can be differentiated into antimicrobial Th17 cells or IL-17+FOXP3+ T cells to overcome the immunosuppressive function of Treg cells, leading to anti-tumor immunity in cancers. The efficacy of DC cancer immunotherapy has been limited due to immunosuppression by tumor-secreted TGF-β and Treg cells with tumor response rates rarely exceeding 15% in many clinical trials [213, 217], though many clinical trials were completed in melanoma (> 1000 patients), renal cell carcinoma (RCC; > 250 patients), glioblastoma (GBM; > 500 patients), prostate cancer (> 750 patients) [217]. Thus, Treg depletion and Th17 booster can be a potent strategy for DC cancer immunotherapy and mature DC vaccines were suggested as a next generation cancer immunotherapy with the standardization and quality of DC vaccines. Here we suggest the cocktail of specialized DC vaccines and Th17 cells by reprogramming Treg cells into Th17 cells [146] or ex vivo expansion of Th17 cells from human PBMCs [218] is suggested as a next generation cellular cancer immunotherapy, which should be further investigated in vivo and clinically (Fig. 4).

Cellular cancer immunotherapy by using Th17 cells and DC vaccine cocktail. Proinflammatory T helper 17 (Th17) cells, one of the CD4+ T cells, can produce IL-17 and protect cells against microbial infection, expressing RORγt (orphan nuclear receptor) [143], while excessive activation of Treg cells suppresses antipathogenic or anticancer immunity by inactivation of Th1, CTL and NK cells [220], leading to chronic infection and tumor progression [144]. Dendritic cells (DCs), the most efficient antigen-presenting cells (APCs) of the innate immune system, can be produced from peripheral blood mononuclear cells (PBMCs) or human pluripotent stem cells (hPSC) including embryonic stem cells and induced pluripotent stem cells [221]. Loading tumor specific antigens on immature DCs is the first step for DC vaccine production and DCs can be activated for maturation by defined cytokine formulation such as IL-1β+ IL-6+ PGE2+ TNF and TLR agonists (IL-2, IFNα/γ, GM-CSF, bacterial toxoids). Combination of TGFβ1 and IL-6 can be used for Th7 differentiation by reprogramming Treg cells into Th17 cells [146] and also a cocktail of TGFβ1, IL-6,IL-23, IL-1β and IL-21 is used for Th17 differentiation expansion from human PBMCs [218, 222]. Next generation cancer immunotherapy by a cocktail of DC vaccines and Th17 cells is suggested for cancer regression, which should be validated in vivo or clinically by intradermal injection or infusion after checking safety in the future
Overall, our review demonstrates that FOXP proteins are critically involved in cancer progression and immunology in concert with other molecules including noncoding RNAs and signaling pathways as potent biomarkers and targets for cancer diagnosis and treatment and also suggests another clinical trial for cellular cancer immunotherapy by DC vaccine and Th17 cells cocktail through Treg depletion, which should be also validated in vitro, in vivo and clinically in the future.
Availability of data and materials
Not applicable.
Abbreviations
- FHD:
-
Forkhead DNA binding domain
- FOXP:
-
Forkhead box P
- HEK293:
-
Human embryonic kidney 293
- IL-17:
-
Interleukin 17
- MALT:
-
Mucosa-associated lymphoid tissue
- MHC:
-
Major Histocompatibility Complex
- Myc:
-
MYC proto-oncogene
- NFAT1:
-
Nuclear factor of activated T-cells 1
- NF-κB:
-
nuclear factor kappa-light-chain-enhancer of activated B cells
- RUNX1:
-
Runt-related transcription factor 1
- STAT3:
-
Signal transducer and activator of transcription 3
- Th17:
-
T helper 17
- Treg:
-
regulatory T
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147:275–92.
Santos ME, Athanasiadis A, Leitao AB, DuPasquier L, Sucena E. Alternative splicing and gene duplication in the evolution of the FoxP gene subfamily. Mol Biol Evol. 2011;28:237–47.
Jackson BC, Carpenter C, Nebert DW, Vasiliou V. Update of human and mouse forkhead box (FOX) gene families. Hum Genomics. 2010;4:345–52.
Myatt SS, Lam EW. The emerging roles of forkhead box (Fox) proteins in cancer. Nat Rev Cancer. 2007;7:847–59.
Uva V, Sfondrini L, Triulzi T, Casalini P, Tagliabue E, Balsari A. FOXP3 expression in tumor cells and its role in cancer progression. Atlas Genet Cytogenet Oncol Haematol. 2015;19:234–9.
Kaur G, Busch R, Gaston HJ. The role of Foxp3 in regulatory T cell differentiation and function. Curr Immunol Rev. 2009;5:89–101.
Chiu YC, Li MY, Liu YH, Ding JY, Yu JY, Wang TW. Foxp2 regulates neuronal differentiation and neuronal subtype specification. Dev Neurobiol. 2014;74:723–38.
Grundmann S, Lindmayer C, Hans FP, Hoefer I, Helbing T, Pasterkamp G, Bode C, de Kleijn D, Moser M. FoxP1 stimulates angiogenesis by repressing the inhibitory guidance protein semaphorin 5B in endothelial cells. PLoS One. 2013;8:e70873.
Carvalho MI, Pires I, Prada J, Gregorio H, Lobo L, Queiroga FL. Intratumoral FoxP3 expression is associated with angiogenesis and prognosis in malignant canine mammary tumors. Vet Immunol Immunopathol. 2016;178:1–9.
Patzelt T, Keppler SJ, Gorka O, Thoene S, Wartewig T, Reth M, Forster I, Lang R, Buchner M, Ruland J. Foxp1 controls mature B cell survival and the development of follicular and B-1 B cells. Proc Natl Acad Sci U S A. 2018;115:3120–5.
MacDermot KD, Bonora E, Sykes N, Coupe AM, Lai CS, Vernes SC, Vargha-Khadem F, McKenzie F, Smith RL, Monaco AP, Fisher SE. Identification of FOXP2 truncation as a novel cause of developmental speech and language deficits. Am J Hum Genet. 2005;76:1074–80.
Teufel A, Wong EA, Mukhopadhyay M, Malik N, Westphal H. FoxP4, a novel forkhead transcription factor. Biochim Biophys Acta. 1627;2003:147–52.
Lu L, Barbi J, Pan F. The regulation of immune tolerance by FOXP3. Nat Rev Immunol. 2017;17:703–17.
Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, Bates DL, Guo L, Han A, Ziegler SF, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell. 2006;126:375–87.
Fleskens V, van Boxtel R. Forkhead box P family members at the crossroad between tolerance and immunity: a balancing act. Int Rev Immunol. 2013;33:94–109.
Lakshmi Narendra B, Eshvendar Reddy K, Shantikumar S, Ramakrishna S. Immune system: a double-edged sword in cancer. Inflamm Res. 2013;62:823–34.
Oskay Halacli S. FOXP1 enhances tumor cell migration by repression of NFAT1 transcriptional activity in MDA-MB-231 cells. Cell Biol Int. 2017;41:102–10.
Gascoyne DM, Spearman H, Lyne L, Puliyadi R, Perez-Alcantara M, Coulton L, Fisher SE, Croucher PI, Banham AH. The Forkhead transcription factor FOXP2 is required for regulation of p21WAF1/CIP1 in 143B osteosarcoma cell growth arrest. PLoS One. 2015;10:e0128513.
Hou F, Li Z, Ma D, Zhang W, Zhang Y, Zhang T, Kong B, Cui B. Distribution of Th17 cells and Foxp3-expressing T cells in tumor-infiltrating lymphocytes in patients with uterine cervical cancer. Clin Chim Acta. 2012;413:1848–54.
Recouvreux MS, Grasso EN, Echeverria PC, Rocha-Viegas L, Castilla LH, Schere-Levy C, Tocci JM, Kordon EC, Rubinstein N. RUNX1 and FOXP3 interplay regulates expression of breast cancer related genes. Oncotarget. 2016;7:6552–65.
Hossain DM, Panda AK, Manna A, Mohanty S, Bhattacharjee P, Bhattacharyya S, Saha T, Chakraborty S, Kar RK, Das T, et al. FoxP3 acts as a cotranscription factor with STAT3 in tumor-induced regulatory T cells. Immunity. 2013;39:1057–69.
Liu R, Yi B, Wei S, Yang WH, Hart KM, Chauhan P, Zhang W, Mao X, Liu X, Liu CG, Wang L. FOXP3-miR-146-NF-kappaB Axis and therapy for precancerous lesions in prostate. Cancer Res. 2015;75:1714–24.
Xie Z, Guo Z, Liu J. Whey acidic protein/four-disulfide Core domain 21 regulate Sepsis pathogenesis in a mouse model and a macrophage cell line via the Stat3/toll-like receptor 4 (TLR4) signaling pathway. Med Sci Monit. 2018;24:4054–63.
Akimova T, Zhang T, Negorev D, Singhal S, Stadanlick J, Rao A, Annunziata M, Levine MH, Beier UH, Diamond JM, et al. Human lung tumor FOXP3+ Tregs upregulate four "Treg-locking" transcription factors. JCI Insight. 2017;2:e94075.
Huang W, Solouki S, Koylass N, Zheng SG, August A. ITK signalling via the Ras/IRF4 pathway regulates the development and function of Tr1 cells. Nat Commun. 2017;8:15871.
Yasuda K, Kitagawa Y, Kawakami R, Isaka Y, Watanabe H, Kondoh G, Kohwi-Shigematsu T, Sakaguchi S, Hirota K. Satb1 regulates the effector program of encephalitogenic tissue Th17 cells in chronic inflammation. Nat Commun. 2019;10:549.
Wang N, Gu Y, Li L, Wang F, Lv P, Xiong Y, Qiu X. Circular RNA circMYO9B facilitates breast cancer cell proliferation and invasiveness via upregulating FOXP4 expression by sponging miR-4316. Arch Biochem Biophys. 2018;653:63–70.
Yin Z, Ding H, He E, Chen J, Li M. Up-regulation of microRNA-491-5p suppresses cell proliferation and promotes apoptosis by targeting FOXP4 in human osteosarcoma. Cell Prolif. 2016;50:e12308.
Wang G, Sun Y, He Y, Ji C, Hu B. MicroRNA-338-3p inhibits cell proliferation in hepatocellular carcinoma by target forkhead box P4 (FOXP4). Int J Clin Exp Pathol. 2015;8:337–44.
Clark KL, Halay ED, Lai E, Burley SK. Co-crystal structure of the HNF-3/fork head DNA-recognition motif resembles histone H5. Nature. 1993;364:412–20.
Chen Y, Chen C, Zhang Z, Liu CC, Johnson ME, Espinoza CA, Edsall LE, Ren B, Zhou XJ, Grant SF, et al. DNA binding by FOXP3 domain-swapped dimer suggests mechanisms of long-range chromosomal interactions. Nucleic Acids Res. 2015;43:1268–82.
Li S, Weidenfeld J, Morrisey EE. Transcriptional and DNA binding activity of the Foxp1/2/4 family is modulated by heterotypic and homotypic protein interactions. Mol Cell Biol. 2004;24:809–22.
Wang B, Lin D, Li C, Tucker P. Multiple domains define the expression and regulatory properties of Foxp1 forkhead transcriptional repressors. J Biol Chem. 2003;278:24259–68.
Perumal K, Dirr HW, Fanucchi S. A single amino acid in the hinge loop region of the FOXP Forkhead domain is significant for dimerisation. Protein J. 2015;34:111–21.
Stroud JC, Wu Y, Bates DL, Han A, Nowick K, Paabo S, Tong H, Chen L. Structure of the forkhead domain of FOXP2 bound to DNA. Structure. 2006;14:159–66.
Chu YP, Chang CH, Shiu JH, Chang YT, Chen CY, Chuang WJ. Solution structure and backbone dynamics of the DNA-binding domain of FOXP1: insight into its domain swapping and DNA binding. Protein Sci. 2011;20:908–24.
Obsil T, Obsilova V. Structure/function relationships underlying regulation of FOXO transcription factors. Oncogene. 2008;27:2263–75.
Mendoza E, Scharff C. Protein-protein interaction among the FoxP family members and their regulation of two target genes, VLDLR and CNTNAP2 in the Zebra finch Song system. Front Mol Neurosci. 2017;10:112.
Lu MM, Li S, Yang H, Morrisey EE. Foxp4: a novel member of the Foxp subfamily of winged-helix genes co-expressed with Foxp1 and Foxp2 in pulmonary and gut tissues. Gene Expr Patterns. 2002;2:223–8.
Godfrey VL, Wilkinson JE, Rinchik EM, Russell LB. Fatal lymphoreticular disease in the scurfy (sf) mouse requires T cells that mature in a sf thymic environment: potential model for thymic education. Proc Natl Acad Sci U S A. 1991;88:5528–32.
Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, Wilkinson JE, Galas D, Ziegler SF, Ramsdell F. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001;27:68–73.
Taams LS, Smith J, Rustin MH, Salmon M, Poulter LW, Akbar AN. Human anergic/suppressive CD4(+)CD25(+) T cells: a highly differentiated and apoptosis-prone population. Eur J Immunol. 2001;31:1122–31.
Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–64.
Shu W, Yang H, Zhang L, Lu MM, Morrisey EE. Characterization of a new subfamily of winged-helix/forkhead (Fox) genes that are expressed in the lung and act as transcriptional repressors. J Biol Chem. 2001;276:27488–97.
Banham AH, Beasley N, Campo E, Fernandez PL, Fidler C, Gatter K, Jones M, Mason DY, Prime JE, Trougouboff P, et al. The FOXP1 winged helix transcription factor is a novel candidate tumor suppressor gene on chromosome 3p. Cancer Res. 2001;61:8820–9.
Fox SB, Brown P, Han C, Ashe S, Leek RD, Harris AL, Banham AH. Expression of the forkhead transcription factor FOXP1 is associated with estrogen receptor alpha and improved survival in primary human breast carcinomas. Clin Cancer Res. 2004;10:3521–7.
Streubel B, Vinatzer U, Lamprecht A, Raderer M, Chott A. T(3;14)(p14.1;q32) involving IGH and FOXP1 is a novel recurrent chromosomal aberration in MALT lymphoma. Leukemia. 2005;19:652–8.
Jia WZ, Yu T, An Q, Yang H, Zhang Z, Liu X, Xiao G. MicroRNA-190 regulates FOXP2 genes in human gastric cancer. Onco Targets Ther. 2016;9:3643–51.
Yan X, Zhou H, Zhang T, Xu P, Zhang S, Huang W, Yang L, Gu X, Ni R. Downregulation of FOXP2 promoter human hepatocellular carcinoma cell invasion. Tumour Biol. 2015;36:9611–9.
Lai CS, Fisher SE, Hurst JA, Vargha-Khadem F, Monaco AP. A forkhead-domain gene is mutated in a severe speech and language disorder. Nature. 2001;413:519–23.
Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–61.
Schubert LA, Jeffery E, Zhang Y, Ramsdell F, Ziegler SF. Scurfin (FOXP3) acts as a repressor of transcription and regulates T cell activation. J Biol Chem. 2001;276:37672–9.
Maruyama T, Kono K, Mizukami Y, Kawaguchi Y, Mimura K, Watanabe M, Izawa S, Fujii H. Distribution of Th17 cells and FoxP3(+) regulatory T cells in tumor-infiltrating lymphocytes, tumor-draining lymph nodes and peripheral blood lymphocytes in patients with gastric cancer. Cancer Sci. 2010;101:1947–54.
Afzali B, Lombardi G, Lechler RI, Lord GM. The role of T helper 17 (Th17) and regulatory T cells (Treg) in human organ transplantation and autoimmune disease. Clin Exp Immunol. 2007;148:32–46.
Fantini MC, Rizzo A, Fina D, Caruso R, Becker C, Neurath MF, Macdonald TT, Pallone F, Monteleone G. IL-21 regulates experimental colitis by modulating the balance between Treg and Th17 cells. Eur J Immunol. 2007;37:3155–63.
Xu L, Kitani A, Fuss I, Strober W. Cutting edge: regulatory T cells induce CD4+CD25-Foxp3- T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J Immunol. 2007;178:6725–9.
Homey B. After TH1/TH2 now comes Treg/TH17: significance of T helper cells in immune response organization. Hautarzt. 2006;57:730–2.
Herrero MJ, Gitton Y. The untold stories of the speech gene, the FOXP2 cancer gene. Genes Cancer. 2018;9:11–38.
Halacli SO, Dogan AL. FOXP1 regulation via the PI3K/Akt/p70S6K signaling pathway in breast cancer cells. Oncol Lett. 2015;9:1482–8.
Wlodarska I, Veyt E, De Paepe P, Vandenberghe P, Nooijen P, Theate I, Michaux L, Sagaert X, Marynen P, Hagemeijer A, De Wolf-Peeters C. FOXP1, a gene highly expressed in a subset of diffuse large B-cell lymphoma, is recurrently targeted by genomic aberrations. Leukemia. 2005;19:1299–305.
Barrans SL, Fenton JA, Banham A, Owen RG, Jack AS. Strong expression of FOXP1 identifies a distinct subset of diffuse large B-cell lymphoma (DLBCL) patients with poor outcome. Blood. 2004;104:2933–5.
Banham AH, Connors JM, Brown PJ, Cordell JL, Ott G, Sreenivasan G, Farinha P, Horsman DE, Gascoyne RD. Expression of the FOXP1 transcription factor is strongly associated with inferior survival in patients with diffuse large B-cell lymphoma. Clin Cancer Res. 2005;11:1065–72.
Sagaert X, de Paepe P, Libbrecht L, Vanhentenrijk V, Verhoef G, Thomas J, Wlodarska I, De Wolf-Peeters C. Forkhead box protein P1 expression in mucosa-associated lymphoid tissue lymphomas predicts poor prognosis and transformation to diffuse large B-cell lymphoma. J Clin Oncol. 2006;24:2490–7.
Hoefnagel JJ, Mulder MM, Dreef E, Jansen PM, Pals ST, Meijer CJ, Willemze R, Vermeer MH. Expression of B-cell transcription factors in primary cutaneous B-cell lymphoma. Mod Pathol. 2006;19:1270–6.
Espinet B, Garcia-Herrera A, Gallardo F, Baro C, Salgado R, Servitje O, Estrach T, Colomo L, Romagosa V, Barranco C, et al. FOXP1 molecular cytogenetics and protein expression analyses in primary cutaneous large B cell lymphoma, leg-type. Histol Histopathol. 2010;26:213–21.
Brown P, Marafioti T, Kusec R, Banham AH. The FOXP1 transcription factor is expressed in the majority of follicular lymphomas but is rarely expressed in classical and lymphocyte predominant Hodgkin's lymphoma. J Mol Histol. 2005;36:249–56.
Han SL, Wu XL, Wan L, Zeng QQ, Li JL, Liu Z. FOXP1 expression predicts polymorphic histology and poor prognosis in gastric mucosa-associated lymphoid tissue lymphomas. Dig Surg. 2009;26:156–62.
Wang X, Sun J, Cui M, Zhao F, Ge C, Chen T, Yao M, Li J. Downregulation of FOXP1 inhibits cell proliferation in hepatocellular carcinoma by inducing G1/S phase cell cycle arrest. Int J Mol Sci. 2016;17:1501.
Brown PJ, Wong KK, Felce SL, Lyne L, Spearman H, Soilleux EJ, Pedersen LM, Moller MB, Green TM, Gascoyne DM, Banham AH. FOXP1 suppresses immune response signatures and MHC class II expression in activated B-cell-like diffuse large B-cell lymphomas. Leukemia. 2015;30:605–16.
Walker MP, Stopford CM, Cederlund M, Fang F, Jahn C, Rabinowitz AD, Goldfarb D, Graham DM, Yan F, Deal AM, et al. FOXP1 potentiates Wnt/beta-catenin signaling in diffuse large B cell lymphoma. Sci Signal. 2015;8:ra12.
Bates GJ, Fox SB, Han C, Launchbury R, Leek RD, Harris AL, Banham AH. Expression of the forkhead transcription factor FOXP1 is associated with that of estrogen receptor-beta in primary invasive breast carcinomas. Breast Cancer Res Treat. 2008;111:453–9.
Zhang Y, Zhang S, Wang X, Liu J, Yang L, He S, Chen L, Huang J. Prognostic significance of FOXP1 as an oncogene in hepatocellular carcinoma. J Clin Pathol. 2012;65:528–33.
Koon HB, Ippolito GC, Banham AH, Tucker PW. FOXP1: a potential therapeutic target in cancer. Expert Opin Ther Targets. 2007;11:955–65.
Xue L, Yue S, Zhang J. FOXP1 has a low expression in human gliomas and its overexpression inhibits proliferation, invasion and migration of human glioma U251 cells. Mol Med Rep. 2014;10:467–72.
Sheng H, Li X, Xu Y. Knockdown of FOXP1 promotes the development of lung adenocarcinoma. Cancer Biol Ther. 2018;20:1–9.
Takayama K, Suzuki T, Tsutsumi S, Fujimura T, Takahashi S, Homma Y, Urano T, Aburatani H, Inoue S. Integrative analysis of FOXP1 function reveals a tumor-suppressive effect in prostate cancer. Mol Endocrinol. 2014;28:2012–24.
Bieche I, Lidereau R. Genetic alterations in breast cancer. Genes Chromosom Cancer. 1995;14:227–51.
Bai S, Kerppola TK. Opposing roles of FoxP1 and Nfat3 in transcriptional control of cardiomyocyte hypertrophy. Mol Cell Biol. 2011;31:3068–80.
Cuiffo BG, Karnoub AE. Silencing FOXP2 in breast cancer cells promotes cancer stem cell traits and metastasis. Mol Cell Oncol. 2016;3:e1019022.
Morris G, Pahad N, Dirr HW, Fanucchi S. A conserved cation binding site in the DNA binding domain of forkhead box transcription factors regulates DNA binding by FOXP2. Arch Biochem Biophys. 2018;657:56–64.
Meredith LJ, Wang CM, Nascimento L, Liu R, Wang L, Yang WH. The key regulator for language and speech development, FOXP2, is a novel substrate for SUMOylation. J Cell Biochem. 2015;117:426–38.
Campbell AJ, Lyne L, Brown PJ, Launchbury RJ, Bignone P, Chi J, Roncador G, Lawrie CH, Gatter KC, Kusec R, Banham AH. Aberrant expression of the neuronal transcription factor FOXP2 in neoplastic plasma cells. Br J Haematol. 2010;149:221–30.
Stumm L, Burkhardt L, Steurer S, Simon R, Adam M, Becker A, Sauter G, Minner S, Schlomm T, Sirma H, Michl U. Strong expression of the neuronal transcription factor FOXP2 is linked to an increased risk of early PSA recurrence in ERG fusion-negative cancers. J Clin Pathol. 2013;66:563–8.
Merlo A, Casalini P, Carcangiu ML, Malventano C, Triulzi T, Menard S, Tagliabue E, Balsari A. FOXP3 expression and overall survival in breast cancer. J Clin Oncol. 2009;27:1746–52.
Ladoire S, Arnould L, Mignot G, Coudert B, Rebe C, Chalmin F, Vincent J, Bruchard M, Chauffert B, Martin F, et al. Presence of Foxp3 expression in tumor cells predicts better survival in HER2-overexpressing breast cancer patients treated with neoadjuvant chemotherapy. Breast Cancer Res Treat. 2010;125:65–72.
Szylberg L, Karbownik D, Marszalek A. The role of FOXP3 in human cancers. Anticancer Res. 2016;36:3789–94.
Zuo T, Wang L, Morrison C, Chang X, Zhang H, Li W, Liu Y, Wang Y, Liu X, Chan MW, et al. FOXP3 is an X-linked breast cancer suppressor gene and an important repressor of the HER-2/ErbB2 oncogene. Cell. 2007;129:1275–86.
Zuo T, Liu R, Zhang H, Chang X, Liu Y, Wang L, Zheng P. FOXP3 is a novel transcriptional repressor for the breast cancer oncogene SKP2. J Clin Invest. 2007;117:3765–73.
Nakahira K, Morita A, Kim NS, Yanagihara I. Phosphorylation of FOXP3 by LCK downregulates MMP9 expression and represses cell invasion. PLoS One. 2013;8:e77099.
Bettelli E, Dastrange M, Oukka M. Foxp3 interacts with nuclear factor of activated T cells and NF-kappa B to repress cytokine gene expression and effector functions of T helper cells. Proc Natl Acad Sci U S A. 2005;102:5138–43.
Ono M, Yaguchi H, Ohkura N, Kitabayashi I, Nagamura Y, Nomura T, Miyachi Y, Tsukada T, Sakaguchi S. Foxp3 controls regulatory T-cell function by interacting with AML1/Runx1. Nature. 2007;446:685–9.
Yang T, Li H, Thakur A, Chen T, Xue J, Li D, Chen M. FOXP4 modulates tumor growth and independently associates with miR-138 in non-small cell lung cancer cells. Tumour Biol. 2015;36:8185–91.
Li XH, Xu Y, Yang K, Shi JJ, Zhang X, Yang F, Yuan H, Zhu X, Zhang YH, Wang JY, Yang Z. Association of THADA, FOXP4, GPRC6A/RFX6 genes and 8q24 risk alleles with prostate cancer in northern Chinese men. J BUON. 2015;20:1223–8.
Liu M, Shi X, Wang J, Xu Y, Wei D, Zhang Y, Yang K, Wang X, Liang S, Chen X, et al. Association of FOXP4 gene with prostate Cancer and the cumulative effects of rs4714476 and 8q24 in Chinese men. Clin Lab. 2015;61:1491–9.
Li J, Lian Y, Yan C, Cai Z, Ding J, Ma Z, Peng P, Wang K. Long non-coding RNA FOXP4-AS1 is an unfavourable prognostic factor and regulates proliferation and apoptosis in colorectal cancer. Cell Prolif. 2016;50:e12312.
Yang L, Ge D, Chen X, Qiu J, Yin Z, Zheng S, Jiang C. FOXP4-AS1 participates in the development and progression of osteosarcoma by downregulating LATS1 via binding to LSD1 and EZH2. Biochem Biophys Res Commun. 2018;502:493–500.
Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–6.
McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M, Byrne MC. CD4(+)CD25(+) immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 2002;16:311–23.
Chen X, Du Y, Lin X, Qian Y, Zhou T, Huang Z. CD4+CD25+ regulatory T cells in tumor immunity. Int Immunopharmacol. 2016;34:244–9.
Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol. 2007;8:1353–62.
Peng J, Yu Z, Xue L, Wang J, Li J, Liu D, Yang Q, Lin Y. The effect of foxp3-overexpressing Treg cells on non-small cell lung cancer cells. Mol Med Rep. 2018;17:5860–8.
Magg T, Mannert J, Ellwart JW, Schmid I, Albert MH. Subcellular localization of FOXP3 in human regulatory and nonregulatory T cells. Eur J Immunol. 2012;42:1627–38.
Grimmig T, Kim M, Germer CT, Gasser M, Waaga-Gasser AM. The role of FOXP3 in disease progression in colorectal cancer patients. Oncoimmunology. 2013;2:e24521.
Feng LL, Wang X. Targeting Foxp3+ regulatory T cells-related immunosuppression for cancer immunotherapy. Chin Med J. 2010;123:3334–42.
Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. 2010;10:490–500.
Adeegbe DO, Nishikawa H. Natural and induced T regulatory cells in cancer. Front Immunol. 2013;4:190.
Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, Parizot C, Taflin C, Heike T, Valeyre D, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity. 2009;30:899–911.
Kaczorowski M, Jutel M. Human T regulatory cells: on the way to cognition. Arch Immunol Ther Exp. 2013;61:229–36.
Whiteside TL. What are regulatory T cells (Treg) regulating in cancer and why? Semin Cancer Biol. 2012;22:327–34.
Workman CJ, Szymczak-Workman AL, Collison LW, Pillai MR, Vignali DA. The development and function of regulatory T cells. Cell Mol Life Sci. 2009;66:2603–22.
Facciabene A, Motz GT, Coukos G. T-regulatory cells: key players in tumor immune escape and angiogenesis. Cancer Res. 2012;72:2162–71.
Hinz S, Pagerols-Raluy L, Oberg HH, Ammerpohl O, Grussel S, Sipos B, Grutzmann R, Pilarsky C, Ungefroren H, Saeger HD, et al. Foxp3 expression in pancreatic carcinoma cells as a novel mechanism of immune evasion in cancer. Cancer Res. 2007;67:8344–50.
Flammiger A, Weisbach L, Huland H, Tennstedt P, Simon R, Minner S, Bokemeyer C, Sauter G, Schlomm T, Trepel M. High tissue density of FOXP3+ T cells is associated with clinical outcome in prostate cancer. Eur J Cancer. 2013;49:1273–9.
Zhang L, Xu J, Zhang X, Zhang Y, Wang L, Huang X, Xu Z. The role of Tumoral FOXP3 on cell proliferation, migration, and invasion in gastric Cancer. Cell Physiol Biochem. 2017;42:1739–54.
Takeuchi Y, Nishikawa H. Roles of regulatory T cells in cancer immunity. Int Immunol. 2016;28:401–9.
Wei H, Geng J, Shi B, Liu Z, Wang YH, Stevens AC, Sprout SL, Yao M, Wang H, Hu H. Cutting edge: Foxp1 controls naive CD8+ T cell quiescence by simultaneously repressing key pathways in cellular metabolism and cell cycle progression. J Immunol. 2016;196:3537–41.
Bi E, Ma X, Lu Y, Yang M, Wang Q, Xue G, Qian J, Wang S, Yi Q. Foxo1 and Foxp1 play opposing roles in regulating the differentiation and antitumor activity of TH9 cells programmed by IL-7. Sci Signal. 2017;10:eaak9741.
Feng X, Wang H, Takata H, Day TJ, Willen J, Hu H. Transcription factor Foxp1 exerts essential cell-intrinsic regulation of the quiescence of naive T cells. Nat Immunol. 2011;12:544–50.
Chen C, Liu X, Ren Y. Interleukin 21 treatment in a murine model as a novel potential cytokine immunotherapy for colon cancer. Adv Clin Exp Med. 2018;27:583–9.
De Silva P, Garaud S, Solinas C, de Wind A, Van den Eyden G, Jose V, Gu-Trantien C, Migliori E, Boisson A, Naveaux C, et al. FOXP1 negatively regulates tumor infiltrating lymphocyte migration in human breast cancer. EBioMedicine. 2019;39:226–38.
Mancini M, Toker A. NFAT proteins: emerging roles in cancer progression. Nat Rev Cancer. 2009;9:810–20.
Gabriel CH, Gross F, Karl M, Stephanowitz H, Hennig AF, Weber M, Gryzik S, Bachmann I, Hecklau K, Wienands J, et al. Identification of novel nuclear factor of activated T cell (NFAT)-associated proteins in T cells. J Biol Chem. 2016;291:24172–87.
Shou J, Jing J, Xie J, You L, Jing Z, Yao J, Han W, Pan H. Nuclear factor of activated T cells in cancer development and treatment. Cancer Lett. 2015;361:174–84.
Liu JF, Zhao SH, Wu SS. Depleting NFAT1 expression inhibits the ability of invasion and migration of human lung cancer cells. Cancer Cell Int. 2013;13:41.
Yiu GK, Toker A. NFAT induces breast cancer cell invasion by promoting the induction of cyclooxygenase-2. J Biol Chem. 2006;281:12210–7.
Kim TH, Kim HI, Soung YH, Shaw LA, Chung J. Integrin (alpha6beta4) signals through Src to increase expression of S100A4, a metastasis-promoting factor: implications for cancer cell invasion. Mol Cancer Res. 2009;7:1605–12.
Yiu GK, Kaunisto A, Chin YR, Toker A. NFAT promotes carcinoma invasive migration through glypican-6. Biochem J. 2011;440:157–66.
Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, Baer R, Gu W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell. 2012;149:1269–83.
Jung DJ, Jin DH, Hong SW, Kim JE, Shin JS, Kim D, Cho BJ, Hwang YI, Kang JS, Lee WJ. Foxp3 expression in p53-dependent DNA damage responses. J Biol Chem. 2010;285:7995–8002.
Kim JE, Shin JS, Moon JH, Hong SW, Jung DJ, Kim JH, Hwang IY, Shin YJ, Gong EY, Lee DH, et al. Foxp3 is a key downstream regulator of p53-mediated cellular senescence. Oncogene. 2017;36:219–30.
Rao DS, O'Connell RM, Chaudhuri AA, Garcia-Flores Y, Geiger TL, Baltimore D. MicroRNA-34a perturbs B lymphocyte development by repressing the forkhead box transcription factor Foxp1. Immunity. 2010;33:48–59.
El-Deiry WS. p21(WAF1) mediates cell-cycle inhibition, relevant to Cancer suppression and therapy. Cancer Res. 2016;76:5189–91.
Liu RZ, Vo TM, Jain S, Choi WS, Garcia E, Monckton EA, Mackey JR, Godbout R. NFIB promotes cell survival by directly suppressing p21 transcription in TP53-mutated triple-negative breast cancer. J Pathol. 2019;247:186–98.
Bellido T, O'Brien CA, Roberson PK, Manolagas SC. Transcriptional activation of the p21(WAF1,CIP1,SDI1) gene by interleukin-6 type cytokines. A prerequisite for their pro-differentiating and anti-apoptotic effects on human osteoblastic cells. J Biol Chem. 1998;273:21137–44.
Cerna K, Mraz M. p53 limits B cell receptor (BCR) signalling: a new role for miR-34a and FOXP1. Oncotarget. 2018;9:36409–10.
Zhao J, Li H, Zhou R, Ma G, Dekker JD, Tucker HO, Yao Z, Guo X. Foxp1 regulates the proliferation of hair follicle stem cells in response to oxidative stress during hair cycling. PLoS One. 2015;10:e0131674.
Khalife E, Khodadadi A, Talaeizadeh A, Rahimian L, Nemati M, Jafarzadeh A. Overexpression of regulatory T cell-related markers (FOXP3, CTLA-4 and GITR) by peripheral blood mononuclear cells from patients with breast Cancer. Asian Pac J Cancer Prev. 2018;19:3019–25.
Nagase H, Takeoka T, Urakawa S, Morimoto-Okazawa A, Kawashima A, Iwahori K, Takiguchi S, Nishikawa H, Sato E, Sakaguchi S, et al. ICOS(+) Foxp3(+) TILs in gastric cancer are prognostic markers and effector regulatory T cells associated with helicobacter pylori. Int J Cancer. 2017;140:686–95.
Jang JE, Hajdu CH, Liot C, Miller G, Dustin ML, Bar-Sagi D. Crosstalk between regulatory T cells and tumor-associated dendritic cells negates anti-tumor immunity in pancreatic Cancer. Cell Rep. 2017;20:558–71.
Xu T, Lu J, An H. The relative change in regulatory T cells / T helper lymphocytes ratio as parameter for prediction of therapy efficacy in metastatic colorectal cancer patients. Oncotarget. 2017;8:109079–93.
Chen C, Chen D, Zhang Y, Chen Z, Zhu W, Zhang B, Wang Z, Le H. Changes of CD4+CD25+FOXP3+ and CD8+CD28- regulatory T cells in non-small cell lung cancer patients undergoing surgery. Int Immunopharmacol. 2014;18:255–61.
Ren J, Li B. The functional stability of FOXP3 and RORgammat in Treg and Th17 and their therapeutic applications. Adv Protein Chem Struct Biol. 2017;107:155–89.
Najafi M, Farhood B, Mortezaee K. Contribution of regulatory T cells to cancer: a review. J Cell Physiol. 2019;234:7983–93.
Wang D, Huang S, Yuan X, Liang J, Xu R, Yao G, Feng X, Sun L. The regulation of the Treg/Th17 balance by mesenchymal stem cells in human systemic lupus erythematosus. Cell Mol Immunol. 2017;14:423–31.
Du R, Zhao H, Yan F, Li H. IL-17+Foxp3+ T cells: an intermediate differentiation stage between Th17 cells and regulatory T cells. J Leukoc Biol. 2014;96:39–48.
Beyer M. Interleukin-2 treatment of tumor patients can expand regulatory T cells. Oncoimmunology. 2012;1:1181–2.
Sharma MD, Hou DY, Liu Y, Koni PA, Metz R, Chandler P, Mellor AL, He Y, Munn DH. Indoleamine 2,3-dioxygenase controls conversion of Foxp3+ Tregs to TH17-like cells in tumor-draining lymph nodes. Blood. 2009;113:6102–11.
Baban B, Chandler PR, Sharma MD, Pihkala J, Koni PA, Munn DH, Mellor AL. IDO activates regulatory T cells and blocks their conversion into Th17-like T cells. J Immunol. 2009;183:2475–83.
Rueda CM, Presicce P, Jackson CM, Miller LA, Kallapur SG, Jobe AH, Chougnet CA. Lipopolysaccharide-induced Chorioamnionitis promotes IL-1-dependent inflammatory FOXP3+ CD4+ T cells in the fetal rhesus macaque. J Immunol. 2016;196:3706–15.
Konkel JE, Zhang D, Zanvit P, Chia C, Zangarle-Murray T, Jin W, Wang S, Chen W. Transforming growth factor-beta signaling in regulatory T cells controls T Helper-17 cells and tissue-specific immune responses. Immunity. 2017;46:660–74.
Li L, Boussiotis VA. The role of IL-17-producing Foxp3+ CD4+ T cells in inflammatory bowel disease and colon cancer. Clin Immunol. 2013;148:246–53.
Huang C, Fu ZX. Localization of IL-17+Foxp3+ T cells in esophageal cancer. Immunol Investig. 2011;40:400–12.
Kryczek I, Wu K, Zhao E, Wei S, Vatan L, Szeliga W, Huang E, Greenson J, Chang A, Rolinski J, et al. IL-17+ regulatory T cells in the microenvironments of chronic inflammation and cancer. J Immunol. 2011;186:4388–95.
Okui T, Aoki Y, Ito H, Honda T, Yamazaki K. The presence of IL-17+/FOXP3+ double-positive cells in periodontitis. J Dent Res. 2012;91:574–9.
Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, Tanaka S, Bluestone JA, Takayanagi H. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med. 2014;20:62–8.
Wong WF, Kohu K, Chiba T, Sato T, Satake M. Interplay of transcription factors in T-cell differentiation and function: the role of Runx. Immunology. 2011;132:157–64.
Xing S, Shao P, Li F, Zhao X, Seo W, Wheat JC, Ramasamy S, Wang J, Li X, Peng W, et al. Tle corepressors are differentially partitioned to instruct CD8(+) T cell lineage choice and identity. J Exp Med. 2018;215:2211–26.
Khosravi M, Bidmeshkipour A, Moravej A, Hojjat-Assari S, Naserian S, Karimi MH. Induction of CD4(+)CD25(+)Foxp3(+) regulatory T cells by mesenchymal stem cells is associated with RUNX complex factors. Immunol Res. 2018;66:207–18.
Liu SX, Xiao HR, Wang GB, Chen XW, Li CG, Mai HR, Yuan XL, Liu GS, Wen FQ. Preliminary investigation on the abnormal mechanism of CD4(+)FOXP3(+)CD25(high) regulatory T cells in pediatric B-cell acute lymphoblastic leukemia. Exp Ther Med. 2018;16:1433–41.
Li L, Patsoukis N, Petkova V, Boussiotis VA. Runx1 and Runx3 are involved in the generation and function of highly suppressive IL-17-producing T regulatory cells. PLoS One. 2012;7:e45115.
Wang CQ, Motoda L, Satake M, Ito Y, Taniuchi I, Tergaonkar V, Osato M. Runx3 deficiency results in myeloproliferative disorder in aged mice. Blood. 2013;122:562–6.
Kortylewski M, Yu H. Role of Stat3 in suppressing anti-tumor immunity. Curr Opin Immunol. 2008;20:228–33.
Wang R, Han G, Wang J, Song L, Chen G, Xu R, Yu M, Qian J, Shen B, Li Y. The role of STAT3 in antigen-IgG inducing regulatory CD4(+)Foxp3(+)T cells. Cell Immunol. 2007;246:103–9.
Lam E, Choi SH, Pareek TK, Kim BG, Letterio JJ. Cyclin-dependent kinase 5 represses Foxp3 gene expression and Treg development through specific phosphorylation of Stat3 at serine 727. Mol Immunol. 2015;67:317–24.
Rani A, Murphy JJ. STAT5 in Cancer and immunity. J Interf Cytokine Res. 2016;36:226–37.
Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J Immunol. 2007;178:280–90.
Yao Z, Kanno Y, Kerenyi M, Stephens G, Durant L, Watford WT, Laurence A, Robinson GW, Shevach EM, Moriggl R, et al. Nonredundant roles for Stat5a/b in directly regulating Foxp3. Blood. 2007;109:4368–75.
Wang W, Edington HD, Rao UN, Jukic DM, Radfar A, Wang H, Kirkwood JM. Effects of high-dose IFNalpha2b on regional lymph node metastases of human melanoma: modulation of STAT5, FOXP3, and IL-17. Clin Cancer Res. 2008;14:8314–20.
Kim CG, Lee H, Gupta N, Ramachandran S, Kaushik I, Srivastava S, Kim SH, Srivastava SK. Role of Forkhead box class O proteins in cancer progression and metastasis. Semin Cancer Biol. 2018;50:142–51.
Harada Y, Harada Y, Elly C, Ying G, Paik JH, DePinho RA, Liu YC. Transcription factors Foxo3a and Foxo1 couple the E3 ligase Cbl-b to the induction of Foxp3 expression in induced regulatory T cells. J Exp Med. 2010;207:1381–91.
Ouyang W, Beckett O, Ma Q, Paik JH, DePinho RA, Li MO. Foxo proteins cooperatively control the differentiation of Foxp3+ regulatory T cells. Nat Immunol. 2010;11:618–27.
Vahl JC, Drees C, Heger K, Heink S, Fischer JC, Nedjic J, Ohkura N, Morikawa H, Poeck H, Schallenberg S, et al. Continuous T cell receptor signals maintain a functional regulatory T cell pool. Immunity. 2014;41:722–36.
Wallis CJ, Gordanpour A, Bendavid JS, Sugar L, Nam RK, Seth A. MiR-182 is associated with growth, migration and invasion in prostate Cancer via suppression of FOXO1. J Cancer. 2015;6:1295–305.
Bothur E, Raifer H, Haftmann C, Stittrich AB, Brustle A, Brenner D, Bollig N, Bieringer M, Kang CH, Reinhard K, et al. Antigen receptor-mediated depletion of FOXP3 in induced regulatory T-lymphocytes via PTPN2 and FOXO1. Nat Commun. 2015;6:8576.
Du X, Shi H, Li J, Dong Y, Liang J, Ye J, Kong S, Zhang S, Zhong T, Yuan Z, et al. Mst1/Mst2 regulate development and function of regulatory T cells through modulation of Foxo1/Foxo3 stability in autoimmune disease. J Immunol. 2014;192:1525–35.
van Boxtel R, Gomez-Puerto C, Mokry M, Eijkelenboom A, van der Vos KE, Nieuwenhuis EE, Burgering BM, Lam EW, Coffer PJ. FOXP1 acts through a negative feedback loop to suppress FOXO-induced apoptosis. Cell Death Differ. 2013;20:1219–29.
Baeuerle PA, Henkel T. Function and activation of NF-kappa B in the immune system. Annu Rev Immunol. 1994;12:141–79.
Hao Q, Zhang C, Gao Y, Wang S, Li J, Li M, Xue X, Li W, Zhang W, Zhang Y. FOXP3 inhibits NF-kappaB activity and hence COX2 expression in gastric cancer cells. Cell Signal. 2013;26:564–9.
Hao Q, Li W, Zhang C, Qin X, Xue X, Li M, Shu Z, Xu T, Xu Y, Wang W, et al. TNFalpha induced FOXP3-NFkappaB interaction dampens the tumor suppressor role of FOXP3 in gastric cancer cells. Biochem Biophys Res Commun. 2013;430:436–41.
Wang X, Liu Y, Dai L, Liu Q, Jia L, Wang H, An L, Jing X, Liu M, Li P, Cheng Z. Foxp3 downregulation in NSCLC mediates epithelial-mesenchymal transition via NF-kappaB signaling. Oncol Rep. 2016;36:2282–8.
Chu R, Liu SY, Vlantis AC, van Hasselt CA, Ng EK, Fan MD, Ng SK, Chan AB, Du J, Wei W, et al. Inhibition of Foxp3 in cancer cells induces apoptosis of thyroid cancer cells. Mol Cell Endocrinol. 2015;399:228–34.
Jia T, Fu H, Sun J, Zhang Y, Yang W, Li Y. Foxp3 expression in A549 cells is regulated by toll-like receptor 4 through nuclear factor-kappaB. Mol Med Rep. 2012;6:167–72.
Yehya AHS, Asif M, Petersen SH, Subramaniam AV, Kono K, Majid A, Oon CE. Angiogenesis: Managing the Culprits behind Tumorigenesis and Metastasis. Medicina (Kaunas). 2018;54:8.
Li X, Gao Y, Li J, Zhang K, Han J, Li W, Hao Q, Zhang W, Wang S, Zeng C, et al. FOXP3 inhibits angiogenesis by downregulating VEGF in breast cancer. Cell Death Dis. 2018;9:744.
Li S, Wang Y, Feng C, Wu G, Ye Y, Tian J. Calycosin inhibits the migration and invasion of human breast Cancer cells by Down-regulation of Foxp3 expression. Cell Physiol Biochem. 2017;44:1775–84.
Tang J, Yang Z, Wang Z, Li Z, Li H, Yin J, Deng M, Zhu W, Zeng C. Foxp3 is correlated with VEGF-C expression and lymphangiogenesis in cervical cancer. World J Surg Oncol. 2017;15:173.
Wan L, Huang J, Chen J, Wang R, Dong C, Lu S, Wu X. Expression and significance of FOXP1, HIF-1a and VEGF in renal clear cell carcinoma. J buon. 2015;20:188–95.
Chen D, Li L, Tu X, Yin Z, Wang Q. Functional characterization of Klippel-Trenaunay syndrome gene AGGF1 identifies a novel angiogenic signaling pathway for specification of vein differentiation and angiogenesis during embryogenesis. Hum Mol Genet. 2013;22:963–76.
He Q, Zhao L, Liu Y, Liu X, Zheng J, Yu H, Cai H, Ma J, Liu L. Wang P, et al: circ-SHKBP1 regulates the angiogenesis of U87 Glioma-exposed endothelial cells through miR-544a/FOXP1 and miR-379/FOXP2 pathways. Mol Ther Nucleic Acids. 2018;10:331–48.
Gibb EA, Brown CJ, Lam WL. The functional role of long non-coding RNA in human carcinomas. Mol Cancer. 2011;10:38.
Gomez GG, Volinia S, Croce CM, Zanca C, Li M, Emnett R, Gutmann DH, Brennan CW, Furnari FB, Cavenee WK. Suppression of microRNA-9 by mutant EGFR signaling upregulates FOXP1 to enhance glioblastoma tumorigenicity. Cancer Res. 2014;74:1429–39.
Craig VJ, Cogliatti SB, Imig J, Renner C, Neuenschwander S, Rehrauer H, Schlapbach R, Dirnhofer S, Tzankov A, Muller A. Myc-mediated repression of microRNA-34a promotes high-grade transformation of B-cell lymphoma by dysregulation of FoxP1. Blood. 2011;117:6227–36.
Cuiffo BG, Campagne A, Bell GW, Lembo A, Orso F, Lien EC, Bhasin MK, Raimo M, Hanson SE, Marusyk A, et al. MSC-regulated microRNAs converge on the transcription factor FOXP2 and promote breast cancer metastasis. Cell Stem Cell. 2014;15:762–74.
Chen H, Xiao Z, Yu R, Wang Y, Xu R. Zhu X: miR-181d-5p-FOXP1 feedback loop modulates the progression of osteosarcoma. Biochem Biophys Res Commun. 2018;503:1434–41.
Li H, Liang J, Qin F, Zhai Y. MiR-374b-5p-FOXP1 feedback loop regulates cell migration, epithelial-mesenchymal transition and chemosensitivity in ovarian cancer. Biochem Biophys Res Commun. 2018;505:554–60.
Kumar S, Batra A, Kanthaje S, Ghosh S, Chakraborti A. Crosstalk between microRNA-122 and FOX family genes in HepG2 cells. Exp Biol Med (Maywood). 2017;242:436–40.
Musilova K, Devan J, Cerna K, Seda V, Pavlasova G, Sharma S, Oppelt J, Pytlik R, Prochazka V. Prouzova Z, et al: miR-150 downregulation contributes to the high-grade transformation of follicular lymphoma by upregulating FOXP1 levels. Blood. 2018;132:2389–400.
Cui R, Guan Y, Sun C, Chen L, Bao Y, Li G, Qiu B, Meng X, Pang C, Wang Y. A tumor-suppressive microRNA, miR-504, inhibits cell proliferation and promotes apoptosis by targeting FOXP1 in human glioma. Cancer Lett. 2016;374:1–11.
Yao S, Liu Y, Yao Z, Zhao Y, Wang H, Xu Y, Zhang J, Li J, Yang S. MicroRNA-376a regulates cell proliferation and apoptosis by targeting forkhead box protein P2 in lymphoma. Oncol Lett. 2018;16:3169–76.
Zhong C, Liu J, Zhang Y, Luo J, Zheng J. MicroRNA-139 inhibits the proliferation and migration of osteosarcoma cells via targeting forkhead-box P2. Life Sci. 2017;191:68–73.
Liu R, Liu C, Chen D, Yang WH, Liu X, Liu CG, Dugas CM, Tang F, Zheng P, Liu Y, Wang L. FOXP3 controls an miR-146/NF-kappaB negative feedback loop that inhibits apoptosis in breast Cancer cells. Cancer Res. 2015;75:1703–13.
McInnes N, Sadlon TJ, Brown CY, Pederson S, Beyer M, Schultze JL, McColl S, Goodall GJ, Barry SC. FOXP3 and FOXP3-regulated microRNAs suppress SATB1 in breast cancer cells. Oncogene. 2012;31:1045–54.
Xiao L, Zhou H, Li XP, Chen J, Fang C, Mao CX, Cui JJ, Zhang W, Zhou HH, Yin JY, Liu ZQ. MicroRNA-138 acts as a tumor suppressor in non small cell lung cancer via targeting YAP1. Oncotarget. 2016;7:40038–46.
Gu Y, Xiao X, Yang S. LncRNA MALAT1 acts as an oncogene in multiple myeloma through sponging miR-509-5p to modulate FOXP1 expression. Oncotarget. 2017;8:101984–93.
Sun Y, Liu J, Chu L, Yang W, Liu H, Li C, Yang J. Long noncoding RNA SNHG12 facilitates the tumorigenesis of glioma through miR-101-3p/FOXP1 axis. Gene. 2018;676:315–21.
Xi J, Feng J, Zeng S, Huang P. Long noncoding RNA UFC1 is activated by E2F1 and exerts oncogenic properties by functioning as a ceRNA of FOXP3. Cancer Med. 2018;7:3301–10.
Yang Y, Cheng J, Ren H, Zhao H, Gong W, Shan C. Tumor FOXP3 represses the expression of long noncoding RNA 7SL. Biochem Biophys Res Commun. 2016;472:432–6.
Xiong Y, Zhang J, Song C. CircRNA ZNF609 functions as a competitive endogenous RNA to regulate FOXP4 expression by sponging miR-138-5p in renal carcinoma. J Cell Physiol. 2018;234:10646–54.
Garaud S, Roufosse F, De Silva P, Gu-Trantien C, Lodewyckx JN, Duvillier H, Dedeurwaerder S, Bizet M, Defrance M, Fuks F, et al. FOXP1 is a regulator of quiescence in healthy human CD4(+) T cells and is constitutively repressed in T cells from patients with lymphoproliferative disorders. Eur J Immunol. 2017;47:168–79.
Steinman RM. Decisions about dendritic cells: past, present, and future. Annu Rev Immunol. 2012;30:1–22.
Sabado RL, Balan S, Bhardwaj N. Dendritic cell-based immunotherapy. Cell Res. 2017;27:74–95.
Jung NC, Lee JH, Chung KH, Kwak YS, Lim DS. Dendritic cell-based immunotherapy for solid tumors. Transl Oncol. 2018;11:686–90.
Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–33.
Garg AD, Coulie PG, Van den Eynde BJ, Agostinis P. Integrating next-generation dendritic cell vaccines into the current Cancer immunotherapy landscape. Trends Immunol. 2017;38:577–93.
Wilgenhof S, Corthals J, Heirman C, van Baren N, Lucas S, Kvistborg P, Thielemans K, Neyns B. Phase II study of autologous monocyte-derived mRNA Electroporated dendritic cells (TriMixDC-MEL) plus Ipilimumab in patients with pretreated advanced melanoma. J Clin Oncol. 2016;34:1330–8.
Bol KF, Schreibelt G, Gerritsen WR, de Vries IJ, Figdor CG. Dendritic cell-based immunotherapy: state of the art and beyond. Clin Cancer Res. 2016;22:1897–906.
Revu S, Wu J, Henkel M, Rittenhouse N, Menk A, Delgoffe GM, Poholek AC, McGeachy MJ. IL-23 and IL-1beta drive human Th17 cell differentiation and metabolic reprogramming in absence of CD28 Costimulation. Cell Rep. 2018;22:2642–53.
Luckey MA, Kimura MY, Waickman AT, Feigenbaum L, Singer A, Park JH. The transcription factor ThPOK suppresses Runx3 and imposes CD4(+) lineage fate by inducing the SOCS suppressors of cytokine signaling. Nat Immunol. 2014;15:638–45.
Yamamoto M, Kamigaki T, Yamashita K, Hori Y, Hasegawa H, Kuroda D, Moriyama H, Nagata M, Ku Y, Kuroda Y. Enhancement of anti-tumor immunity by high levels of Th1 and Th17 with a combination of dendritic cell fusion hybrids and regulatory T cell depletion in pancreatic cancer. Oncol Rep. 2009;22:337–43.
Li Y, Liu M, Yang ST. Dendritic cells derived from pluripotent stem cells: potential of large scale production. World J Stem Cells. 2014;6:1–10.
Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, Wu C, Kleinewietfeld M, Kunder S, Hafler DA, et al. Induction and molecular signature of pathogenic TH17 cells. Nat Immunol. 2012;13:991–9.
Acknowledgments
Not applicable.
Disclosures
The authors disclose no conflicts.
Funding
This work was funded by the Korea Science and Engineering Foundation (KOSEF) grant funded by the Korea government (MEST) (no.2017R1A2A1A17069297).
Author information
Authors and Affiliations
Contributions
J-HK and JH documented papers and summarized data. JHJ and H-JL helped to write manuscript and DYL gave some comments to Figure preparation. J-HK mainly drafted manuscript and four Figures and JH prepared for Table 1. S-HK supervised and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kim, JH., Hwang, J., Jung, J.H. et al. Molecular networks of FOXP family: dual biologic functions, interplay with other molecules and clinical implications in cancer progression. Mol Cancer 18, 180 (2019). https://doi.org/10.1186/s12943-019-1110-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-019-1110-3