
Abstract
Bruton’s tyrosine kinase (BTK) is a non-receptor kinase that plays a crucial role in oncogenic signaling that is critical for proliferation and survival of leukemic cells in many B cell malignancies. BTK was initially shown to be defective in the primary immunodeficiency X-linked agammaglobulinemia (XLA) and is essential both for B cell development and function of mature B cells. Shortly after its discovery, BTK was placed in the signal transduction pathway downstream of the B cell antigen receptor (BCR). More recently, small-molecule inhibitors of this kinase have shown excellent anti-tumor activity, first in animal models and subsequently in clinical studies. In particular, the orally administered irreversible BTK inhibitor ibrutinib is associated with high response rates in patients with relapsed/refractory chronic lymphocytic leukemia (CLL) and mantle-cell lymphoma (MCL), including patients with high-risk genetic lesions. Because ibrutinib is generally well tolerated and shows durable single-agent efficacy, it was rapidly approved for first-line treatment of patients with CLL in 2016. To date, evidence is accumulating for efficacy of ibrutinib in various other B cell malignancies. BTK inhibition has molecular effects beyond its classic role in BCR signaling. These involve B cell-intrinsic signaling pathways central to cellular survival, proliferation or retention in supportive lymphoid niches. Moreover, BTK functions in several myeloid cell populations representing important components of the tumor microenvironment. As a result, there is currently a considerable interest in BTK inhibition as an anti-cancer therapy, not only in B cell malignancies but also in solid tumors. Efficacy of BTK inhibition as a single agent therapy is strong, but resistance may develop, fueling the development of combination therapies that improve clinical responses. In this review, we discuss the role of BTK in B cell differentiation and B cell malignancies and highlight the importance of BTK inhibition in cancer therapy.
Similar content being viewed by others

Background
Protein kinases represent classes of enzymes that catalyze phosphorylation of proteins and thereby alter their substrate’s activity or capacity to interact with other proteins. Kinase signaling pathways represent the most common form of reversible post-translational modifications that control many aspects of cellular function. Aberrant activation of protein kinases drive major hallmarks of malignancies, including alterations in cellular proliferation, survival, motility and metabolism, as well as angiogenesis and evasion of the anti-tumor immune response [1, 2].
One such kinase that plays a crucial role in oncogenic signaling is Bruton’s tyrosine kinase (BTK), which is critical for the survival of leukemic cells in various B cell malignancies. BTK was initially shown to be mutated in the primary immunodeficiency X-linked agammaglobulinemia (XLA) and is essential at various stages of B lymphocyte development [3, 4]. XLA is an inherited immunodeficiency disease originally described by the pediatrician Ogdon Bruton in 1952 and characterized by recurrent bacterial infections. Due to a severe block of B cell development in the bone marrow, XLA patients have very low numbers of B cells in the circulation and antibodies are almost completely absent in the serum. A milder phenotype of the disease is present in CBA/N mice, which harbor the loss-of-function mutation R28C BTK [5, 6]. These mice, known as xid (X-linked immunodeficiency) mice, manifest only minor defects in B cell development in the bone marrow, but instead the differentiation and survival of mature peripheral B cells is severely impaired [7,8,9,10]. Importantly, BTK has received large interest since small-molecule inhibitors of this kinase have shown excellent anti-tumor activity in clinical studies [11, 12]. In particular, the orally administered BTK inhibitor ibrutinib, which forms a covalent bond with a cysteine residue in the BTK active site, was also approved for first-line treatment of patients with chronic lymphocytic leukemia (CLL) and small lymphocytic leukemia (SLL) in 2016 [13].
Shortly after its discovery as the non-receptor tyrosine kinase defective in XLA [3, 4], BTK was placed in the signal transduction pathway downstream of the B cell receptor (BCR). This receptor is expressed on the B cell surface and has the unique capacity to specifically recognize antigens due to hypervariable regions present in the immunoglobulin heavy (IGH) and light (IGL) chains that together form the BCR [14]. BTK is also involved in many other signaling pathways in B cells, including chemokine receptor, Toll-like receptor (TLR) and Fc receptor signaling. Expression of BTK is not restricted to B cells, as also cells of the myeloid lineage express BTK. In these cells, BTK acts also downstream of TLRs and e.g. the FcεR in mast cells [15, 16] and the FcyRI in macrophages [17, 18]. In addition, BTK is involved in various other pathways, including Receptor activator of nuclear factor-κB (RANK) in osteoclasts [19], collagen and CD32 signaling in platelets [20] and the NLRP3 inflammasome in macrophages and neutrophils [21]. Since myeloid cells are important components of the tumor microenvironment and particularly tumor-associated macrophages contribute to cancer progression [22, 23], there is currently a considerable interest in BTK inhibition as an anti-cancer therapy not only in B cell leukemias but also in other hematological malignancies and solid tumors [24,25,26,27].
In this review, we describe the importance of BTK in multiple signaling pathways. We discuss the crucial function of BTK in different stages of normal B cell development. In addition, we discuss its role in oncogenic signaling in B cell malignancies associated with genetic events that result in increased BTK activity. We describe clinical benefits of targeting BTK with small molecule inhibitors in B cell malignancies. Finally, we discuss the effects of BTK inhibitors on tumor growth in solid malignancies in the context of the function of myeloid cells in the tumor environment.
BTK structure
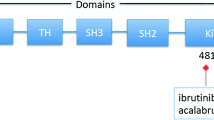
BTK is one of the five members of the TEC family of non-receptor tyrosine kinases - along with tyrosine kinase expressed in hepatocellular carcinoma (TEC), interleukin-2-inducible T cell kinase (ITK), resting lymphocyte kinase (RLK) and bone marrow expressed kinase (BMX) - which are strongly conserved throughout evolution [28]. BTK, TEC and ITK are most similar and both contain five different protein interaction domains (Fig. 1a). These domains include an amino terminal pleckstrin homology (PH) domain, a proline-rich TEC homology (TH) domain, SRC homology (SH) domains SH2 and SH3, as well as kinase domain with enzymatic activity [28, 29]. BTK is essentially cytoplasmic and is only transiently recruited to the membrane through interaction of its PH domain with phosphatidylinositol-3,4,5-triphosphate (PIP3), which is generated by phosphatidylinositol-3 kinase (PI3K) (Fig. 1b) [14]. BTK activation occurs in two steps upon its recruitment to the cell membrane. First, BTK is phosphorylated at position Y551 in the kinase domain by SYK or SRC family kinases [30]. Phosphorylation of BTK at Y551 promotes its catalytic activity and subsequently results in its autophosphorylation at position Y223 in the SH3 domain [31]. Phosphorylation at Y223 is thought to stabilize the active conformation and fully activate BTK kinase activity [32]. Nevertheless, a Y223F mutation did not significantly affect the function of BTK during B cell development in vivo, since B-cell specific transgenic expression of Y223F-BTK could still rescue the xid phenotype of Btk-deficient mice [33]. Therefore, the function of the Y223 BTK autophosphorylation site remains unclear in B cells and to date is unexplored in vivo in myeloid cells.

Domain structure of TEC kinase family members and key interacting partners of Bruton’s tyrosine kinase. a Schematic overview of the protein structure of BTK and other TEC kinase family members. Shown are five different domains, as explained in text, the Y223 autophosphorylation site, the Y551 phosphorylation site that activates BTK, and the C481 binding site of ibrutinib. b Schematic overview of the protein structure of key interacting partners of BTK. PH, pleckstrin homology; TH, TEC homology; BH, BTK homology; PRR, proline rich domain; SH2/SH3, SRC homology domains 2 and 3; Cys, cysteine-string motif
BTK in B cell receptor signaling
The IgM BCR is essential for survival of peripheral B cells [34]. In the absence of BTK B cells have a high rate of apoptosis, which correlates with strongly reduced BCR-mediated induction of the anti-apoptotic protein Bcl-xL [35, 36]. Upon stimulation with anti-IgM, cell size enlargement and degradation of the cyclin inhibitor p27Kip1 occurs normally, indicating that BTK is not essential for several G1 events [37]. BTK-deficient B cells enter early G1, but not S phase of the cell cycle, because they fail to induce cyclin D2 expression [38]. Apart from B cell survival and proliferation, the BCR controls integrin α4β1 (VLA-4)-mediated adhesion of B cells to vascular cell adhesion molecule-1 (VCAM-1) and fibronectin via BTK [39].
BCR cross-linking activates four families of non-receptor protein tyrosine kinases and these are transducers of signaling events including phospholipase Cγ (PLCγ), mitogen-activated protein kinase (MAPK) activation, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-кB) pathway components and activation of the serine/threonine kinase AKT (or protein kinase B, PKB).
The IgM BCR has a very short cytoplasmic domain and thus cannot signal directly, but associates with the disulphide-linked Ig-α/Ig-β(CD79a/CD79b) heterodimers. These transmembrane proteins contain immunoreceptor tyrosine based activation motifs (ITAMs) in their cytoplasmic domain (Fig. 2). BCR engagement by antigen induces ITAM phosphorylation by Src-family protein tyrosine kinases such as LYN, thereby creating docking sites for spleen tyrosine kinase (SYK)(Fig. 1b) [40]. In addition, LYN and SYK also phosphorylate tyrosine residues in the cytoplasmic tail of the B-cell co-receptor CD19 and/or the adaptor protein B-cell PI3K adaptor (BCAP), which facilitates recruitment and activation of PI3K and the guanine nucleotide exchange factor VAV [41, 42]. VAV further enhances enzymatic activity of PI3K through activation of RAC1, a member of Rho family of GTPases [43]. PI3K phosphorylates PIP2 to generate PIP3, which acts as a critical secondary messenger for activating downstream pathways. PIP3 interacts with the BTK PH-domain, resulting in its recruitment to the plasma membrane [44].

Role of Bruton’s tyrosine kinase downstream of the B cell receptor. Signaling cascade showing important events downstream of B cell receptor (BCR). Antigen engagement by the BCR results in the formation of a micro-signalosome whereby BTK activates four families of non-receptor protein tyrosine kinases that transduce key signaling events including phospholipase Cγ, mitogen-activated protein kinase (MAPK) activation, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-кB) pathway components and activation of the serine/threonine kinase AKT (PKB). In addition, BTK mediated signaling events are regulated by various phosphatases that can be recruited to the cell membrane, following crosslinking of inhibitory receptors, e.g., FcγRIIB that is exclusively expressed on B cells and signals upon immune complex binding. See text for details
In addition, Ig-α contains a conserved non-ITAM tyrosine residue, Y204, that upon activation by SYK recruits and phosphorylates the central B cell-linker molecule SH2-domain-containing leukocyte protein of 65 kDa (SLP65/BLNK) [45] (Fig. 2). Hereby, the adaptor molecule Cbl-interacting protein of 85 kD (CIN85) functions to oligomerize SLP65 and assembles intracellular signaling clusters for B cell activation [46]. SLP65 serves as a scaffold for various signaling molecules, including BTK and its substrate PLCγ2 [47,48,49,50]. In this micro-signalosome BTK is activated through Y551 phosphorylation by SYK or LYN and subsequently at Y223, as described above [30,31,32]. Fully activated BTK phosphorylates PLCγ2 at Y753 and Y759, which is important for its lipase activity [51]. Activated PLCγ2 hydrolyses PIP2 into inositol triphosphate (IP3) and diacylglycerol (DAG). IP3 regulates intracellular calcium levels and thereby activates nuclear factor of activated T cells (NFAT) transcription, via calcineurin and calmodulin. DAG mediates activation of protein kinase Cβ (PKCβ), which induces activation of several members of the MAPK family, including extracellular signal-regulated kinases 1 and 2 (ERK1/ERK2) and other MAPK targets, such as Jun N-terminal kinase (JNK), p38, and NF-кB pathway components [52] (Fig. 2). Hereby, BTK links the BCR to NF-кB activation [53, 54].
Another important branching point is induced more upstream in the BCR signaling cascade: in addition to BTK, PIP3 also interacts with PH-domain of AKT, resulting in its recruitment to the plasma membrane. Full activation of AKT requires phosphorylation at position T308, induced by 3-phosphoinositide-dependent protein kinase-1 (PDK1), and at S473, phosphorylated by mechanistic target of rapamycin (mTOR) complex 2 (See Ref [55] for an excellent review). Fully activated AKT then returns to the cytoplasm to enable a pro-survival signaling program that involves NFAT, forkhead transcription factors (FOXOs) and NF-кB-mediated pathways. Importantly, phosphorylation of AKT is positively regulated by BTK [56]. The BTK family member TEC, which can partly compensate for BTK [57], may on the other hand limit the capacity of BTK to activate AKT [58].
Upon activation in germinal centers (GCs), B cells can perform IGH chain class switching, by which it changes Ig expression from one isotype to another with different effector function, e.g. from IgM to IgG. In this process, the IGH constant (C) region is changed, but the variable (V) region remains the same. Interestingly, in contrast to IgM, the IgG BCR contains a cytoplasmic domain of considerable length with an Ig tail tyrosine (ITT) motif, which amplifies signaling [59]. SYK is required for ITT phosphorylation followed by recruitment of BTK through the adapter protein Grb2, leading to enhancement of IgG BCR-induced calcium mobilization. This amplification loop is thought to represent a cell-intrinsic mechanism for rapid activation of class-switched memory B cells.
Regulation of BTK activity and expression
Consistent with its crucial role in B cell differentiation, proliferation and survival, proper control of BTK activity is important for B cell homeostasis. Several mechanisms for its regulation have been identified to date.
The recruitment of BTK to the plasma membrane and its subsequent activation is regulated by various phosphatases that can be recruited to the cell membrane, similar to BTK. For example, the FcγRIIB is an inhibitory receptor that is exclusively expressed on B cells [60]. In contrast to the Igα/Ig-β ITAM motifs, FcγRIIB has immune tyrosine inhibitory motifs (ITIMs) in its cytoplasmic domain [61, 62] (Fig. 2). The binding of IgG antibodies to FcγRIIB results in LYN-mediated phosphorylation of ITIMs and recruitment of protein phosphatases such as SH2-domain containing inositol polyphosphate 5’phosphatase-1 (SHIP1) [63,64,65]. SHIP1 catalyzes the dephosphorylation of PIP3 and thereby inhibits recruitment of PH-domain containing proteins, such as BTK and PLCγ2 to the cell membrane. As a result, the downstream increase in intracellular calcium levels is diminished. Another phosphatase, SH2 domain containing protein tyrosine phosphatase-1 (SHP1), has the capacity to dephosphorylate tyrosine on BTK [65]. SHP1 acts downstream of CD22, a lectin molecule, and the glycoprotein CD5, both of which are on the B cell surface and function as negative regulators of BCR signaling.
In addition, several negative regulators of BTK have been identified. The iBTK protein directly binds to the BTK PH domain and thereby inhibits its activity [66]; PKCβ phosphorylates BTK on residue S180 in TH domain, modulating its membrane localization [67]; microRNA-185 reduces BTK mRNA levels and thereby downregulates BTK expression [68]. Likewise, expression of other microRNAs, including miR-210 and miR-425, significantly reduce BTK expression [69]. In this context, it was shown that treatment of primary CLL samples with histone deacetylase (HDAC) inhibitors resulted in increased expression of these miRs and decreased BTK protein. On the other hand, BTK itself can initiate a proteasome-dependent positive autoregulatory feedback loop by stimulating transcription from its own promoter through a pathway involving NF-кB [70].
BTK in other signaling pathways
Chemokine receptors
These receptors are G-protein coupled receptors that consist of seven transmembrane spanning domains and intracellular hetero-trimeric G-proteins composed of α, β, and y subunits (Gα, Gβ, and Gy) [71]. The chemokine receptors CXCR4 and CXCR5 are expressed on B cells in different stages of their development and play important roles in trafficking, homing and homeostasis [72]. Chemokine binding to the extracellular domain of its receptor induces conformational changes that result in dissociation of Gα and Gβy subunits (Fig. 3a). Both Gα and Gβy subunits can independently activate PI3K, which results in activation of BTK, AKT and MAPK dependent pathways [73, 74]. In addition, both Gα and Gβy subunits can directly bind BTK via the PH and TH domain [74, 75]. It has been shown that the Gα subunit directly stimulates the activity of BTK [76]. Due to its function downstream of chemokine receptors including CXCR4 and CXCR5, BTK is important for positioning of B cells in various lymphoid tissue compartments. This was first demonstrated by adoptive transfer experiments with BTK-deficient B cells, which exhibited impaired in vivo migration and homing to lymph nodes [77].

Role of Bruton’s tyrosine kinase downstream of chemokine receptors, Toll-like receptors and activating Fcγ receptors. Signaling cascade showing important events downstream of (a) Chemokine receptors (e.g. CXCR4): upon chemokine binding to the extracellular domain Gα and Gβy subunits can independently activate PI3K, which results in activation of BTK, AKT and MAPK-dependent pathways. b Toll-like receptors: upon ligand recognition TLRs recruit different proteins including TIR, MYD88, IRAK1 and TIRAP/MAL, all of which interact with BTK and induce downstream activation of the transcription factor NF-κB. c Activating Fc receptors (e.g. FcγRI): Following FcγRI cross-linking, Src-kinases, SYK, PI3K-γ and BTK are activated. In contrast, inhibitory Fc-receptors (FcγRIIB) containing ITIM domains recruit phosphatases and reduce BTK activation (Fig. 2). See text for details
Toll-like receptors (TLRs)
These extracellular or intracellular pattern recognition receptors are characterized by leucine-rich repeats and Toll/interleukin-1 receptor (TIR) domains (Fig. 3b). TLRs, expressed in B cells or myeloid cells, recognize structurally conserved molecules derived from bacteria and viruses. Upon activation most TLRs recruit the adaptor myeloid differentiation primary response 88 (MYD88) [78]. MYD88 activates interleukin-1 receptor-associated kinase1 (IRAK1), either on its own or in combination with an adaptor molecule, TIR domain containing adaptor protein (TIRAP, also known as MyD88 adapter-like (MAL)). BTK interacts with four different proteins downstream of TLR signaling including TIR, MYD88, IRAK1 and TIRAP/MAL) [79,80,81]. TLR signaling induces transcription factors including NF-кB, activator protein-1 (AP-1) and interferon regulatory factor 3 (IRF3), which results in activation, proliferation, antibody secretion, class switch recombination and pro-inflammatory cytokine production in B cells.
Fc receptor signaling
BTK is involved in signaling of both activating (ITAM-containing) and inhibitory (ITIM-containing) Fc-receptors, whose balance regulates several myeloid cell processes including activation, polarization and phagocytosis (Fig. 3c) [60, 82]. BTK is rapidly activated upon FcεRI cross-linking in mast cells [15]. In parallel to BCR signaling, following activating Fc-receptor cross-linking, SRC-kinases, SYK, PI3K-γ and BTK are activated [60]. In contrast, inhibitory Fc-receptors (FcγRIIB) containing ITIM domains recruit phosphatases and reduce BTK activation (see above).
BTK and B cell development in the bone marrow
Even before the gene involved in XLA was identified, X-chromosome inactivation studies showed that the defect in XLA patients was intrinsic to the B cell lineage and that myeloid cells had no developmental defects [83, 84]. B cells are generated from hematopoietic stem cells in the bone marrow throughout life by the ordered rearrangement of IGH and IGL chain gene segments (Fig. 4). After productive recombination of the IGH V, D and J genes, the IGH μ protein is expressed on the cell surface in association with the two invariant surrogate light chain (SLC) proteins VpreB and λ5 [85, 86],as the pre-BCR. Pre-BCR signaling marks a crucial checkpoint (checkpoint 1) to test the functionality of the IGH μ protein (Fig. 4) [87, 88]. To date, the mechanisms that initiate pre-BCR-mediated signaling are not fully resolved as both cell-autonomous and ligand-mediated signaling has been described [89,90,91,92]. An important function of pre-BCR signaling is to inhibit further IGH VDJ recombination, a phenomenon known as allelic exclusion [88]. Pre-BCR signaling leads to proliferation of pre-B cells and at the same time downregulation of SLC expression [88]. This is important for the exit of pre-B cells from the cell cycle to undergo the transition from large, cycling cells into small resting pre-B cells, in which IGL chain recombination occurs. In XLA patients B cell development is almost completely arrested at the pre-B cell stage. Although pre-B cells expressing intracellular IGH μ are present, they are small in size, indicating that BTK is essential for pre-BCR-dependent proliferation. BTK-deficient mice have only a mild pre-B cell defect, whereby pre-B cells show impaired developmental progression into immature B-cells [9, 10]. Nevertheless, an almost complete block is only found in mice that are double-deficient for e.g. BTK and SLP65 or BTK and TEC [57, 93, 94]. Interestingly SLP65-deficient mice, which also have a mild arrest at the pre-B cell stage, develop pre-B cell leukemia resembling pre-B ALL in humans [93, 94]. In this regard, BTK cooperates with SLP65 as a tumor suppressor independent of its kinase activity [95, 96]. SLP65 also mediates downregulation of SLC expression [97]. Analyses in wild-type, BTK and SLP65 deficient pre-B cells demonstrated that pre-BCR signaling induces IGL κ locus accessibility by functional redistribution of enhancer-mediated chromatin interactions [98]. BTK and SLP65 are important for the induction of IGL chain germ-line transcripts that are associated with locus accessibility. Moreover, BTK-deficient mice exhibit a ~ 50% reduction of IGL κ chain usage [98, 99]. Transcriptome analyses showed that BTK/SLP65deficient pre-B cells fail to efficiently upregulate many genes involved in IGL chain recombination, including Aiolos, Ikaros, Spib, Irf4, Oct2, polymerase-μ, and Mbp-1 [98].

Stages of B cell differentiation and associated malignancies. Model of B cell development indicating different stages of B cell differentiation and important immune checkpoints where BTK plays a key role. Various B-cell malignancies are indicated, which are associated with abnormal BTK signaling at distinct stages of B-cell differentiation and activation. Note that the cellular origin of U-CLL is thought to be CD5+ mature B cells. Somatic hypermutation status of BCR and gene expression profiling indicates post-germinal center (GC) origin of M-CLL. See text for detailed information. CLP, common lymphoid progenitor; CSR, class switch recombination; FDC, follicular dendritic cell; SHM, somatic hypermutation
If IGL chain recombination is not productive or the resulting BCR is autoreactive (checkpoint 2) (Fig. 4), developing B cells will undergo secondary IGL chain rearrangements, a process termed receptor editing [100,101,102]. Many autoreactive B cells are lost during development to the immature IgM+ B cell stage (central B cell tolerance), but it has been estimated that ~ 40% of the newly formed B cells that leave the bone marrow have self-reactivity [92].
BTK and peripheral B cell development and activation
Immature B cells from the bone marrow migrate to the spleen, where selection and maturation is continued within the transitional B cell compartment containing T1 and T2 B cells. In mice, T1 B cells, but not T2 B cells, are very sensitive to BCR-mediated apoptosis, indicating that the T1 to T2 differentiation marks a peripheral tolerance checkpoint (checkpoint 3) [103, 104]. In the absence of BTK, T2 cells do not generate survival responses and peripheral B cells are reduced by ~ 50%. As a result, BTK-deficient B cells exhibit an impaired transition from IgMhighIgDlow into IgMlowIgDhigh mature B cells. BTK-deficient mice lack the population of innate-like CD5+ B-1 cells, present in the peritoneal and pleural cavities and in small proportions in the spleen [7,8,9]. Consistent with the finding that these cells are important for IgM and IgG3 levels in the serum, in BTK-deficient mice IgM and IgG3 levels in serum are severely reduced, but the other isotypes are largely normal.
Marginal zone B cells are present in an area at the outermost portion of the white pulp in the spleen and are phenotypically defined as IgMhiIgDloCD21highCD23low B cells that respond to polysaccharide antigens independently of T cell help (Fig. 4). BCR and NOTCH2 signaling determine whether T1 B cells expressing surface ADAM10 are committed to becoming MZ B cells in vivo in the spleen [105, 106]. Although contradictory findings on the numbers of MZ B cells in BTK-deficient mice have been reported, it is clear that developing BTK-deficient MZ B cells have a selective disadvantage [107, 108].
Upon antigen recognition, activated B cells may either go into an extrafollicular response or develop into GC B cells [109, 110]. In the GCs B cells strongly proliferate and undergo somatic hypermutation (SHM) induced by activation induced cytidine deaminase (AID). GC B cells are selected involving follicular dendritic cells (FDCs) and T-follicular helper (TFH) cells (checkpoint 4) based on their antigen affinity [109]. Although BTK-deficient mice show normal T-cell dependent responses to model antigens, such as TNP-KLH [7, 8], there is a significant reduction in GC B cell numbers in physiological models, e.g. influenza virus infection [108]. In this context, it is of note that mice expressing the constitutively active BTK mutant E41K fail to form GCs [111, 112], whereas overexpression of wild-type BTK induces spontaneous GC formation [113, 114]. Consequently, BTK-overexpressing mice develop autoimmunity involving B cell-induced disruption of T cell homeostasis [113, 114].
BTK in B cell malignancies
BTK activity is crucial for survival and proliferation of leukemic B cells and for their interactions with cells in the tumor microenvironment. Below, we discuss the role of BTK in various B cell malignancies (Fig. 4).
CLL
This is the most common leukemia in the western world, primarily affecting the elderly, and is characterized by the accumulation of mature circulating IgMlow CD5+ B cells [115]. Several genetic aberrations with prognostic value and impact on treatment decisions in CLL have been described. These include deletions of the chromosomal regions 17p13 (containing the TP53 tumor suppressor gene), 11q23 (containing DNA damage checkpoint protein ATM), or 13q14 (miR-15a, miR-16-1), and trisomy of chromosome 12 [116, 117]. Furthermore, > 80% of cases harboring del(17p) also carry TP53 mutations in the remaining allele [118]. Such patients with TP53 defects are classified as ‘high-risk’ and often respond poorly to therapy [119]. Moreover, a significant proportion of CLL patients carry a TP53 mutation in the absence of a 17p deletion [120, 121].
On the basis of SHM status of IGHV, CLL can be grouped into mutated CLL (M-CLL) and unmutated CLL (U-CLL). M-CLL have a more favorable prognosis and are derived from post-GC B cells. The origin of U-CLL appeared less clear and several cellular origins of CLL were suggested, including MZ B cells, CD5+ B cells, and regulatory B cells [122,123,124,125,126]. Although initial gene expression profiling indicated that M-CLL and U-CLL were quite homogeneous and related to memory B cells derived from T cell-dependent and T-cell independent responses, respectively [123], more recent gene expression profiling studies have provided evidence for a different origin [124]. This study by Seifert et al. shows that U-CLL derives from unmutated mature CD5+ B cells. Moreover, it was concluded that M-CLL originate from a distinct and previously unrecognized post-GC B cell subset with a CD5 + CD27+ surface phenotype.
Several lines of evidence establish a role of chronic BCR-mediated signaling in CLL pathogenesis [127]. (i) Prognosis is correlated with the BCR SHM status [128]; (ii) The BCR repertoire is highly restricted [129, 130], suggesting a role for antigenic selection in the initiation or progression of CLL. Antigens binding to CLL BCRs include self-antigens, such as non-muscle myosin IIA, vimentin, apoptotic cells and oxidized low-density lipoprotein [131,132,133,134,135,136], as well as foreign antigens (bacterial polysaccharides and β-(1,6)-glucan, a major antigenic determinant on fungi [132,133,134,135,136,137]); Interestingly, evidence was provided in mice that pathogens may drive CLL pathogenesis by selecting and expanding pathogen-specific B cells that cross-react with self-antigens [138]; (iii) CLL cells were reported to display cell-autonomous Ca2+ mobilization in the absence of exogenous ligands, by virtue of recognizing a single conserved BCR-internal epitope in the IGHV second framework region [139]; very recently, it was found that the internal epitopes recognized by CLL BCRs from distinct subgroups are different [140]. Moreover, the avidity of the BCR-BCR interactions that can lead to receptor declustering influences the clinical course of the disease [139, 140].
In line with chronic BCR-mediated signaling, CLL cells show constitutive activation of various BCR pathway associated kinases. Hereby, BTK is essential for constitutively active pathways implicated in CLL cell survival, including AKT, ERK and NF-кB, both in patient cells and mouse models [133, 141,142,143]. CLL cells are thought to interact with the tissue microenvironment and lymph node resident CLL cells show gene expression signatures indicative of BCR activation [144, 145]. Moreover, BTK is critical for BCR- and chemokine-controlled integrin-mediated retention and/or homing of CLL B cells in their microenvironment [146].
Mantle cell lymphoma (MCL)
This disease results from malignant transformation of B lymphocytes in the mantle zones surrounding GCs (Fig. 4) and has a remarkably biased BCR repertoire [147]. Approximately 85% of the patients harbor the hallmark chromosomal translocation t(11:14)(q13;32). This event juxtaposes the CCND1 gene to an enhancer in the Ig heavy chain locus [148], resulting in constitutively cyclin-D1 expression and abnormal proliferation. In a fraction of MCL patients lymphoma cells express the SOX11 transcription factor, which is associated with minimal Ig SHM, higher genetic instability and a more aggressive clinical course [149, 150]. Primary MCL cells show strong expression and Y223-phosphoryation of BTK [151] and in a subset of patients constitutive phosphorylation of LYN, SLP65, SYK and PKCβ [152, 153]. Similar to CLL, the tumor microenvironment plays an important role in MCL pathogenesis. BTK is essential for retention of MCL cells in lymphoid tissues, since BTK inhibition induces an egress of malignant cells into peripheral blood [154].
Waldenström’s Macroglobulinemia (WM)
This indolent B-cell malignancy is characterized by IgM-secreting lymphoma cells in the bone marrow. The majority of WM patients have a somatic leucine to proline substitution at position 265 of MyD88 (MyD88L265P) [155]. This activating mutation has also been reported in low frequencies in activated B-cell-like diffuse large B-cell lymphoma (14%–29%) (see below), primary central nervous system lymphoma (PCNSL; 33%), mucosa-associated lymphoid tissue (MALT) lymphoma (9%), and CLL (2.9%) [156,157,158,159]. The mutated MyD88L265P protein binds phosphorylated-BTK and triggers NF-кB signaling [160]. In addition, ~ 30% of WM patients show the CXCR4 S338X somatic mutation, leading to enhanced CXCL12-triggered activation of AKT and ERK [161]. In this regard, CXCR4 and VLA-4 interactions have been shown to regulate trafficking and adhesion of WM cells to the bone marrow [162].
ABC-DLBCL
DLBCL is the most common form of B cell non-Hodgkin lymphomas (B-NHLs) representing ~ 30–40% of all cases. Patients most often present with a rapidly growing tumor in single or multiple, nodal or extranodal sites. Based on gene expression profiling, three major molecular subtypes have been identified: GC B-cell-like (GCB-DLBLCL), activated-B-cell-like (ABC-DLBCL) and primary mediastinal B-cell lymphoma (PMBL) [163]. Whereas GCB-DLBCL and ABC-DLBCL make up the majority of cases at roughly equal frequency, PMBL accounts for up to 10% of cases of DLBCL [164]. GCB-DLBCL tumors express many genes found in normal GC B cells and have typically switched to an IgG BCR, while gene expression in ABC-DLBCL, which are predominantly IgM+, resembles that of antigen-activated plasmablasts [165, 166]. ABC-DLBCL has an inferior clinical outcome than GCB-DLBCL with a three-year overall survival of ~ 45% [167].
ABC-DLBCL are dependent on constitutive NF-кB signaling for their survival and proliferation [168,169,170]. Approximately 50% of ABC-DLBCL harbor mutations in CARD11 or other NF-кB pathway components, including the MyD88L265P mutation [169,170,171]. In addition, ~ 20% of patients carry an activating mutation in CD79A/B. Consistent with a role of NF-кB downstream of the BCR (Fig. 2), it was found that knockdown of BCR components, CD79A/B and downstream signaling molecules, induced cell death in ABC-DLBCL lines with unmutated CARD11 [172]. Moreover, RNAi experiments demonstrated that ABC-DLBCL lines are dependent on MyD88 and its associated kinase IRAK1 for their survival in line with NF-kB function in the TLR pathway (Fig. 3b). In addition, SYK amplification and deletion of PTEN, a phosphatase that dephosphorylated PIP3, are also selective genetic alterations identified in ABC-DLBCL [173].
In contrast to ABC-DLBCL, GCB DLBCLs do not acquire highly recurrent mutations in CD79A/B or NF-κB components. Whereas ABC-DLBCL frequently respond to BTK inhibition (see below), GC-DLBCL do not respond and exhibit tonic BCR signaling that does not affect their calcium flux, but acts primarily to activate AKT [174]. Accordingly, forced activation of AKT rescued GCB-DLBCL lines from knockout of the BCR or SYK and CD19, two mediators of tonic BCR signaling [174]. The importance of the oncogenic AKT/PI3K pathway in GCB-DLBCL is evident from the finding that in ~ 55% of patients the tumor suppressor phosphatase and tensin homolog (PTEN), a negative regulator of PI3K, is inactivated. The mechanisms of PTEN inactivation include mutation, deletion or amplification of the miR17–92 microRNA cluster that downregulates PTEN expression [175, 176].
Primary CNS lymphoma (PCNSL), another DLBCL subtype, is an aggressive brain tumor that has a complete response rate of < 40% with methotrexate-based regimens and is subject to late recurrences. Patients showed mutations in the MYD88, CD79B and CARD11 genes in ~ 58%, ~ 41% and ~ 13% of cases, respectively [177].
Other B cell malignancies
The hallmark of follicular lymphoma (FL), the (14;18) translocation resulting in BCL2 overexpression, is found in up to ~ 85% of patients. The pathogenesis of FL is complex and involves additional cell-intrinsic genetic changes, frequently including mutations in histone-encoding genes (in ~ 40% of cases), the SWI/SNF complex or the interconnected BCR and CXCR4 chemokine receptor signaling pathways, as well as alterations within the FL microenvironment [178]. The importance of BCR and NF-κB signaling is underscored by the finding of recurrent mutations in the genes encoding CD22, SLP65/BLNK, PLCγ2, SYK, PKCβ, BCL10, the NF-κB p100 subunit and the deubiquitinating enzyme A20/TNFAIP3, which is a negative regulator of NF-κB signaling. In addition, the HVCN1 gene (coding for a hydrogen voltage-gated proton channel that acts downstream of the BCR and is downregulated in proliferating B cells) is frequently mutated in FL. Interestingly, BTK mutations were found that suggest activation, e.g. the L528 W mutation in the kinase domain, which is associated with resistance to BTK inhibition in CLL (described below), and an in-frame deletion that also alters this amino acid and the adjacent C527. Moreover, two loss-of-function BTK mutations were identified, T117P and R562W, which are also found in XLA patients, but it remains unclear how these mutations contribute to FL pathogenesis [178].
In multiple myeloma (MM), a malignancy of plasma cells in the bone marrow, BTK was shown to be overexpressed, whereby BTK activated AKT signaling, leading to down-regulation of P27 expression and upregulation of key stemness genes [179, 180]. MM cells originate from plasma cells, which do not express surface BCR, and rely for their survival and proliferation on signals from the microenvironment in the bone marrow. BTK may be critical in the MM microenvironment, in particular for secretion of cytokines and chemokines by osteoclasts [181].
Finally, BCR and TLR are thought to be key activation pathways in marginal zone lymphoma (MZL), often associated with chronic inflammation in the context of autoimmunity and/or infection [182], implicating BTK as a potential target. In this context, whole exome sequencing identified recurrent inactivating mutations in Kruppel-like factor 2 (KLF2) which impeded its capacity to suppress NF-κB activation. In addition, recurrent mutations in the TLR/NF-κB pathway were found, affecting e.g. the MYD88, TRAF3, CARD11, A20/TNFAIP3 and CARD11 genes [183].
The BTK inhibitor ibrutinib in clinical studies
Ibrutinib (PCI-32765) is an oral irreversible BTK inhibitor that covalently binds to cysteine at position 481 in the kinase domain and thereby blocks kinase activity [184]. As a result BTK has lost its kinase activity, but Y551 phosphorylation by SYK is not affected. The in vivo effect of ibrutinib was first confirmed in a mouse model of autoimmune disease and in dogs with spontaneous B-cell non-Hodgkin lymphoma, in which it induced objective clinical responses [185].
Efficacy of ibrutinib in a clinical study was first reported in patients with various relapsed/refractory B-cell malignancies, showing clinical safety and promising durable objective responses particularly in CLL and MCL [186]. Responding patients showed sustained reduction in lymphadenopathy, accompanied by transient rise in absolute lymphocyte count, a phenomenon known as lymphocytosis [186]. The next phase Ib/II multicenter trial, with a continuous ibrutinib regimen in relapsed/refractory CLL patients also showed lymphocytosis in the first weeks of treatment, but lymphocyte counts normalized or dropped below baseline after prolonged treatment [11]. Importantly, the overall response rate was ~ 71%, independent of clinical or genomic risk factors.
In a phase II study, patients with relapsed or refractory MCL were treated orally with ibrutinib, resulting in a response rate of ~ 68% [187]. It was subsequently demonstrated that Ibrutinib was also highly active and associated with durable responses in pretreated patients with Waldenström’s macroglobulinemia, whereby MYD88 and CXCR4 mutation status affected the response [188]. Ibrutinib very rapidly received breakthrough designation and was subsequently approved by the Food and Drug Administration (FDA) for the treatment of MCL, CLL and WM between November 2013 and January 2017.
In addition, ibrutinib has also been tested in other B cell malignancies. In line with the possible role of BTK in FL, 6 out of 16 (38%) relapsed/refractory FL patients show response upon ibrutinib treatment [186]. In a phase II study ibrutinib induced durable remissions in ~ 50% of the MZL patients [189]. In a phase I study the majority (77%) of patients with PCNSL show clinical responses to ibrutinib [177]. Table 1 summarizes the data from current clinical trials in various B-cell malignancies.
Several studies were performed to explain the therapeutic mode of action of ibrutinib. In CD40- or BCR-activated CLL cells, ibrutinib reduced survival by abrogating downstream pathways including ERK, PI3K and NF-кB [141]. Ibrutinib inhibited migration of CLL cells towards chemokines such as CXCL12 and CXCL13, suggesting that treatment inhibits homing and retention of malignant cells in their survival niches [77]. Ibrutinib was also found to reduce secretion of BCR-dependent chemokines CCL3 and CCL4 [142]. Another key effect was that it inhibited integrin α4β1-mediated adhesion of CLL cells to fibronectin and VCAM1 [146] and thus interaction with the tumor microenvironment [146]. Therefore, ibrutinib apparently works by a dual mechanism, by inhibiting intrinsic B cell signaling pathways to compromise their proliferation and survival as well as by disrupting tumor-microenvironment interactions. Importantly, both in CLL and MCL ibrutinib treatment induces a redistribution lymphocytosis, a transient rise of leukemic cells in the circulation and a concomitant rapid reduction of these cells at the affected tissue sites. In contrast to classical cytotoxic chemotherapy, ibrutinib does not cause tumor lysis syndrome, which is a common complication of cancer therapy because of metabolic disturbances when large numbers of tumor cells die quickly. Therefore, most likely the displacement of B cells from nurturing tissue niches because of inhibition of integrin-mediated retention of leukemic cells, is an important mechanism of action of ibrutinib, rather than robust inhibition of survival of malignant B cells [190]. As a result, leukemic cells undergo ‘death by neglect’, because their mobilization induces ‘homelessness’ (anoikis), a form of programmed cell death [191, 192].
Despite impressive clinical success of ibrutinib, its curative potential in B cell malignancies is not established yet, as ibrutinib is often prescribed as life-long therapy. Importantly, continuous therapy may lead to selection or outgrowth of resistant clones, as described in a subset of patients who relapse upon ibrutinib therapy. Two important therapy-associated resistance mechanisms have been identified, involving BTK C481S mutation (the site of action of Ibrutinib) or activating mutations in PLCy2 (R665W, S707Y and L845F) [193, 194]. Recently another BTK mutation, T316A in the SH2 domain, was described, as well as clonal evolution underlying leukemia progression in patients with ibrutinib-relapsed CLL [195]. In addition, missense mutation within the coiled-coil domain of CARD11 (R179Q) have been shown to promote BTK-independent activation of NF-κB and thus ibrutinib resistance in DLBCL, MCL and PCNSL [177, 196, 197]. Furthermore, an activating mutation in BTK (L528 W) that confers resistance to ibrutinib treatment has been found in CLL and FL [178, 198].
In clinical trials the adverse events were mostly limited to grade 1 or 2 in severity, but in some cases side-effects led to discontinuation of the therapy [199,200,201]. Because ibrutinib treatment has a considerable high risk of bleeding in treated patients, concomitant anti-coagulation (~ 11%) and antiplatelet (~ 34) use is common and ~ 3% of the patients were reported to have major bleeding events [202]. Atrial fibrillation has been reported in up to 16% of patients taking ibrutinib, whereby stroke prevention poses a challenge because of the increased bleeding risk. Therefore, close monitoring is recommended, especially during the first 6 months of ibrutinib therapy [203]. Although the occurrence of atrial fibrillation might possibly be related to inhibition of the BTK-regulated PI3K/AKT pathway in cardiac myocytes [204], the mechanisms involved remain largely unidentified.
Three year follow-up of ibrutinib-treated CLL patients showed that prolonged treatment was associated with improvement in response quality (the ORR increased to > 90%) and durable remission, while toxicity including cytopenia, fatigue, and infection diminished. Moreover, progression remains uncommon [205]. Findings from the longest follow-up reported to date, evaluating up to 5 years of ibrutinib in CLL patients, show that it is relatively safe and effective, with ~ 89% of treatment-naïve and relapsed patients experiencing a response to the therapy [206].
Part of the toxicities and side effects of ibrutinib can be explained by its non-specific nature: ibrutinib is not an exclusive inhibitor of BTK and off-target inhibition includes kinases that contain a cysteine residue aligning with Cys-481 in BTK. These include other TEC-family kinases (ITK, BMX, TEC), as well as epidermal growth factor receptor (EGFR), T-cell X chromosome kinase (TXK) and Janus Kinase 3 (JAK3) [12, 185, 207]. In this context, it is of note that the bleeding risk in patients receiving ibrutinib was thought to relate to off-target inhibition of TEC [12]. BTK is expressed in platelets where it is important for signaling via the collagen receptor glycoprotein VI (GPVI); platelets from XLA patients display diminished aggregation, dense granule secretion and calcium mobilization in response to collagen and C-reactive protein [208]. Nevertheless, XLA patients do not have an increased risk of bleeding [209]. Findings by Bye et al. indicated that both BTK and TEC – although required for GPVI-mediated platelet aggregation – are redundant for platelet adhesion to collagen and thrombus formation [210]. Rather, ibrutinib but not the more selective BTK inhibitor acalabrutinib (see below) inhibits SRC family kinases that have a critical role in platelet function [210]. These findings explain why in contrast to ibrutinib, treatment with acalabrutinib was not associated with major bleeding events [12].
A recent systematic review of infectious events with ibrutinib in the treatment of B cell malignancies provided evidence for infection-related complications in ~ 50% of patients taking ibrutinib, whereby ~ 20% of patients developed pneumonia due to opportunistic pathogens [211]. Hereby, data suggest that these events may involve inhibition of both BTK and its closely related family member ITK. On the other hand, it was shown that ibrutinib treatment increased the in vivo persistence of both CD4+ and CD8+ activated T cells and diminished the immune-suppressive properties of CLL cells. As these effects were not seen with more specific BTK inhibitor acalabrutinib that lacks ITK inhibitory activity (see below), it was concluded that the T cell expansion is unlikely to be caused by BTK inhibition [212]. Rather, ibrutinib treatment of activated T cells diminishes activation-induced cell death by targeting ITK, a finding also reported in murine models of ITK deficiency. However, both inhibitors reduced the expression of the inhibitory co-receptors programmed cell death protein 1 (PD-1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA4) on T cells, as well as expression of the immunosuppressive molecules CD200, B- and T-lymphocyte attenuator (BTLA) and IL-10 by CLL cells [212]. Therefore, ibrutinib likely diminishes the immune-suppressive properties of CLL cells through both BTK-dependent and ITK-dependent mechanisms.
Inhibition of BTK and ITK with ibrutinib was shown to be effective in the prevention of chronic graft-versus-host (GvH) disease following allogeneic hematopoietic stem cell transplantation (SCT) in several mouse models [213, 214]. Accordingly, also studies in patients with relapsed CLL following SCT support that ibrutinib augments the GvH versus-leukemia (GVL) benefit likely through ITK inhibition [215]. In particular, it was shown that ibrutinib selectively targeted pre–germinal B cells and depleted Th2 helper cells, whereby these effects persisted after drug discontinuation.
Taken together, these findings provide a rationale for combination immunotherapy approaches with ibrutinib in CLL and other cancers.
Ibrutinib in combination therapies and second generation BTK inhibitors
The finding of ibrutinib resistance, together with multiple modes of action and the microenvironmental dependence of B-cell malignancies, has fueled the development of novel combination strategies. With the aim to achieve deeper remissions within a short treatment time, many ibrutinib combination therapies are currently considered (Table 2). Hereby, ibrutinib treatment forces egress of malignant B cells out of their protective niches into circulation, where they become vulnerable to direct cytotoxic activity of either chemotherapy, an inhibitor of the pro-survival protein Bcl-2, or antibody mediated cytotoxicity (ADCC) of anti-CD20 antibody therapy.
Side-effects associated with off-target kinase inhibition may limit the use of ibrutinib as therapeutic agent (as discussed above). Ibrutinib can antagonize rituximab-induced ADCC due to inhibition of its family member ITK in NK cells, further limiting its use in combination regimens [216]. Therefore, many efforts have focused on developing highly selective BTK inhibitors, of which three have reached advanced stages of clinical development [217].
Acalabrutinib (ACP-196)
This highly selective irreversible BTK inhibitor has significantly less off-target kinase activity [207]. Acalabrutinib also binds C481 and lacks irreversible targeting to alternative kinases, such as EGFR, ITK, TXK, SRC family kinases and JAK3. The first pre-clinical study in canine models of Non-Hodgkin B-cell lymphoma demonstrated enhanced in vivo potency compared to ibrutinib [218]. In a phase I/II clinical trial in patients with relapsed/refractory CLL the overall response rate was ~ 95% and in patients with del(17)(p13.1) this was 100%, with a median follow-up up ~ 14 months [12]. No dose-limiting toxicities, episodes of atrial fibrillation, or bleeding-related events have been reported to date. To investigate the superiority of either inhibitor, a phase III trial for direct comparison of ibrutinib with acalabrutinib in R/R CLL patients is currently ongoing (NCT02477696). Additionally, in a phase II trial in patients with relapsed/refractory MCL, acalabrutinib induced an overall response of ~ 81% with ~ 40% patients achieving a complete response [219]. This led to accelerated FDA approval of acalabrutinib in MCL [220].
BGB-3111
Another selective inhibitor of BTK kinase activity with superior oral bioavailability and higher selectivity than ibrutinib is BGB-3111, which was shown to inhibit proliferation of several MCL and DLBCL cell lines. Due to weaker ITK inhibition, BGB-3111 was at least 10-fold weaker than ibrutinib in inhibiting rituximab induced ADCC. When 45 CLL patients were treated on a phase I/II study, therapy was well tolerated and was associated with a response rate of ~ 90% after a follow-up of 7.5 months and no cases of disease progression or Richter’s transformation [221] (see also Table 1).
Ono/GS-4059
In vivo efficacy of this compound was initially described in an ABC-DLBCL xenograft model and in vitro anti-proliferative effects in DLBCL, FL, MCL and CLL cell lines were described [222]. Early-phase clinical trial data in patients with several B-cell malignancies include clinical responses in patients with high-risk CLL genetics (Table 1).
Role of BTK in the tumor microenvironment
Inhibition of BTK has now also extended into the field of solid tumors, following insights into the role of BTK in various cells of the tumor microenvironment and in non-hematological tumor cells when ectopically expressed. An understanding of the diverse roles of BTK in non-lymphocytic cells will be pivotal in the development of novel treatment combinations for haematopoietic and solid tumors.
BTK is involved in TLR- and Fc-receptor mediated activation, maturation, migration and survival of myeloid cells [223, 224]. However, the role of BTK identified is dependent on cell type investigated, the nature of activating stimuli, the model used (in vivo or in vitro) and the species investigated, i.e. mouse or human. Analyses in various mouse models and in vitro studies with myeloid cells from XLA-patients clearly implicate BTK in TLR4/8/9-signaling, and possibly others [79, 225,226,227]. However, data are often conflicting, e.g. TLR8-induced IL-6 production by BTK-deficient DCs were reported to be impaired [226], enhanced [228], or unaffected [229]. Also TLR4/7/8-induced TNFα was reported to be reduced [226, 229] or enhanced [228].
Of further relevance in the context of the tumor microenvironment is the polarization status of macrophages, with M1 macrophages displaying a pro-inflammatory anti-tumor phenotype and M2 macrophages being immunosuppressive [22]. Whereas one study indicated an M2-skewing of BTK-deficient macrophages [230], recently in a pancreatic cancer mouse model an M1-skewing of intratumoral macrophages was found following ibrutinib treatment [231]. In contrast, ibrutinib induced M1 to M2-skewing of nurse-like cells, which show properties of tumor-associated macrophages, accompanied by impaired phagocytosis, increased IL-10 production mediating pro-survival signals in CLL [232]. It remains unknown what causes these incongruences in BTK-dependent myeloid polarization, however it is conceivable that the different roles of BTK in a complex ecology of tumor-infiltrating cells and the limited specificity of ibrutinib contribute to the conflicting findings.
In solid tumors, chronic deposition of immune complexes foster carcinogenesis due to chronic inflammation, angiogenesis and M2 macrophage polarization in response to activating Fc-receptor ligation on myeloid cells [231, 233, 234]. Interestingly, inhibiting BTK during Fc-receptor stimulation of macrophages in vitro using Ibrutinib prevented M2-skewing [231].
Granulocytes and their immature immune-suppressive counterparts, myeloid derived suppressor cells (MDSC), are strongly implicated in tumor progression, rendering them important candidates for therapy [235]. Although loss of BTK in XLA neutrophils does not impair functional TLR responses [236], the numbers of circulating granulocytes are reduced in XLA-patients and BTK-deficient mice [237,238,239]. Moreover, BTK-deficient neutrophils manifest increased sensitivity to apoptosis, decreased maturation, differentiation, trafficking and impaired functionality including reactive oxygen species (ROS) production [238,239,240,241]. Likewise, ibrutinib treatment inhibited the generation, migration, TNFα and ROS-production of MDSCs both in vitro and in solid tumor mouse models [242]. Ibrutinib treatment partially alleviated MDSC-mediated CD8+ T-cell suppression and enhanced anti-PD-L1 therapy efficacy in a breast cancer model. BTK inhibition in granulocytes and MDSCs in solid tumors may therefore be important in the development of effective combination therapies.
BTK inhibition in solid malignancies
Ectopic BTK expression has been observed in various solid tumors, whereby evidence is accumulating for its involvement in oncogenesis [24,25,26,27]. These pre-clinical findings have led to the initiation of several early phase I/II clinical trials in which BTK inhibition monotherapy is evaluated in advanced ovarian, colorectal, prostate and brain cancer patients(Table 3).
Also in BTK-negative solid tumors that do not express BTK, its inhibition may hold promise as multiple cell types in the tumor microenvironment are regulated by BTK. Inhibition of BTK in pre-clinical models of pancreatic cancer, breast cancer and BTK-negative colon cancer have shown only marginal improvement of survival as monotherapy, but when combined with chemo- or immunotherapy, survival was greatly enhanced [231, 242, 243]. This has sparked the emergence of several trials investigating the safety and efficacy of ibrutinib or acalabrutinib, in combination with conventional PD-1/PD-L1 checkpoint inhibition therapy (Table 3).
Given that ibrutinib shows off-target inhibition of JAK3, ITK and EGFR [185, 207], it can be used to target oncogenic pathways other than BTK in tumor cells and as a T-cell modulator in combination immunotherapy [243,244,245,246]. Hereby, ibrutinib treatment increased cellular persistence and decreased expression of co-inhibitory surface molecules on Chimeric antigen receptor (CAR) T cells in models of CLL and MCL [245, 246]. Whether in these studies ibrutinib acts on ITK in (CAR) T cells, on BTK in the malignant cells or other kinases remains undetermined. Paradoxically, inhibiting ITK in T cells may be efficacious in cancer, as this may enhance Th1-skewing of CD4+ T-cells and thereby improved memory formation and functionality of CD8+ T-cells, potentially leading to improved anti-tumor immunity [243, 247]. These potentially beneficial off-target effects of ibrutinib may be lost in the highly specific BTK-inhibitors that are currently being evaluated.
Conclusions
Targeting of BTK, which has a central role in several signaling pathways in B cells, particularly the BCR, has shown impressive efficacy as therapeutic option for various B cell malignancies in clinical trials. Much progress has been made in recent years in defining the complex mechanisms of action of BTK inhibition. These involve intrinsic signaling pathways in leukemic cells that are central to cellular survival, proliferation and - most importantly - retention in supportive microenvironments. Moreover, BTK inhibition shows promise as a therapy that influences crucial immune cells in the tumor microenvironment. Because data from BTK-deficient or inhibitor-treated myeloid cells in the context of cancer are scarce, it is not clear whether BTK inhibition by e.g. ibrutinib is based on its specificity for BTK in particular myeloid cells and/or due to off-target effects in signaling pathways in CD4+ or CD8+ T cells. Of note, because in CLL ibrutinib treatment diminished the immunosuppressive properties of malignant cells through BTK-dependent and BTK-independent mechanisms (probably via ITK inhibition) [212], it will be interesting to observe whether the same level of anti-tumor efficacy is maintained by specific BTK inhibition alone. It is very well conceivable that for particular malignancies it may be advantageous to use BTK inhibitors that show additional specificity for related kinases.
Although the efficacy of BTK inhibition as a single agent therapy is strong, it has been shown that resistance may develop and now a broad range of studies focus on development of effective combination therapies to improve clinical responses. The identification of differences in efficacy and toxicity profiles between available BTK inhibitors awaits direct comparative studies. In this context, design of treatment strategies will depend on detailed analyses of clinical responses, resistance development, toxicity and quality of life for individual BTK inhibitors in combination therapies in relation to the various malignancies and patient subgroups.
Change history
03 April 2019
Following publication of the original article [1], the authors reported an error in Table 1.
Abbreviations
- ABC-DLBCL:
-
Activate B-cell diffuse large B-cell lymphoma
- BCR:
-
B cell receptor
- BTK:
-
Bruton’s tyrosine kinase
- CLL:
-
Chronic lymphocytic leukemia
- CR:
-
Complete response
- DLBCL:
-
Diffuse large B cell lymphoma
- ERK:
-
Extracellular signal-regulated kinase
- GC:
-
Germinal center
- ITAM:
-
Immunoreceptor tyrosine-based activation motif
- ITIM:
-
Immunoreceptor tyrosine-based inhibitory motif
- MCL:
-
Mantle cell lymphoma
- M-CLL:
-
Mutated chronic lymphocytic leukemia
- MYD88:
-
Myeloid differentiation primary response 88
- NFAT:
-
Nuclear factor of activated T cells
- NF-κB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- ORR:
-
Overall response rate
- OS:
-
Overall survival
- PCNSL:
-
Primary central nervous system lymphoma
- PD-1:
-
Programmed cell death protein 1
- PI3K:
-
Phosphatidyl-inositol-3-kinase
- PIP3:
-
Phosphatidylinositol-3,4,5-triphosphate
- PLCγ:
-
Phospholipase C γ
- PR:
-
Partial response
- SHIP1:
-
SH2-domain containing inositol polyphosphate 5’phosphatase-1
- SHM:
-
Somatic hypermutation
- SHP1:
-
SH2 domain containing protein tyrosine phosphatase-1
- SYK:
-
Spleen tyrosine kinase
- TLR:
-
Toll-like receptor
- U-CLL:
-
Unmutated chronic lymphocytic leukemia
- WM:
-
Waldenström’s Macroglobulinemia
References
Gross S, Rahal R, Stransky N, Lengauer C, Hoeflich KP. Targeting cancer with kinase inhibitors. J Clin Invest. 2015;125:1780–9.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Vetrie D, Vorechovsky I, Sideras P, Holland J, Davies A, Flinter F, Hammarstrom L, Kinnon C, Levinsky R, Bobrow M, et al. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature. 1993;361:226–33.
Tsukada S, Saffran DC, Rawlings DJ, Parolini O, Allen RC, Klisak I, Sparkes RS, Kubagawa H, Mohandas T, Quan S, et al. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell. 1993;72:279–90.
Rawlings DJ, Saffran DC, Tsukada S, Largaespada DA, Grimaldi JC, Cohen L, Mohr RN, Bazan JF, Howard M, Copeland NG, et al. Mutation of unique region of Bruton's tyrosine kinase in immunodeficient XID mice. Science. 1993;261:358–61.
Thomas JD, Sideras P, Smith CI, Vorechovsky I, Chapman V, Paul WE. Colocalization of X-linked agammaglobulinemia and X-linked immunodeficiency genes. Science. 1993;261:355–8.
Scher I. The CBA/N mouse strain: an experimental model illustrating the influence of the X-chromosome on immunity. Adv Immunol. 1982;33:1–71.
Khan WN, Alt FW, Gerstein RM, Malynn BA, Larsson I, Rathbun G, Davidson L, Muller S, Kantor AB, Herzenberg LA, et al. Defective B cell development and function in Btk-deficient mice. Immunity. 1995;3:283–99.
Hendriks RW, de Bruijn MF, Maas A, Dingjan GM, Karis A, Grosveld F. Inactivation of Btk by insertion of lacZ reveals defects in B cell development only past the pre-B cell stage. EMBO J. 1996;15:4862–72.
Middendorp S, Dingjan GM, Hendriks RW. Impaired precursor B cell differentiation in Bruton's tyrosine kinase-deficient mice. J Immunol. 2002;168:2695–703.
Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, Grant B, Sharman JP, Coleman M, Wierda WG, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369:32–42.
Byrd JC, Harrington B, O'Brien S, Jones JA, Schuh A, Devereux S, Chaves J, Wierda WG, Awan FT, Brown JR, et al. Acalabrutinib (ACP-196) in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374:323–32.
Burger JA, Tedeschi A, Barr PM, Robak T, Owen C, Ghia P, Bairey O, Hillmen P, Bartlett NL, Li J, et al. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N Engl J Med. 2015;373:2425–37.
Hendriks RW, Yuvaraj S, Kil LP. Targeting Bruton's tyrosine kinase in B cell malignancies. Nat Rev Cancer. 2014;14:219–32.
Kawakami Y, Yao L, Miura T, Tsukada S, Witte ON, Kawakami T. Tyrosine phosphorylation and activation of Bruton tyrosine kinase upon fc epsilon RI cross-linking. Mol Cell Biol. 1994;14:5108–13.
Hata D, Kawakami Y, Inagaki N, Lantz CS, Kitamura T, Khan WN, Maeda-Yamamoto M, Miura T, Han W, Hartman SE, et al. Involvement of Bruton's tyrosine kinase in FcepsilonRI-dependent mast cell degranulation and cytokine production. J Exp Med. 1998;187:1235–47.
Jongstra-Bilen J, Puig Cano A, Hasija M, Xiao H, Smith CI, Cybulsky MI. Dual functions of Bruton's tyrosine kinase and Tec kinase during Fcgamma receptor-induced signaling and phagocytosis. J Immunol. 2008;181:288–98.
Wang D, Feng J, Wen R, Marine JC, Sangster MY, Parganas E, Hoffmeyer A, Jackson CW, Cleveland JL, Murray PJ, Ihle JN. Phospholipase Cgamma2 is essential in the functions of B cell and several fc receptors. Immunity. 2000;13:25–35.
Shinohara M, Koga T, Okamoto K, Sakaguchi S, Arai K, Yasuda H, Takai T, Kodama T, Morio T, Geha RS, et al. Tyrosine kinases Btk and Tec regulate osteoclast differentiation by linking RANK and ITAM signals. Cell. 2008;132:794–806.
Oda A, Ikeda Y, Ochs HD, Druker BJ, Ozaki K, Handa M, Ariga T, Sakiyama Y, Witte ON, Wahl MI. Rapid tyrosine phosphorylation and activation of Bruton's tyrosine/Tec kinases in platelets induced by collagen binding or CD32 cross-linking. Blood. 2000;95:1663–70.
Ito M, Shichita T, Okada M, Komine R, Noguchi Y, Yoshimura A, Morita R. Bruton's tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat Commun. 2015;6:7360.
Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49–61.
Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14:399–416.
Kokabee L, Wang X, Sevinsky CJ, Wang WL, Cheu L, Chittur SV, Karimipoor M, Tenniswood M, Conklin DS. Bruton's tyrosine kinase is a potential therapeutic target in prostate cancer. Cancer Biol Ther. 2015;16:1604–15.
Grassilli E, Pisano F, Cialdella A, Bonomo S, Missaglia C, Cerrito MG, Masiero L, Ianzano L, Giordano F, Cicirelli V, et al. A novel oncogenic BTK isoform is overexpressed in colon cancers and required for RAS-mediated transformation. Oncogene. 2016;35:4368–78.
Zucha MA, Wu AT, Lee WH, Wang LS, Lin WW, Yuan CC, Yeh CT. Bruton's tyrosine kinase (Btk) inhibitor ibrutinib suppresses stem-like traits in ovarian cancer. Oncotarget. 2015;6:13255–68.
Wei L, Su YK, Lin CM, Chao TY, Huang SP, Huynh TT, Jan HJ, Whang-Peng J, Chiou JF, Wu AT, Hsiao M. Preclinical investigation of ibrutinib, a Bruton's kinase tyrosine (Btk) inhibitor, in suppressing glioma tumorigenesis and stem cell phenotypes. Oncotarget. 2016;7:69961–75.
Bradshaw JM. The Src, Syk, and Tec family kinases: distinct types of molecular switches. Cell Signal. 2010;22:1175–84.
Hyvonen M, Saraste M. Structure of the PH domain and Btk motif from Bruton's tyrosine kinase: molecular explanations for X-linked agammaglobulinaemia. EMBO J. 1997;16:3396–404.
Rawlings DJ, Scharenberg AM, Park H, Wahl MI, Lin S, Kato RM, Fluckiger AC, Witte ON, Kinet JP. Activation of BTK by a phosphorylation mechanism initiated by SRC family kinases. Science. 1996;271:822–5.
Park H, Wahl MI, Afar DE, Turck CW, Rawlings DJ, Tam C, Scharenberg AM, Kinet JP, Witte ON. Regulation of Btk function by a major autophosphorylation site within the SH3 domain. Immunity. 1996;4:515–25.
Marcotte DJ, Liu YT, Arduini RM, Hession CA, Miatkowski K, Wildes CP, Cullen PF, Hong V, Hopkins BT, Mertsching E, et al. Structures of human Bruton's tyrosine kinase in active and inactive conformations suggest a mechanism of activation for TEC family kinases. Protein Sci. 2010;19:429–39.
Middendorp S, Dingjan GM, Maas A, Dahlenborg K, Hendriks RW. Function of Bruton's tyrosine kinase during B cell development is partially independent of its catalytic activity. J Immunol. 2003;171:5988–96.
Lam KP, Kuhn R, Rajewsky K. In Vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death. Cell. 1997;90:1073–83.
Anderson JS, Teutsch M, Dong Z, Wortis HH. An essential role for Bruton's [corrected] tyrosine kinase in the regulation of B-cell apoptosis. Proc Natl Acad Sci U S A. 1996;93:10966–71.
Solvason N, Wu WW, Kabra N, Lund-Johansen F, Roncarolo MG, Behrens TW, Grillot DA, Nunez G, Lees E, Howard M. Transgene expression of bcl-xL permits anti-immunoglobulin (Ig)-induced proliferation in xid B cells. J Exp Med. 1998;187:1081–91.
Brorson K, Brunswick M, Ezhevsky S, Wei DG, Berg R, Scott D, Stein KE. Xid affects events leading to B cell cycle entry. J Immunol. 1997;159:135–43.
Glassford J, Soeiro I, Skarell SM, Banerji L, Holman M, Klaus GG, Kadowaki T, Koyasu S, Lam EW. BCR targets cyclin D2 via Btk and the p85alpha subunit of PI3-K to induce cell cycle progression in primary mouse B cells. Oncogene. 2003;22:2248–59.
Spaargaren M, Beuling EA, Rurup ML, Meijer HP, Klok MD, Middendorp S, Hendriks RW, Pals ST. The B cell antigen receptor controls integrin activity through Btk and PLCgamma2. J Exp Med. 2003;198:1539–50.
Rolli V, Gallwitz M, Wossning T, Flemming A, Schamel WW, Zurn C, Reth M. Amplification of B cell antigen receptor signaling by a Syk/ITAM positive feedback loop. Mol Cell. 2002;10:1057–69.
O'Rourke LM, Tooze R, Turner M, Sandoval DM, Carter RH, Tybulewicz VL, Fearon DT. CD19 as a membrane-anchored adaptor protein of B lymphocytes: costimulation of lipid and protein kinases by recruitment of Vav. Immunity. 1998;8:635–45.
Okada T, Maeda A, Iwamatsu A, Gotoh K, Kurosaki T. BCAP: the tyrosine kinase substrate that connects B cell receptor to phosphoinositide 3-kinase activation. Immunity. 2000;13:817–27.
Inabe K, Ishiai M, Scharenberg AM, Freshney N, Downward J, Kurosaki T. Vav3 modulates B cell receptor responses by regulating phosphoinositide 3-kinase activation. J Exp Med. 2002;195:189–200.
Saito K, Scharenberg AM, Kinet JP. Interaction between the Btk PH domain and phosphatidylinositol-3,4,5-trisphosphate directly regulates Btk. J Biol Chem. 2001;276:16201–6.
Engels N, Konig LM, Heemann C, Lutz J, Tsubata T, Griep S, Schrader V, Wienands J. Recruitment of the cytoplasmic adaptor Grb2 to surface IgG and IgE provides antigen receptor-intrinsic costimulation to class-switched B cells. Nat Immunol. 2009;10:1018–25.
Kuhn J, Wong LE, Pirkuliyeva S, Schulz K, Schwiegk C, Funfgeld KG, Keppler S, Batista FD, Urlaub H, Habeck M, et al. The adaptor protein CIN85 assembles intracellular signaling clusters for B cell activation. Sci Signal. 2016;9:ra66.
Guo B, Kato RM, Garcia-Lloret M, Wahl MI, Rawlings DJ. Engagement of the human pre-B cell receptor generates a lipid raft-dependent calcium signaling complex. Immunity. 2000;13:243–53.
Su YW, Zhang Y, Schweikert J, Koretzky GA, Reth M, Wienands J. Interaction of SLP adaptors with the SH2 domain of Tec family kinases. Eur J Immunol. 1999;29:3702–11.
Chiu CW, Dalton M, Ishiai M, Kurosaki T, Chan AC. BLNK: molecular scaffolding through 'cis'-mediated organization of signaling proteins. EMBO J. 2002;21:6461–72.
Weber M, Treanor B, Depoil D, Shinohara H, Harwood NE, Hikida M, Kurosaki T, Batista FD. Phospholipase C-gamma2 and Vav cooperate within signaling microclusters to propagate B cell spreading in response to membrane-bound antigen. J Exp Med. 2008;205:853–68.
Kim YJ, Sekiya F, Poulin B, Bae YS, Rhee SG. Mechanism of B-cell receptor-induced phosphorylation and activation of phospholipase C-gamma2. Mol Cell Biol. 2004;24:9986–99.
Hashimoto A, Okada H, Jiang A, Kurosaki M, Greenberg S, Clark EA, Kurosaki T. Involvement of guanosine triphosphatases and phospholipase C-gamma2 in extracellular signal-regulated kinase, c-Jun NH2-terminal kinase, and p38 mitogen-activated protein kinase activation by the B cell antigen receptor. J Exp Med. 1998;188:1287–95.
Petro JB, Rahman SM, Ballard DW, Khan WN. Bruton’s tyrosine kinase is required for activation of IkappaB kinase and nuclear factor kappaB in response to B cell receptor engagement. J Exp Med. 2000;191:1745–54.
Bajpai UD, Zhang K, Teutsch M, Sen R, Wortis HH. Bruton’s tyrosine kinase links the B cell receptor to nuclear factor kappaB activation. J Exp Med. 2000;191:1735–44.
Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell. 2017;169:381–405.
Craxton A, Jiang A, Kurosaki T, Clark EA. Syk and Bruton's tyrosine kinase are required for B cell antigen receptor-mediated activation of the kinase Akt. J Biol Chem. 1999;274:30644–50.
Ellmeier W, Jung S, Sunshine MJ, Hatam F, Xu Y, Baltimore D, Mano H, Littman DR. Severe B cell deficiency in mice lacking the tec kinase family members Tec and Btk. J Exp Med. 2000;192:1611–24.
de Bruijn MJ, Rip J, van der Ploeg EK, van Greuningen LW, Ta VT, Kil LP, Langerak AW, Rimmelzwaan GF, Ellmeier W, Hendriks RW, Corneth OB. Distinct and overlapping functions of TEC kinase and BTK in B cell receptor signaling. J Immunol. 2017;198:3058–68.
Engels N, Konig LM, Schulze W, Radtke D, Vanshylla K, Lutz J, Winkler TH, Nitschke L, Wienands J. The immunoglobulin tail tyrosine motif upgrades memory-type BCRs by incorporating a Grb2-Btk signalling module. Nat Commun. 2014;5:5456.
Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. 2008;8:34–47.
Amigorena S, Bonnerot C, Drake JR, Choquet D, Hunziker W, Guillet JG, Webster P, Sautes C, Mellman I, Fridman WH. Cytoplasmic domain heterogeneity and functions of IgG fc receptors in B lymphocytes. Science. 1992;256:1808–12.
Muta T, Kurosaki T, Misulovin Z, Sanchez M, Nussenzweig MC, Ravetch JV. A 13-amino-acid motif in the cytoplasmic domain of fc gamma RIIB modulates B-cell receptor signalling. Nature. 1994;369:340.
Bolland S, Ravetch JV. Inhibitory pathways triggered by ITIM-containing receptors. Adv Immunol. 1999;72:149–77.
Ono M, Bolland S, Tempst P, Ravetch JV. Role of the inositol phosphatase SHIP in negative regulation of the immune system by the receptor fc(gamma)RIIB. Nature. 1996;383:263–6.
Ono M, Okada H, Bolland S, Yanagi S, Kurosaki T, Ravetch JV. Deletion of SHIP or SHP-1 reveals two distinct pathways for inhibitory signaling. Cell. 1997;90:293–301.
Liu W, Quinto I, Chen X, Palmieri C, Rabin RL, Schwartz OM, Nelson DL, Scala G. Direct inhibition of Bruton's tyrosine kinase by IBtk, a Btk-binding protein. Nat Immunol. 2001;2:939–46.
Kang SW, Wahl MI, Chu J, Kitaura J, Kawakami Y, Kato RM, Tabuchi R, Tarakhovsky A, Kawakami T, Turck CW, et al. PKCbeta modulates antigen receptor signaling via regulation of Btk membrane localization. EMBO J. 2001;20:5692–702.
Belver L, de Yebenes VG, Ramiro AR. MicroRNAs prevent the generation of autoreactive antibodies. Immunity. 2010;33:713–22.
Bottoni A, Rizzotto L, Lai TH, Liu C, Smith LL, Mantel R, Reiff S, El-Gamal D, Larkin K, Johnson AJ, et al. Targeting BTK through microRNA in chronic lymphocytic leukemia. Blood. 2016;128:3101–12.
Yu L, Mohamed AJ, Simonson OE, Vargas L, Blomberg KE, Bjorkstrand B, Arteaga HJ, Nore BF, Smith CI. Proteasome-dependent autoregulation of Bruton tyrosine kinase (Btk) promoter via NF-kappaB. Blood. 2008;111:4617–26.
Ritter SL, Hall RA. Fine-tuning of GPCR activity by receptor-interacting proteins. Nat Rev Mol Cell Biol. 2009;10:819–30.
Okada T, Ngo VN, Ekland EH, Forster R, Lipp M, Littman DR, Cyster JG. Chemokine requirements for B cell entry to lymph nodes and Peyer's patches. J Exp Med. 2002;196:65–75.
Servant G, Weiner OD, Herzmark P, Balla T, Sedat JW, Bourne HR. Polarization of chemoattractant receptor signaling during neutrophil chemotaxis. Science. 2000;287:1037–40.
Lowry WE, Huang XY, Protein G. Beta gamma subunits act on the catalytic domain to stimulate Bruton's agammaglobulinemia tyrosine kinase. J Biol Chem. 2002;277:1488–92.
Tsukada S, Simon MI, Witte ON, Katz A. Binding of beta gamma subunits of heterotrimeric G proteins to the PH domain of Bruton tyrosine kinase. Proc Natl Acad Sci U S A. 1994;91:11256–60.
Bence K, Ma W, Kozasa T, Huang XY. Direct stimulation of Bruton's tyrosine kinase by G(q)-protein alpha-subunit. Nature. 1997;389:296–9.
de Gorter DJ, Beuling EA, Kersseboom R, Middendorp S, van Gils JM, Hendriks RW, Pals ST, Spaargaren M. Bruton's tyrosine kinase and phospholipase Cgamma2 mediate chemokine-controlled B cell migration and homing. Immunity. 2007;26:93–104.
Rawlings DJ, Schwartz MA, Jackson SW, Meyer-Bahlburg A. Integration of B cell responses through toll-like receptors and antigen receptors. Nat Rev Immunol. 2012;12:282–94.
Jefferies CA, Doyle S, Brunner C, Dunne A, Brint E, Wietek C, Walch E, Wirth T, O'Neill LA. Bruton's tyrosine kinase is a toll/interleukin-1 receptor domain-binding protein that participates in nuclear factor kappaB activation by toll-like receptor 4. J Biol Chem. 2003;278:26258–64.
Liu X, Zhan Z, Li D, Xu L, Ma F, Zhang P, Yao H, Cao X. Intracellular MHC class II molecules promote TLR-triggered innate immune responses by maintaining activation of the kinase Btk. Nat Immunol. 2011;12:416–24.
Gray P, Dunne A, Brikos C, Jefferies CA, Doyle SL, O'Neill LA. MyD88 adapter-like (mal) is phosphorylated by Bruton's tyrosine kinase during TLR2 and TLR4 signal transduction. J Biol Chem. 2006;281:10489–95.
Bournazos S, Wang TT, Ravetch JV. The role and function of Fcgamma receptors on myeloid cells. Microbiol Spectr. 2016;4
Fearon ER, Winkelstein JA, Civin CI, Pardoll DM, Vogelstein B. Carrier detection in X-linked agammaglobulinemia by analysis of X-chromosome inactivation. N Engl J Med. 1987;316:427–31.
Puck JM, Nussbaum RL, Conley ME. Carrier detection in X-linked severe combined immunodeficiency based on patterns of X chromosome inactivation. J Clin Invest. 1987;79:1395–400.
Melchers F, ten Boekel E, Seidl T, Kong XC, Yamagami T, Onishi K, Shimizu T, Rolink AG, Andersson J. Repertoire selection by pre-B-cell receptors and B-cell receptors, and genetic control of B-cell development from immature to mature B cells. Immunol Rev. 2000;175:33–46.
Hendriks RW, Middendorp S. The pre-BCR checkpoint as a cell-autonomous proliferation switch. Trends Immunol. 2004;25:249–56.
Hendriks RW, Bredius RG, Pike-Overzet K, Staal FJ. Biology and novel treatment options for XLA, the most common monogenetic immunodeficiency in man. Expert Opin Ther Targets. 2011;15:1003–21.
Herzog S, Reth M, Jumaa H. Regulation of B-cell proliferation and differentiation by pre-B-cell receptor signalling. Nat Rev Immunol. 2009;9:195–205.
Ohnishi K, Melchers F. The nonimmunoglobulin portion of lambda5 mediates cell-autonomous pre-B cell receptor signaling. Nat Immunol. 2003;4:849–56.
Ubelhart R, Bach MP, Eschbach C, Wossning T, Reth M, Jumaa H. N-linked glycosylation selectively regulates autonomous precursor BCR function. Nat Immunol. 2010;11:759–65.
ten Boekel E, Yamagami T, Andersson J, Rolink AG, Melchers F. The formation and selection of cells expressing preB cell receptors and B cell receptors. Curr Top Microbiol Immunol. 1999;246:3–9. discussion 9-10
Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science. 2003;301:1374–7.
Jumaa H, Mitterer M, Reth M, Nielsen PJ. The absence of SLP65 and Btk blocks B cell development at the preB cell receptor-positive stage. Eur J Immunol. 2001;31:2164–9.
Jumaa H, Bossaller L, Portugal K, Storch B, Lotz M, Flemming A, Schrappe M, Postila V, Riikonen P, Pelkonen J, et al. Deficiency of the adaptor SLP-65 in pre-B-cell acute lymphoblastic leukaemia. Nature. 2003;423:452–6.
Middendorp S, Zijlstra AJ, Kersseboom R, Dingjan GM, Jumaa H, Hendriks RW. Tumor suppressor function of Bruton tyrosine kinase is independent of its catalytic activity. Blood. 2005;105:259–65.
Kersseboom R, Middendorp S, Dingjan GM, Dahlenborg K, Reth M, Jumaa H, Hendriks RW. Bruton's tyrosine kinase cooperates with the B cell linker protein SLP-65 as a tumor suppressor in pre-B cells. J Exp Med. 2003;198:91–8.
Thompson EC, Cobb BS, Sabbattini P, Meixlsperger S, Parelho V, Liberg D, Taylor B, Dillon N, Georgopoulos K, Jumaa H, et al. Ikaros DNA-binding proteins as integral components of B cell developmental-stage-specific regulatory circuits. Immunity. 2007;26:335–44.
Stadhouders R, de Bruijn MJ, Rother MB, Yuvaraj S, Ribeiro de Almeida C, Kolovos P, Van Zelm MC, van Ijcken W, Grosveld F, Soler E, Hendriks RW. Pre-B cell receptor signaling induces immunoglobulin kappa locus accessibility by functional redistribution of enhancer-mediated chromatin interactions. PLoS Biol. 2014;12:e1001791.
Dingjan GM, Middendorp S, Dahlenborg K, Maas A, Grosveld F, Hendriks RW. Bruton's tyrosine kinase regulates the activation of gene rearrangements at the lambda light chain locus in precursor B cells in the mouse. J Exp Med. 2001;193:1169–78.
Gay D, Saunders T, Camper S, Weigert M. Receptor editing: an approach by autoreactive B cells to escape tolerance. J Exp Med. 1993;177:999–1008.
Nemazee D, Weigert M. Revising B cell receptors. J Exp Med. 2000;191:1813–7.
Rolink A, Grawunder U, Haasner D, Strasser A, Melchers F. Immature surface Ig+ B cells can continue to rearrange kappa and lambda L chain gene loci. J Exp Med. 1993;178:1263–70.
Petro JB, Gerstein RM, Lowe J, Carter RS, Shinners N, Khan WN. Transitional type 1 and 2 B lymphocyte subsets are differentially responsive to antigen receptor signaling. J Biol Chem. 2002;277:48009–19.
Levine MH, Haberman AM, Sant'Angelo DB, Hannum LG, Cancro MP, Janeway CA Jr, Shlomchik MJ. A B-cell receptor-specific selection step governs immature to mature B cell differentiation. Proc Natl Acad Sci U S A. 2000;97:2743–8.
Hammad H, Vanderkerken M, Pouliot P, Deswarte K, Toussaint W, Vergote K, Vandersarren L, Janssens S, Ramou I, Savvides SN, et al. Transitional B cells commit to marginal zone B cell fate by Taok3-mediated surface expression of ADAM10. Nat Immunol. 2017;18:313–20.
Gibb DR, El Shikh M, Kang DJ, Rowe WJ, El Sayed R, Cichy J, Yagita H, Tew JG, Dempsey PJ, Crawford HC, Conrad DH. ADAM10 is essential for Notch2-dependent marginal zone B cell development and CD23 cleavage in vivo. J Exp Med. 2010;207:623–35.
Martin F, Kearney JF. Positive selection from newly formed to marginal zone B cells depends on the rate of clonal production, CD19, and btk. Immunity. 2000;12:39–49.
Corneth OB, Klein Wolterink RG, Hendriks RW. BTK signaling in B cell differentiation and autoimmunity. Curr Top Microbiol Immunol. 2016;393:67–105.
Victora GD. SnapShot: the germinal center reaction. Cell. 2014;159:700–0. e701
Oropallo MA, Cerutti A. Germinal center reaction: antigen affinity and presentation explain it all. Trends Immunol. 2014;35:287–9.
Dingjan GM, Maas A, Nawijn MC, Smit L, Voerman JS, Grosveld F, Hendriks RW, Severe B. Cell deficiency and disrupted splenic architecture in transgenic mice expressing the E41K mutated form of Bruton's tyrosine kinase. EMBO J. 1998;17:5309–20.
Kersseboom R, Kil L, Flierman R, van der Zee M, Dingjan GM, Middendorp S, Maas A, Hendriks RW. Constitutive activation of Bruton's tyrosine kinase induces the formation of autoreactive IgM plasma cells. Eur J Immunol. 2010;40:2643–54.
Kil LP, de Bruijn MJ, van Nimwegen M, Corneth OB, van Hamburg JP, Dingjan GM, Thaiss F, Rimmelzwaan GF, Elewaut D, Delsing D, et al. Btk levels set the threshold for B-cell activation and negative selection of autoreactive B cells in mice. Blood. 2012;119:3744–56.
Corneth OB, de Bruijn MJ, Rip J, Asmawidjaja PS, Kil LP, Hendriks RW. Enhanced expression of Bruton's tyrosine kinase in B cells drives systemic autoimmunity by disrupting T cell homeostasis. J Immunol. 2016;197:58–67.
Ammann EM, Shanafelt TD, Wright KB, McDowell BD, Link BK, Chrischilles EA. Updating survival estimates in patients with chronic lymphocytic leukemia or small lymphocytic lymphoma (CLL/SLL) based on treatment-free interval length. Leuk Lymphoma. 2017:1–7.
Kil LP, Yuvaraj S, Langerak AW, Hendriks RW. The role of B cell receptor stimulation in CLL pathogenesis. Curr Pharm Des. 2012;18:3335–55.
Zenz T, Eichhorst B, Busch R, Denzel T, Habe S, Winkler D, Buhler A, Edelmann J, Bergmann M, Hopfinger G, et al. TP53 mutation and survival in chronic lymphocytic leukemia. J Clin Oncol. 2010;28:4473–9.
Gonzalez D, Martinez P, Wade R, Hockley S, Oscier D, Matutes E, Dearden CE, Richards SM, Catovsky D, Morgan GJ. Mutational status of the TP53 gene as a predictor of response and survival in patients with chronic lymphocytic leukemia: results from the LRF CLL4 trial. J Clin Oncol. 2011;29:2223–9.
Robak P, Robak T. Novel synthetic drugs currently in clinical development for chronic lymphocytic leukemia. Expert Opin Investig Drugs. 2017;26:1249–65.
Malcikova J, Smardova J, Rocnova L, Tichy B, Kuglik P, Vranova V, Cejkova S, Svitakova M, Skuhrova Francova H, Brychtova Y, et al. Monoallelic and biallelic inactivation of TP53 gene in chronic lymphocytic leukemia: selection, impact on survival, and response to DNA damage. Blood. 2009;114:5307–14.
Zenz T, Krober A, Scherer K, Habe S, Buhler A, Benner A, Denzel T, Winkler D, Edelmann J, Schwanen C, et al. Monoallelic TP53 inactivation is associated with poor prognosis in chronic lymphocytic leukemia: results from a detailed genetic characterization with long-term follow-up. Blood. 2008;112:3322–9.
Chiorazzi N, Ferrarini M. Cellular origin(s) of chronic lymphocytic leukemia: cautionary notes and additional considerations and possibilities. Blood. 2011;117:1781–91.
Klein U, Tu Y, Stolovitzky GA, Mattioli M, Cattoretti G, Husson H, Freedman A, Inghirami G, Cro L, Baldini L, et al. Gene expression profiling of B cell chronic lymphocytic leukemia reveals a homogeneous phenotype related to memory B cells. J Exp Med. 2001;194:1625–38.
Seifert M, Sellmann L, Bloehdorn J, Wein F, Stilgenbauer S, Durig J, Kuppers R. Cellular origin and pathophysiology of chronic lymphocytic leukemia. J Exp Med. 2012;209:2183–98.
Griffin DO, Holodick NE, Rothstein TL. Human B1 cells in umbilical cord and adult peripheral blood express the novel phenotype CD20+ CD27+ CD43+ CD70. J Exp Med. 2011;208:67–80.
DiLillo DJ, Weinberg JB, Yoshizaki A, Horikawa M, Bryant JM, Iwata Y, Matsushita T, Matta KM, Chen Y, Venturi GM, et al. Chronic lymphocytic leukemia and regulatory B cells share IL-10 competence and immunosuppressive function. Leukemia. 2013;27:170–82.
Muggen AF, Singh SP, Hendriks RW, Langerak AW. Targeting signaling pathways in chronic lymphocytic leukemia. Curr Cancer Drug Targets. 2016;16:669–88.
Damle RN, Wasil T, Fais F, Ghiotto F, Valetto A, Allen SL, Buchbinder A, Budman D, Dittmar K, Kolitz J, et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood. 1999;94:1840–7.
Agathangelidis A, Darzentas N, Hadzidimitriou A, Brochet X, Murray F, Yan XJ, Davis Z, van Gastel-Mol EJ, Tresoldi C, Chu CC, et al. Stereotyped B-cell receptors in one-third of chronic lymphocytic leukemia: a molecular classification with implications for targeted therapies. Blood. 2012;119:4467–75.
Murray F, Darzentas N, Hadzidimitriou A, Tobin G, Boudjogra M, Scielzo C, Laoutaris N, Karlsson K, Baran-Marzsak F, Tsaftaris A, et al. Stereotyped patterns of somatic hypermutation in subsets of patients with chronic lymphocytic leukemia: implications for the role of antigen selection in leukemogenesis. Blood. 2008;111:1524–33.
Hayakawa K, Formica AM, Colombo MJ, Shinton SA, Brill-Dashoff J, Morse Iii HC, Li YS, Hardy RR. Loss of a chromosomal region with synteny to human 13q14 occurs in mouse chronic lymphocytic leukemia that originates from early-generated B-1 B cells. Leukemia. 2016;30:1510–9.
Chen SS, Batliwalla F, Holodick NE, Yan XJ, Yancopoulos S, Croce CM, Rothstein TL, Chiorazzi N. Autoantigen can promote progression to a more aggressive TCL1 leukemia by selecting variants with enhanced B-cell receptor signaling. Proc Natl Acad Sci U S A. 2013;110:E1500–7.
Singh SP, Pillai SY, de Bruijn MJW, Stadhouders R, Corneth OBJ, van den Ham HJ, Muggen A, van IW SE, Kuil A, et al. Cell lines generated from a chronic lymphocytic leukemia mouse model exhibit constitutive Btk and Akt signaling. Oncotarget. 2017;8:71981–95.
Messmer BT, Albesiano E, Efremov DG, Ghiotto F, Allen SL, Kolitz J, Foa R, Damle RN, Fais F, Messmer D, et al. Multiple distinct sets of stereotyped antigen receptors indicate a role for antigen in promoting chronic lymphocytic leukemia. J Exp Med. 2004;200:519–25.
Herve M, Xu K, Ng YS, Wardemann H, Albesiano E, Messmer BT, Chiorazzi N, Meffre E. Unmutated and mutated chronic lymphocytic leukemias derive from self-reactive B cell precursors despite expressing different antibody reactivity. J Clin Invest. 2005;115:1636–43.
Lanemo Myhrinder A, Hellqvist E, Sidorova E, Soderberg A, Baxendale H, Dahle C, Willander K, Tobin G, Backman E, Soderberg O, et al. A new perspective: molecular motifs on oxidized LDL, apoptotic cells, and bacteria are targets for chronic lymphocytic leukemia antibodies. Blood. 2008;111:3838–48.
Hoogeboom R, van Kessel KP, Hochstenbach F, Wormhoudt TA, Reinten RJ, Wagner K, Kater AP, Guikema JE, Bende RJ, van Noesel CJ. A mutated B cell chronic lymphocytic leukemia subset that recognizes and responds to fungi. J Exp Med. 2013;210:59–70.
Jimenez de Oya N, De Giovanni M, Fioravanti J, Ubelhart R, Di Lucia P, Fiocchi A, Iacovelli S, Efremov DG, Caligaris-Cappio F, Jumaa H, et al. Pathogen-specific B-cell receptors drive chronic lymphocytic leukemia by light-chain-dependent cross-reaction with autoantigens. EMBO Mol Med. 2017;9:1482–90.
Duhren-von Minden M, Ubelhart R, Schneider D, Wossning T, Bach MP, Buchner M, Hofmann D, Surova E, Follo M, Kohler F, et al. Chronic lymphocytic leukaemia is driven by antigen-independent cell-autonomous signalling. Nature. 2012;489:309–12.
Minici C, Gounari M, Ubelhart R, Scarfo L, Duhren-von Minden M, Schneider D, Tasdogan A, Alkhatib A, Agathangelidis A, Ntoufa S, et al. Distinct homotypic B-cell receptor interactions shape the outcome of chronic lymphocytic leukaemia. Nat Commun. 2017;8:15746.
Herman SE, Gordon AL, Hertlein E, Ramanunni A, Zhang X, Jaglowski S, Flynn J, Jones J, Blum KA, Buggy JJ, et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood. 2011;117:6287–96.
Ponader S, Chen SS, Buggy JJ, Balakrishnan K, Gandhi V, Wierda WG, Keating MJ, O'Brien S, Chiorazzi N, Burger JA. The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood. 2012;119:1182–9.
Kil LP, de Bruijn MJ, van Hulst JA, Langerak AW, Yuvaraj S, Hendriks RW. Bruton's tyrosine kinase mediated signaling enhances leukemogenesis in a mouse model for chronic lymphocytic leukemia. Am J Blood Res. 2013;3:71–83.
Ramsay AD, Rodriguez-Justo M. Chronic lymphocytic leukaemia--the role of the microenvironment pathogenesis and therapy. Br J Haematol. 2013;162:15–24.
Burger JA, Gribben JG. The microenvironment in chronic lymphocytic leukemia (CLL) and other B cell malignancies: insight into disease biology and new targeted therapies. Semin Cancer Biol. 2014;24:71–81.
de Rooij MF, Kuil A, Geest CR, Eldering E, Chang BY, Buggy JJ, Pals ST, Spaargaren M. The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood. 2012;119:2590–4.
Cheah CY, Seymour JF, Wang ML. Mantle cell lymphoma. J Clin Oncol. 2016;34:1256–69.
Bertoni F, Rinaldi A, Zucca E, Cavalli F. Update on the molecular biology of mantle cell lymphoma. Hematol Oncol. 2006;24:22–7.
Meggendorfer M, Kern W, Haferlach C, Haferlach T, Schnittger S. SOX11 overexpression is a specific marker for mantle cell lymphoma and correlates with t(11;14) translocation, CCND1 expression and an adverse prognosis. Leukemia. 2013;27:2388–91.
Navarro A, Clot G, Royo C, Jares P, Hadzidimitriou A, Agathangelidis A, Bikos V, Darzentas N, Papadaki T, Salaverria I, et al. Molecular subsets of mantle cell lymphoma defined by the IGHV mutational status and SOX11 expression have distinct biologic and clinical features. Cancer Res. 2012;72:5307–16.
Cinar M, Hamedani F, Mo Z, Cinar B, Amin HM, Alkan S. Bruton tyrosine kinase is commonly overexpressed in mantle cell lymphoma and its attenuation by Ibrutinib induces apoptosis. Leuk Res. 2013;37:1271–7.
Pighi C, Gu TL, Dalai I, Barbi S, Parolini C, Bertolaso A, Pedron S, Parisi A, Ren J, Cecconi D, et al. Phospho-proteomic analysis of mantle cell lymphoma cells suggests a pro-survival role of B-cell receptor signaling. Cell Oncol (Dordr). 2011;34:141–53.
Boyd RS, Jukes-Jones R, Walewska R, Brown D, Dyer MJ, Cain K. Protein profiling of plasma membranes defines aberrant signaling pathways in mantle cell lymphoma. Mol Cell Proteomics. 2009;8:1501–15.
Chang BY, Francesco M, De Rooij MF, Magadala P, Steggerda SM, Huang MM, Kuil A, Herman SE, Chang S, Pals ST, et al. Egress of CD19(+)CD5(+) cells into peripheral blood following treatment with the Bruton tyrosine kinase inhibitor ibrutinib in mantle cell lymphoma patients. Blood. 2013;122:2412–24.
Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao Y, Sheehy P, Manning RJ, Patterson CJ, Tripsas C, et al. MYD88 L265P somatic mutation in Waldenstrom's macroglobulinemia. N Engl J Med. 2012;367:826–33.
Pasqualucci L, Trifonov V, Fabbri G, Ma J, Rossi D, Chiarenza A, Wells VA, Grunn A, Messina M, Elliot O, et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet. 2011;43:830–7.
Puente XS, Pinyol M, Quesada V, Conde L, Ordonez GR, Villamor N, Escaramis G, Jares P, Bea S, Gonzalez-Diaz M, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475:101–5.
Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, Kohlhammer H, Xu W, Yang Y, Zhao H, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;470:115–9.
Montesinos-Rongen M, Godlewska E, Brunn A, Wiestler OD, Siebert R, Deckert M. Activating L265P mutations of the MYD88 gene are common in primary central nervous system lymphoma. Acta Neuropathol. 2011;122:791–2.
Yang G, Zhou Y, Liu X, Xu L, Cao Y, Manning RJ, Patterson CJ, Buhrlage SJ, Gray N, Tai YT, et al. A mutation in MYD88 (L265P) supports the survival of lymphoplasmacytic cells by activation of Bruton tyrosine kinase in Waldenstrom macroglobulinemia. Blood. 2013;122:1222–32.
Hunter ZR, Xu L, Yang G, Zhou Y, Liu X, Cao Y, Manning RJ, Tripsas C, Patterson CJ, Sheehy P, Treon SP. The genomic landscape of Waldenstrom macroglobulinemia is characterized by highly recurring MYD88 and WHIM-like CXCR4 mutations, and small somatic deletions associated with B-cell lymphomagenesis. Blood. 2014;123:1637–46.
Ngo HT, Leleu X, Lee J, Jia X, Melhem M, Runnels J, Moreau AS, Burwick N, Azab AK, Roccaro A, et al. SDF-1/CXCR4 and VLA-4 interaction regulates homing in Waldenstrom macroglobulinemia. Blood. 2008;112:150–8.
Iqbal J, Shen Y, Huang X, Liu Y, Wake L, Liu C, Deffenbacher K, Lachel CM, Wang C, Rohr J, et al. Global microRNA expression profiling uncovers molecular markers for classification and prognosis in aggressive B-cell lymphoma. Blood. 2015;125:1137–45.
Dunleavy K, Wilson WH. Primary mediastinal B-cell lymphoma and mediastinal gray zone lymphoma: do they require a unique therapeutic approach? Blood. 2015;125:33–9.
Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–11.
Lenz G, Nagel I, Siebert R, Roschke AV, Sanger W, Wright GW, Dave SS, Tan B, Zhao H, Rosenwald A, et al. Aberrant immunoglobulin class switch recombination and switch translocations in activated B cell-like diffuse large B cell lymphoma. J Exp Med. 2007;204:633–43.
Roschewski M, Staudt LM, Wilson WH. Diffuse large B-cell lymphoma-treatment approaches in the molecular era. Nat Rev Clin Oncol. 2014;11:12–23.
Davis RE, Brown KD, Siebenlist U, Staudt LM. Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J Exp Med. 2001;194:1861–74.
Ngo VN, Davis RE, Lamy L, Yu X, Zhao H, Lenz G, Lam LT, Dave S, Yang L, Powell J, Staudt LM. A loss-of-function RNA interference screen for molecular targets in cancer. Nature. 2006;441:106–10.
Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, Bertoni F, Ponzoni M, Scandurra M, Califano A, et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature. 2009;459:717–21.
Lenz G, Davis RE, Ngo VN, Lam L, George TC, Wright GW, Dave SS, Zhao H, Xu W, Rosenwald A, et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science. 2008;319:1676–9.
Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, Kohlhammer H, Lamy L, Zhao H, Yang Y, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463:88–92.
Chen L, Monti S, Juszczynski P, Ouyang J, Chapuy B, Neuberg D, Doench JG, Bogusz AM, Habermann TM, Dogan A, et al. SYK inhibition modulates distinct PI3K/AKT- dependent survival pathways and cholesterol biosynthesis in diffuse large B cell lymphomas. Cancer Cell. 2013;23:826–38.
Havranek O, Xu J, Kohrer S, Wang Z, Becker L, Comer JM, Henderson J, Ma W, Man Chun Ma J, Westin JR, et al. Tonic B-cell receptor signaling in diffuse large B-cell lymphoma. Blood. 2017;130:995–1006.
Pfeifer M, Grau M, Lenze D, Wenzel SS, Wolf A, Wollert-Wulf B, Dietze K, Nogai H, Storek B, Madle H, et al. PTEN loss defines a PI3K/AKT pathway-dependent germinal center subtype of diffuse large B-cell lymphoma. Proc Natl Acad Sci U S A. 2013;110:12420–5.
Lenz G, Wright GW, Emre NC, Kohlhammer H, Dave SS, Davis RE, Carty S, Lam LT, Shaffer AL, Xiao W, et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci U S A. 2008;105:13520–5.
Grommes C, Pastore A, Palaskas N, Tang SS, Campos C, Schartz D, Codega P, Nichol D, Clark O, Hsieh WY, et al. Ibrutinib unmasks critical role of Bruton tyrosine kinase in primary CNS lymphoma. Cancer Discov. 2017;7:1018–29.
Krysiak K, Gomez F, White BS, Matlock M, Miller CA, Trani L, Fronick CC, Fulton RS, Kreisel F, Cashen AF, et al. Recurrent somatic mutations affecting B-cell receptor signaling pathway genes in follicular lymphoma. Blood. 2017;129:473–83.
Yang Y, Shi J, Gu Z, Salama ME, Das S, Wendlandt E, Xu H, Huang J, Tao Y, Hao M, et al. Bruton tyrosine kinase is a therapeutic target in stem-like cells from multiple myeloma. Cancer Res. 2015;75:594–604.
Gu C, Peng H, Lu Y, Yang H, Tian Z, Yin G, Zhang W, Lu S, Zhang Y, Yang Y. BTK suppresses myeloma cellular senescence through activating AKT/P27/Rb signaling. Oncotarget. 2017;8:56858–67.
Tai YT, Chang BY, Kong SY, Fulciniti M, Yang G, Calle Y, Hu Y, Lin J, Zhao JJ, Cagnetta A, et al. Bruton tyrosine kinase inhibition is a novel therapeutic strategy targeting tumor in the bone marrow microenvironment in multiple myeloma. Blood. 2012;120:1877–87.
Yan Q, Huang Y, Watkins AJ, Kocialkowski S, Zeng N, Hamoudi RA, Isaacson PG, de Leval L, Wotherspoon A, Du MQ. BCR and TLR signaling pathways are recurrently targeted by genetic changes in splenic marginal zone lymphomas. Haematologica. 2012;97:595–8.
Clipson A, Wang M, de Leval L, Ashton-Key M, Wotherspoon A, Vassiliou G, Bolli N, Grove C, Moody S, Escudero-Ibarz L, et al. KLF2 mutation is the most frequent somatic change in splenic marginal zone lymphoma and identifies a subset with distinct genotype. Leukemia. 2015;29:1177–85.
Pan Z, Scheerens H, Li SJ, Schultz BE, Sprengeler PA, Burrill LC, Mendonca RV, Sweeney MD, Scott KC, Grothaus PG, et al. Discovery of selective irreversible inhibitors for Bruton's tyrosine kinase. ChemMedChem. 2007;2:58–61.
Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, Li S, Pan Z, Thamm DH, Miller RA, Buggy JJ. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci U S A. 2010;107:13075–80.
Advani RH, Buggy JJ, Sharman JP, Smith SM, Boyd TE, Grant B, Kolibaba KS, Furman RR, Rodriguez S, Chang BY, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol. 2013;31:88–94.
Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS, Jurczak W, Advani RH, Romaguera JE, Williams ME, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369:507–16.
Treon SP, Tripsas CK, Meid K, Warren D, Varma G, Green R, Argyropoulos KV, Yang G, Cao Y, Xu L, et al. Ibrutinib in previously treated Waldenstrom's macroglobulinemia. N Engl J Med. 2015;372:1430–40.
Noy A, de Vos S, Thieblemont C, Martin P, Flowers CR, Morschhauser F, Collins GP, Ma S, Coleman M, Peles S, et al. Targeting Bruton tyrosine kinase with ibrutinib in relapsed/refractory marginal zone lymphoma. Blood. 2017;129:2224–32.
Burger JA, Wiestner A. Targeting B cell receptor signalling in cancer: preclinical and clinical advances. Nat Rev Cancer. 2018;
Mason JA, Hagel KR, Hawk MA, Schafer ZT. Metabolism during ECM detachment: Achilles heel of cancer cells? Trends Cancer. 2017;3:475–81.
Pals ST, Kersten MJ, Spaargaren M. Targeting cell adhesion and homing as strategy to cure Waldenstrom's macroglobulinemia. Best Pract Res Clin Haematol. 2016;29:161–8.
Woyach JA, Furman RR, Liu TM, Ozer HG, Zapatka M, Ruppert AS, Xue L, Li DH, Steggerda SM, Versele M, et al. Resistance mechanisms for the Bruton's tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370:2286–94.
Furman RR, Cheng S, Lu P, Setty M, Perez AR, Guo A, Racchumi J, Xu G, Wu H, Ma J, et al. Ibrutinib resistance in chronic lymphocytic leukemia. N Engl J Med. 2014;370:2352–4.
Kadri S, Lee J, Fitzpatrick C, Galanina N, Sukhanova M, Venkataraman G, Sharma S, Long B, Petras K, Theissen M, et al. Clonal evolution underlying leukemia progression and Richter transformation in patients with ibrutinib-relapsed CLL. Blood Adv. 2017;1:715–27.
Wilson WH, Young RM, Schmitz R, Yang Y, Pittaluga S, Wright G, Lih CJ, Williams PM, Shaffer AL, Gerecitano J, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med. 2015;21:922–6.
Wu C, de Miranda NF, Chen L, Wasik AM, Mansouri L, Jurczak W, Galazka K, Dlugosz-Danecka M, Machaczka M, Zhang H, et al. Genetic heterogeneity in primary and relapsed mantle cell lymphomas: impact of recurrent CARD11 mutations. Oncotarget. 2016;7:38180–90.
Maddocks KJ, Ruppert AS, Lozanski G, Heerema NA, Zhao W, Abruzzo L, Lozanski A, Davis M, Gordon A, Smith LL, et al. Etiology of Ibrutinib therapy discontinuation and outcomes in patients with chronic lymphocytic leukemia. JAMA Oncol. 2015;1:80–7.
Coutre SE, Furman RR, Flinn IW, Burger JA, Blum K, Sharman J, Jones J, Wierda W, Zhao W, Heerema NA, et al. Extended treatment with single-agent Ibrutinib at the 420 mg dose leads to durable responses in chronic lymphocytic leukemia/small lymphocytic lymphoma. Clin Cancer Res. 2017;23:1149–55.
Brown JR, Hillmen P, O'Brien S, Barrientos JC, Reddy NM, Coutre SE, Tam CS, Mulligan SP, Jaeger U, Barr PM, et al. Extended follow-up and impact of high-risk prognostic factors from the phase 3 RESONATE study in patients with previously treated CLL/SLL. Leukemia. 2017;
Mulligan SP, Ward CM, Whalley D, Hilmer SN. Atrial fibrillation, anticoagulant stroke prophylaxis and bleeding risk with ibrutinib therapy for chronic lymphocytic leukaemia and lymphoproliferative disorders. Br J Haematol. 2016;175:359–64.
Jones JA, Hillmen P, Coutre S, Tam C, Furman RR, Barr PM, Schuster SJ, Kipps TJ, Flinn IW, Jaeger U, et al. Use of anticoagulants and antiplatelet in patients with chronic lymphocytic leukaemia treated with single-agent ibrutinib. Br J Haematol. 2017;178:286–91.
Wiczer TE, Levine LB, Brumbaugh J, Coggins J, Zhao Q, Ruppert AS, Rogers K, McCoy A, Mousa L, Guha A, et al. Cumulative incidence, risk factors, and management of atrial fibrillation in patients receiving ibrutinib. Blood Adv. 2017;1:1739–48.
McMullen JR, Boey EJ, Ooi JY, Seymour JF, Keating MJ, Tam CS. Ibrutinib increases the risk of atrial fibrillation, potentially through inhibition of cardiac PI3K-Akt signaling. Blood. 2014;124:3829–30.
Byrd JC, Furman RR, Coutre SE, Burger JA, Blum KA, Coleman M, Wierda WG, Jones JA, Zhao W, Heerema NA, et al. Three-year follow-up of treatment-naïve and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood. 2015;125:2497–506.
O'Brien SM, Furman RR, Coutre SE, Flinn IW, Burger J, Blum K, Sharman J, Wierda WG, Jones J, Zhao W, et al. Five-year experience with single-agent ibrutinib in patients with previously untreated and relapsed/refractory chronic lymphocytic leukemia/small lymphocytic leukemia. Blood. 2016;128
Herman SEM, Montraveta A, Niemann CU, Mora-Jensen H, Gulrajani M, Krantz F, Mantel R, Smith LL, McClanahan F, Harrington BK, et al. The Bruton tyrosine kinase (BTK) inhibitor Acalabrutinib demonstrates potent on-target effects and efficacy in two mouse models of chronic lymphocytic leukemia. Clin Cancer Res. 2017;23:2831–41.
Quek LS, Bolen J, Watson SP. A role for Bruton's tyrosine kinase (Btk) in platelet activation by collagen. Curr Biol. 1998;8:1137–40.
Futatani T, Watanabe C, Baba Y, Tsukada S, Ochs HD. Bruton's tyrosine kinase is present in normal platelets and its absence identifies patients with X-linked agammaglobulinaemia and carrier females. Br J Haematol. 2001;114:141–9.
Bye AP, Unsworth AJ, Desborough MJ, Hildyard CAT, Appleby N, Bruce D, Kriek N, Nock SH, Sage T, Hughes CE, Gibbins JM. Severe platelet dysfunction in NHL patients receiving ibrutinib is absent in patients receiving acalabrutinib. Blood Adv. 2017;1:2610–23.
Tillman BF, Pauff JM, Satyanarayana G, Talbott M, Warner JL. Systematic review of infectious events with the BTK inhibitor ibrutinib in the treatment of haematologic malignancies. Eur J Haematol. 2017;
Long M, Beckwith K, Do P, Mundy BL, Gordon A, Lehman AM, Maddocks KJ, Cheney C, Jones JA, Flynn JM, et al. Ibrutinib treatment improves T cell number and function in CLL patients. J Clin Invest. 2017;127:3052–64.
Schutt SD, Fu J, Nguyen H, Bastian D, Heinrichs J, Wu Y, Liu C, DG MD, Pidala J, Yu XZ. Inhibition of BTK and ITK with Ibrutinib is effective in the prevention of chronic graft-versus-host disease in mice. PLoS One. 2015;10:e0137641.
Dubovsky JA, Flynn R, Du J, Harrington BK, Zhong Y, Kaffenberger B, Yang C, Towns WH, Lehman A, Johnson AJ, et al. Ibrutinib treatment ameliorates murine chronic graft-versus-host disease. J Clin Invest. 2014;124:4867–76.
Ryan CE, Sahaf B, Logan AC, O'Brien S, Byrd JC, Hillmen P, Brown JR, Dyer MJ, Mato AR, Keating MJ, et al. Ibrutinib efficacy and tolerability in patients with relapsed chronic lymphocytic leukemia following allogeneic HCT. Blood. 2016;128:2899–908.
Kohrt HE, Sagiv-Barfi I, Rafiq S, Herman SE, Butchar JP, Cheney C, Zhang X, Buggy JJ, Muthusamy N, Levy R, et al. Ibrutinib antagonizes rituximab-dependent NK cell-mediated cytotoxicity. Blood. 2014;123:1957–60.
Wu J, Liu C, Tsui ST, Liu D. Second-generation inhibitors of Bruton tyrosine kinase. J Hematol Oncol. 2016;9:80.
Harrington BK, Gardner HL, Izumi R, Hamdy A, Rothbaum W, Coombes KR, Covey T, Kaptein A, Gulrajani M, Van Lith B, et al. Preclinical evaluation of the novel BTK inhibitor Acalabrutinib in canine models of B-cell non-Hodgkin lymphoma. PLoS One. 2016;11:e0159607.
Wang M, Rule S, Zinzani PL, Goy A, Casasnovas O, Smith SD, Damaj G, Doorduijn J, Lamy T, Morschhauser F, et al. Acalabrutinib in relapsed or refractory mantle cell lymphoma (ACE-LY-004): a single-arm, multicentre, phase 2 trial. Lancet. 2017;
Acalabrutinib approved for MCL. Cancer Discov. 2018;8:OF6.
Thompson PA, Burger JA. Bruton's tyrosine kinase inhibitors: first and second generation agents for patients with chronic lymphocytic leukemia (CLL). Expert Opin Investig Drugs. 2018;27:31–42.
Walter HS, Rule SA, Dyer MJ, Karlin L, Jones C, Cazin B, Quittet P, Shah N, Hutchinson CV, Honda H, et al. A phase 1 clinical trial of the selective BTK inhibitor ONO/GS-4059 in relapsed and refractory mature B-cell malignancies. Blood. 2016;127:411–9.
Koprulu AD, Ellmeier W. The role of Tec family kinases in mononuclear phagocytes. Crit Rev Immunol. 2009;29:317–33.
Mirsafian H, Ripen AM, Leong WM, Chear CT, Bin Mohamad S, Merican AF. Transcriptome profiling of monocytes from XLA patients revealed the innate immune function dysregulation due to the BTK gene expression deficiency. Sci Rep. 2017;7:6836.
Doyle SL, Jefferies CA, Feighery C, O'Neill LA. Signaling by toll-like receptors 8 and 9 requires Bruton's tyrosine kinase. J Biol Chem. 2007;282:36953–60.
Sochorova K, Horvath R, Rozkova D, Litzman J, Bartunkova J, Sediva A, Spisek R. Impaired toll-like receptor 8-mediated IL-6 and TNF-alpha production in antigen-presenting cells from patients with X-linked agammaglobulinemia. Blood. 2007;109:2553–6.
Lougaris V, Baronio M, Vitali M, Tampella G, Cattalini M, Tassone L, Soresina A, Badolato R, Plebani A. Bruton tyrosine kinase mediates TLR9-dependent human dendritic cell activation. J Allergy Clin Immunol. 2014;133:1644–50. e1644
Marron TU, Martinez-Gallo M, Yu JE, Cunningham-Rundles C. Toll-like receptor 4-, 7-, and 8-activated myeloid cells from patients with X-linked agammaglobulinemia produce enhanced inflammatory cytokines. J Allergy Clin Immunol. 2012;129:184–90. e181-184
Taneichi H, Kanegane H, Sira MM, Futatani T, Agematsu K, Sako M, Kaneko H, Kondo N, Kaisho T, Miyawaki T. Toll-like receptor signaling is impaired in dendritic cells from patients with X-linked agammaglobulinemia. Clin Immunol. 2008;126:148–54.
Ni Gabhann J, Hams E, Smith S, Wynne C, Byrne JC, Brennan K, Spence S, Kissenpfennig A, Johnston JA, Fallon PG, Jefferies CA. Btk regulates macrophage polarization in response to lipopolysaccharide. PLoS One. 2014;9:e85834.
Gunderson AJ, Kaneda MM, Tsujikawa T, Nguyen AV, Affara NI, Ruffell B, Gorjestani S, Liudahl SM, Truitt M, Olson P, et al. Bruton tyrosine kinase-dependent immune cell cross-talk drives pancreas cancer. Cancer Discov. 2016;6:270–85.
Fiorcari S, Maffei R, Audrito V, Martinelli S, Ten Hacken E, Zucchini P, Grisendi G, Potenza L, Luppi M, Burger JA, et al. Ibrutinib modifies the function of monocyte/macrophage population in chronic lymphocytic leukemia. Oncotarget. 2016;7:65968–81.
Andreu P, Johansson M, Affara NI, Pucci F, Tan T, Junankar S, Korets L, Lam J, Tawfik D, DeNardo DG, et al. FcRgamma activation regulates inflammation-associated squamous carcinogenesis. Cancer Cell. 2010;17:121–34.
Pucci F, Garris C, Lai CP, Newton A, Pfirschke C, Engblom C, Alvarez D, Sprachman M, Evavold C, Magnuson A, et al. SCS macrophages suppress melanoma by restricting tumor-derived vesicle-B cell interactions. Science. 2016;352:242–6.
Coffelt SB, Wellenstein MD, de Visser KE. Neutrophils in cancer: neutral no more. Nat Rev Cancer. 2016;16:431–46.
Marron TU, Rohr K, Martinez-Gallo M, Yu J, Cunningham-Rundles C. TLR signaling and effector functions are intact in XLA neutrophils. Clin Immunol. 2010;137:74–80.
Farrar JE, Rohrer J, Conley ME. Neutropenia in X-linked agammaglobulinemia. Clin Immunol Immunopathol. 1996;81:271–6.
Fiedler K, Sindrilaru A, Terszowski G, Kokai E, Feyerabend TB, Bullinger L, Rodewald HR, Brunner C. Neutrophil development and function critically depend on Bruton tyrosine kinase in a mouse model of X-linked agammaglobulinemia. Blood. 2011;117:1329–39.
Mangla A, Khare A, Vineeth V, Panday NN, Mukhopadhyay A, Ravindran B, Bal V, George A, Rath S. Pleiotropic consequences of Bruton tyrosine kinase deficiency in myeloid lineages lead to poor inflammatory responses. Blood. 2004;104:1191–7.
Mueller H, Stadtmann A, Van Aken H, Hirsch E, Wang D, Ley K, Zarbock A. Tyrosine kinase Btk regulates E-selectin-mediated integrin activation and neutrophil recruitment by controlling phospholipase C (PLC) gamma2 and PI3Kgamma pathways. Blood. 2010;115:3118–27.
Volmering S, Block H, Boras M, Lowell CA, Zarbock A. The neutrophil Btk Signalosome regulates integrin activation during sterile inflammation. Immunity. 2016;44:73–87.
Stiff A, Trikha P, Wesolowski R, Kendra K, Hsu V, Uppati S, McMichael E, Duggan M, Campbell A, Keller K, et al. Myeloid-derived suppressor cells express Bruton's tyrosine kinase and can be depleted in tumor-bearing hosts by Ibrutinib treatment. Cancer Res. 2016;76:2125–36.
Sagiv-Barfi I, Kohrt HE, Czerwinski DK, Ng PP, Chang BY, Levy R. Therapeutic antitumor immunity by checkpoint blockade is enhanced by ibrutinib, an inhibitor of both BTK and ITK. Proc Natl Acad Sci U S A. 2015;112:E966–72.
Gao W, Wang M, Wang L, Lu H, Wu S, Dai B, Ou Z, Zhang L, Heymach JV, Gold KA, et al. Selective antitumor activity of ibrutinib in EGFR-mutant non-small cell lung cancer cells. J Natl Cancer Inst. 2014;106
Ruella M, Kenderian SS, Shestova O, Fraietta JA, Qayyum S, Zhang Q, Maus MV, Liu X, Nunez-Cruz S, Klichinsky M, et al. The addition of the BTK inhibitor Ibrutinib to anti-CD19 chimeric antigen receptor T cells (CART19) improves responses against mantle cell lymphoma. Clin Cancer Res. 2016;22:2684–96.
Fraietta JA, Beckwith KA, Patel PR, Ruella M, Zheng Z, Barrett DM, Lacey SF, Melenhorst JJ, McGettigan SE, Cook DR, et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood. 2016;127:1117–27.
Dubovsky JA, Beckwith KA, Natarajan G, Woyach JA, Jaglowski S, Zhong Y, Hessler JD, Liu TM, Chang BY, Larkin KM, et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood. 2013;122:2539–49.
Byrd JC, Brown JR, O'Brien S, Barrientos JC, Kay NE, Reddy NM, Coutre S, Tam CS, Mulligan SP, Jaeger U, et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med. 2014;371:213–23.
Dreyling M, Jurczak W, Jerkeman M, Silva RS, Rusconi C, Trneny M, Offner F, Caballero D, Joao C, Witzens-Harig M, et al. Ibrutinib versus temsirolimus in patients with relapsed or refractory mantle-cell lymphoma: an international, randomised, open-label, phase 3 study. Lancet. 2016;387:770–8.
Burger JA, Keating MJ, Wierda WG, Hartmann E, Hoellenriegel J, Rosin NY, de Weerdt I, Jeyakumar G, Ferrajoli A, Cardenas-Turanzas M, et al. Safety and activity of ibrutinib plus rituximab for patients with high-risk chronic lymphocytic leukaemia: a single-arm, phase 2 study. Lancet Oncol. 2014;15:1090–9.
Chanan-Khan A, Cramer P, Demirkan F, Fraser G, Silva RS, Grosicki S, Pristupa A, Janssens A, Mayer J, Bartlett NL, et al. Ibrutinib combined with bendamustine and rituximab compared with placebo, bendamustine, and rituximab for previously treated chronic lymphocytic leukaemia or small lymphocytic lymphoma (HELIOS): a randomised, double-blind, phase 3 study. Lancet Oncol. 2016;17:200–11.
Wang ML, Lee H, Chuang H, Wagner-Bartak N, Hagemeister F, Westin J, Fayad L, Samaniego F, Turturro F, Oki Y, et al. Ibrutinib in combination with rituximab in relapsed or refractory mantle cell lymphoma: a single-centre, open-label, phase 2 trial. Lancet Oncol. 2016;17:48–56.
Tam C, Grigg AP, Opat S, Ku M, Gilbertson M, Anderson MA, Seymour JF, Ritchie DS, Dicorleto C, Dimovski B, et al. The BTK inhibitor, Bgb-3111, is safe, tolerable, and highly active in patients with relapsed/refractory B-cell malignancies: initial report of a phase 1 first-in-human trial. Blood. 2015;126
Noy A, de Vos S, Thieblemont C, Martin P, Flowers C, Morschhauser F, Collins GP, Ma S, Coleman M, Peles S, et al. Single-agent Ibrutinib demonstrates efficacy and safety in patients with relapsed/refractory marginal zone lymphoma: a multicenter, open-label, phase 2 study. Blood. 2016;128:1213.
Secchiero P, Voltan R, Rimondi E, Melloni E, Athanasakis E, Tisato V, Gallo S, Rigolin GM, Zauli G. The gamma-secretase inhibitors enhance the anti-leukemic activity of ibrutinib in B-CLL cells. Oncotarget. 2017;8:59235–45.
Rotin LE, Gronda M, MacLean N, Hurren R, Wang X, Lin FH, Wrana J, Datti A, Barber DL, Minden MD, et al. Ibrutinib synergizes with poly(ADP-ribose) glycohydrolase inhibitors to induce cell death in AML cells via a BTK-independent mechanism. Oncotarget. 2016;7:2765–79.
Kelly PN, Romero DL, Yang Y, Shaffer AL 3rd, Chaudhary D, Robinson S, Miao W, Rui L, Westlin WF, Kapeller R, Staudt LM. Selective interleukin-1 receptor-associated kinase 4 inhibitors for the treatment of autoimmune disorders and lymphoid malignancy. J Exp Med. 2015;212:2189–201.
Goldstein RL, Yang SN, Taldone T, Chang B, Gerecitano J, Elenitoba-Johnson K, Shaknovich R, Tam W, Leonard JP, Chiosis G, et al. Pharmacoproteomics identifies combinatorial therapy targets for diffuse large B cell lymphoma. J Clin Invest. 2015;125:4559–71.
Patel V, Keating MJ, Wierda WG, Gandhi V. Preclinical combination of TP-0903, an AXL inhibitor and B-PAC-1, a procaspase-activating compound with ibrutinib in chronic lymphocytic leukemia. Leuk Lymphoma. 2016;57:1494–7.
Lamothe B, Cervantes-Gomez F, Sivina M, Wierda WG, Keating MJ, Gandhi V. Proteasome inhibitor carfilzomib complements ibrutinib's action in chronic lymphocytic leukemia. Blood. 2015;125:407–10.
Hing ZA, Mantel R, Beckwith KA, Guinn D, Williams E, Smith LL, Williams K, Johnson AJ, Lehman AM, Byrd JC, et al. Selinexor is effective in acquired resistance to ibrutinib and synergizes with ibrutinib in chronic lymphocytic leukemia. Blood. 2015;125:3128–32.
Cervantes-Gomez F, Lamothe B, Woyach JA, Wierda WG, Keating MJ, Balakrishnan K, Gandhi V. Pharmacological and protein profiling suggests Venetoclax (ABT-199) as optimal partner with Ibrutinib in chronic lymphocytic leukemia. Clin Cancer Res. 2015;21:3705–15.
Zhao X, Bodo J, Sun D, Durkin L, Lin J, Smith MR, Hsi ED. Combination of ibrutinib with ABT-199: synergistic effects on proliferation inhibition and apoptosis in mantle cell lymphoma cells through perturbation of BTK, AKT and BCL2 pathways. Br J Haematol. 2015;168:765–8.
Rushworth SA, Bowles KM, Barrera LN, Murray MY, Zaitseva L, MacEwan DJ. BTK inhibitor ibrutinib is cytotoxic to myeloma and potently enhances bortezomib and lenalidomide activities through NF-kappaB. Cell Signal. 2013;25:106–12.
Sagiv-Barfi I, Kohrt HE, Burckhardt L, Czerwinski DK, Levy R. Ibrutinib enhances the antitumor immune response induced by intratumoral injection of a TLR9 ligand in mouse lymphoma. Blood. 2015;125:2079–86.
Xargay-Torrent S, Lopez-Guerra M, Rosich L, Montraveta A, Roldan J, Rodriguez V, Villamor N, Aymerich M, Lagisetti C, Webb TR, et al. The splicing modulator sudemycin induces a specific antitumor response and cooperates with ibrutinib in chronic lymphocytic leukemia. Oncotarget. 2015;6:22734–49.
Yahiaoui A, Meadows SA, Sorensen RA, Cui ZH, Keegan KS, Brockett R, Chen G, Queva C, Li L, Tannheimer SL. PI3Kdelta inhibitor idelalisib in combination with BTK inhibitor ONO/GS-4059 in diffuse large B cell lymphoma with acquired resistance to PI3Kdelta and BTK inhibitors. PLoS One. 2017;12:e0171221.
Funding
S.P.S is partly supported by Netherlands Organization for Scientific Research.
Author information
Authors and Affiliations
Contributions
All authors contributed to literature search and writing of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Pal Singh, S., Dammeijer, F. & Hendriks, R.W. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol Cancer 17, 57 (2018). https://doi.org/10.1186/s12943-018-0779-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-018-0779-z