
Abstract
As an intelligent disease, tumors apply several pathways to evade the immune system. It can use alternative routes to bypass intracellular signaling pathways, such as nuclear factor-κB (NF-κB), Wnt, and mitogen-activated protein (MAP)/phosphoinositide 3-kinase (PI3K)/mammalian target of rapamycin (mTOR). Therefore, these mechanisms lead to therapeutic resistance in cancer. Also, these pathways play important roles in the proliferation, survival, migration, and invasion of cells. In most cancers, these signaling pathways are overactivated, caused by mutation, overexpression, etc. Since numerous molecules share these signaling pathways, the identification of key molecules is crucial to achieve favorable consequences in cancer therapy. One of the key molecules is the mesenchymal-epithelial transition factor (MET; c-Met) and its ligand hepatocyte growth factor (HGF). Another molecule is the epithelial cell adhesion molecule (EpCAM), which its binding is hemophilic. Although both of them are involved in many physiologic processes (especially in embryonic stages), in some cancers, they are overexpressed on epithelial cells. Since they share intracellular pathways, targeting them simultaneously may inhibit substitute pathways that tumor uses to evade the immune system and resistant to therapeutic agents.
Similar content being viewed by others


Introduction
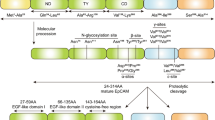
The mesenchymal–epithelial transition factor (MET) gene is expressed on the membrane that is bound to receptor tyrosine kinase (RTK). Besides, epithelial cells express essentially the MET receptor [1]. The hepatocyte growth factor (HGF), as a serum ligand, activates MET/RTK. It has been known as a mitotic factor for hepatocytes. Followed by binding it to c-Met from tumor cells, a signaling pathway is formed, which leads to proliferation, metastasis, and angiogenesis, for example in brain, gastric, and head and neck cancers [2, 3]. Also, stromal cells and fibroblasts are the main source of HGF production. Thus, HGF/MET activation can lead to numerous intracellular events, such as proliferation, survival, and inflammation pathways. Therefore, various molecules such as extracellular signal-regulated kinase 1 and 2 (ERK1/2)/mitogen-activated protein kinases (MAPKs), phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt), signal transducer and activator of transcription (STAT), and nuclear factor-κB (NF-κB) are involved in it. Of note, over-activation of the HGF/MET pathway through germline MET and sporadic MET mutations or even protein over-expression increases tumorigenesis and tumor progressions in numerous cancer forms, such as renal cell carcinoma, metaplasia-dysplasia-adenocarcinoma evolution in esophageal cancer, osteosarcomas, and melanomas, as well as brain, gastric, gliomas, breast, and head and neck cancers [1, 4,5,6]. Moreover, the literature has shown that changes in MET are related to anti-cancer resistance in some cancers, for example, non-small cell lung cancer (NSCLC) and colorectal cancer (CRC); also, it is associated with worse prognosis and aggressiveness [7, 8]. On the other hand, various epithelial tissues expressed the epithelial cell adhesion molecule (EpCAM). It is a 40-kD transmembrane glycoprotein that consists of 341 amino acids. Its structure includes the extracellular domain (EpEX), single transmembrane domain, and intracellular domain (EpICD) [9]. Also, EpCAM is a cell surface marker on many kinds of stem cells and progenitor cells [10, 11]. Previous studies have demonstrated that EpCAM is involved in cell junction via interacting with several important cell adhesion molecule (CAM) junctions [9]. In the 1970s, after administration of cancer cells to the mic, EpCAM was identified as a novel tumor-specific cell surface antigen. Also, it is highly expressed in many kinds of epithelial carcinomas. Thus, it correlates with tumorigenesis, metastasis, and cancer stem cells [12]. Also, some antibodies considering that can target EpCAM are developed [13]. Since EpCAM and HGF/c-Met are involved in important signaling pathways in various cancers, the purpose of this study is to elucidate the interplay between these molecules, tumor microenvironment, and intracellular pathways. The reason is that simultaneously targeting these two molecules may boost the efficacy of cancer therapies.
Interplay HGF/c-Met and tumor stroma
Tumor stroma consists of extracellular matrix (ECM) and various cells, such as fibroblasts, inflammatory and endothelial cells. This composition significantly impacts tumor initiation and progression [14]. Cross talking between tumor and stromal cells results in a suitable microenvironment for tumor growth and metastasis [14, 15]. Fibroblasts are the most frequent cells in the tumor stroma. They have important roles in the maintenance of ECM and adjacent epithelial homeostasis via direct stromal-epithelial contact and the secretion of cytokines [15]. Normal fibroblasts turn into cancer-associated fibroblasts (CAFs), followed by the creation of neoplastic transformation of epithelia. Then, they boost their capacity to promote the malignant process through the production of growth factors and inflammation factors [16, 17]. HGF and MET are expressed by stromal and malignant cells, respectively. Thus, when HGF is coupled with its proto-oncogene receptor, c-Met leads to epithelial phenotype transformation and the acquisition of a migratory phenotype of noncancerous cells. It is a critical, widely documented phenomenon in the transformation of neoplastic features regarding the progression of various cancers [18]. Therefore, HGF creates a microenvironment through interaction between cancerous cells and adjacent stroma, increasing the further development and invasiveness of cancer [19]. HGF facilities cell detachment from the primary tumor. Then, they are infiltrated via the surrounding stroma favoring the pathways, leading to degradation of ECM [18].
Cross talk between HGF/c-Met and immune responses
Previous results have demonstrated that HGF/MET axis impacts immune responses [1, 20]. Although its effects are unclear, the migration of T and B lymphocytes is controlled by HGF. Also, it can counteract the anti-inflammatory effect of transforming growth factor (TGF) [21,22,23,24]. In an experimental animal model of auto-inflammatory disease [experimental autoimmune myocarditis (EAM)], for instance, a greater amount of HGF is conversely associated with inflammation and fibrosis [1, 23]. Furthermore, HGF, in cooperation with other hematopoietic stimuli, is able to increase all types of precursors [25]. Its receptor, MET, is known as a tumor-associated antigen (TAA). Thus, MET can be recognized by CD8 cytotoxic T cells. This mechanism can initiate immune system activation against cancer cells overexpressing MET [26]. MET has significant effects on the immune system via dendritic cells (DCs). DCs, which present TAA to T cells, can induce the activation of regulatory T cells (CD4+), controlling cytotoxic CD8+ T cells. Thus, the HGF/MET axis can boost this mechanism. It showed that this pathway could be targeted for cancer immunotherapy [1]. DCs affected by HGF can induce an increase in T regulatory, interleukin 10 (IL-10), and transforming growth factor β (TGF-β). Also, they increase IL-17-producing lymphocytes [27, 28]. Therefore, this process leads to the inhibition of the immune response [1]. Furthermore, HGF/MET can affect the immune system via granulocytes. The literature has shown that MET deletion in neutrophils leads to the enhancement of tumor growth and metastasis. Thus, MET can play an essential role in chemoattraction and neutrophil-mediated cytotoxicity. The tumor-derived tumor necrosis factor (TNF) or other inflammatory factors can induce MET in human neutrophils, leading to transmigration neutrophils across an activated endothelium, and free radical production results in cancer cell killing. Therefore, it should be taken into consideration that treating cancer patients with MET inhibitors can lead to defective chemotaxis of neutrophils, and tumor cells can escape from tumor killing [29]. Indeed, in some cases, the HGF/MET axis is crucial for cancer cell survival, and, in other cases, it has anti-cancer effects [1]. Thus, it is complicated to target just the HGF/MET pathway in cancer therapy (Fig. 1).

Controversial roles of HGF/c-Met in the immune system: (1) chemoattraction of neutrophils to tumor site; (2) activated Dc presenting TAA, boost TCD4+ regulatory cells, and anti-tumoral immune response decreased; and (3) TCD8+ activation, which led to anti-tumoral immune response
Interplay HGF/c-Met with cytokines
Tumor cells and CAfs, in tumor stromal, produce several cytokines such as IL-6, IL-8, IL-10, monocyte chemoattractant protein 1 (MCP-1), and regulated on activation, normal T cell expressed and secreted (RANTES) [30]. IL-6 is one of the important cytokines, which is well known as a pro-inflammatory cytokine. It plays key roles in some processes, such as B and T differentiation, induction of acute-phase mediators, hematopoiesis, tumor cell proliferation, and increased angiogenesis [31, 32]. There are direct and indirect correlations between disease progression and response to therapeutic agents, respectively, with IL-6 levels [33]. The IL-6/JAK2/STAT3 pathway is activated by coupling IL-6 with its cell-surface receptor (IL-6R) and a common cytokine-receptor signal-transducing subunit (gp130) [34,35,36]. Also, in various solid tumors, HGF and IL-6 play key roles in the phenotype modulation of cancer cells [14, 37]. Ding et al. demonstrated that HGF cooperated with IL-6; also, they showed that the increased level of MET led to the differentiation of normal fibroblast to CAFs in gastric cancer (GC) [14]. To et al. showed that the interaction between HGF and IL-6 was conducted via two ways involved in the invasion of a lung cancer cell line in vitro [38]. They demonstrated that IL-6 was able to stimulate A549 lung adenocarcinoma and increase messenger RNA (mRNA) expression of c-Met/HGF. Also, their results demonstrated that the production of matrix metallopeptidase 2 (MMP-2) and MMP-9 was increased when there was co-stimulation with HGF and IL-6. Thus, it led to an extra effect on tissue invasion [38]. On the other hand, another pro-inflammatory cytokine is IL-8, which has numerous activities such as the migration of neutrophils, monocytes, tumor cell proliferation, and metastasis [32, 39]. Additionally, it is known as an important cytokine involved in the angiogenesis process [40]. Furthermore, RANTES and MCP-1 are other chemokines that have important roles in the migration of normal and malignant cells [33, 41, 42]. Also, IL-10 is one of the cytokines involved in the immunosuppressive process. Thus, it is considered a protecting cancer cell agent; also, it is frequently produced by tumor cells [33]. Previous studies have shown that in bone marrow stromal cells (BMSCs), production of HGF results in the production of IL-11, IL-10, IL-6, IL-8, stromal cell-derived factor (SDF)-1α, and vascular endothelial growth factor (VEGF) [43]. Another role of HGF/c-Met is to decrease the expression of interferon γ (IFN-γ), TGF-β, and TNF-α in a dose-dependent manner [44]. Boissinot et al. demonstrated that in the serum and bone marrow plasma of polycythemia vera (PV) patients, the levels of HGF, IL-11, and tissue inhibitor of metalloproteinase 1 (TIMP-1) increased. Also, they showed that paracrine and autocrine feedback loops were the main ways in which BMSCs and glycophorin A+ (GPA) erythroblasts are involved, and HGF and IL-11 directly affected the production of each other [45].
In conclusion, these network complex connections of signals and mediators are in ECM and have great impacts on cell proliferation and function deviation. Focusing on the management of immunosurveillance, angiogenesis, and key factors produced by tumor cells and CAFs (as prominent cells in the tumor microenvironment) can open ways to increase the efficacy of tumor therapy.
Interplay EpCAM with cancers
EpCAM (or CD326A) is a well-known pan-epithelial differentiation antigen expressed on a vast number of epithelial tissues. It is also involved in cell signaling, proliferation, differentiation, and migration. EpCAM is overexpressed on the basolateral in the epithelial malignancies’ surface of all human carcinomas of various origins [46, 47]. For example, in hepatic malignancies, overexpression of EpCAM relates to poor prognosis because it activates proto-oncogene myelocytomatosis (c-Myc). Thus, it leads to tumor progression [48]. Also, CD326 is considered as a promising target for anti-cancer therapy because of being a part of the molecular network of oncogenic receptors. Also, EpCAM plays an important role in the suppression of anti-tumor immunity. Thus, immune-based therapies are applied to target EpCAM [46].
Cross talk between EpCAM/EpCAM and immune responses
Since EpCAM is a common TAA and can affect T-cell immune responses, Ziegler et al. demonstrated that it could be considered an immune target in colon cancer [49]. EpCAM also leads to IL-4 dominated T helper 2 (Th2) responses. Therefore, Th1-inducing conditions are rarely dominant. They also showed that intra-tumoral expression of cytokines of the IL-12 family and IFN-γ (which are caused by induction Th1 and lead to inhibition of tumor growth) diminished. In return, EpCAM as a human TAA can cause tumor immune evasion via Th2 responses’ development [49]. For example, in ovarian cancer progression, the immune system plays pivotal roles throughout cytokine and chemokine signaling pathways in drug resistance [50, 51].
Interplay EpCAM with cytokines
As mentioned above, the two most important cytokines, IL-6 and IL-8 (CXCL8/IL-8), are involved in various spectrum cellular pathways responsible for the proliferation, metastasis, or tumor cell survival. Also, the data showed that IL-6 and IL-8 affect the expression of EpCAM. Bonneau et al. demonstrated that IL-8 could impact epithelial-mesenchymal transition (EMT) in ovarian and breast cancer (BC) cells. Also, in patients with ovarian cancer, the level of EpCAM can be considered as a predictor of poor prognosis [46]. Additionally, according to the type and/or signaling pathway, AP-1, NF-κB, and C/EBPb transcription factors are involved in IL-8 regulation [52]. In an experiment conducted by Narendra et al. [53], it was illustrated that EpCAM has a correlation with IL-8 in primary BC. They also showed that when EpCAM was downregulated in BC cell lines, IL-8 expression decreased. Consequently, phosphorylation of NF-κB family member RELA increased, while IκBα protein expression decreased. Therefore, EpCAM induces activation of NF-κB, followed by modulation of IL-8 expression at baseline and IL-1β stimulation. Although the data showed that the EpCAM signaling has roles in the modulation of BC invasion, to clarify the molecular mechanism of EpCAM, further study should be conducted to apply appropriate molecular therapies to boost efficacy targeting of EpCAM [53].
In conclusion, IL-8 as a CXC chemokine produced by various cell types is well-known as a potent angiogenic factor through paracrine and autocrine routes in tumorigenesis. Thus, it can be considered as an intervention in cancer metastasis [54]. Furthermore, it plays roles in diverse normal physiological processes, including wound healing and abnormal processes such as cancer metastasis. IL-8 is produced by a large number of solid tumor types and related to inflammatory cells such as neutrophils. On the other hand, the literature has indicated that endothelial cells secret IL-6 that leads to tumor growth enhancement [55]. For example, Shi et al. demonstrated that IL-6 could contribute to the regulation of VEGF and angiogenesis in GC. They showed that IL-6 was an inducer for VEGF expression, which boosted angiogenesis in GC [56]. As mentioned above, both HGF/c-Met and EPCAM signaling pathways are involved in the production of IL-6 and IL-8. Therefore, targeting both of them simultaneously may have significant effects on cancer therapy via direct and indirect effects on angiogenesis. Consequently, cancer patients might benefit from approaches targeting HGF/c-Met and EpCAM (Table 1).
NF-κB signaling pathway in cancer
NF-κB is considered as a pleiotropic transcription factor. Also, numerous processes, inflammation, innate immunity, apoptosis, and cell proliferation, for instance, are mediated by it [57]. Furthermore, NF-κB is involved in various cancers, such as BC, via several stimulators, including various pro-inflammatory cytokines (IL-1β and TNF-α), growth factors (epidermal growth factor [EGF]), DNA-damaging agents (radiation), and oncogenes (rat sarcoma [RAS]) [57,58,59]. Also, NF-κB is a key regulator of several genes, including MMP-9, COX2, c-Myc, cyclin-D, etc. [60]. Thus, it promotes proliferation, migration, and metastasis in cancer cells [1]. IKK and IKB are positive and negative regulators of NF-κB, respectively (Fig. 2) [60]. In some cancers, such as BC, NF-κB is overexpressed [61]; NF-κB not only is involved in the proliferation and development of BC cells but also leads to resistance to some drugs used in cancer therapy such as anti-epidermal growth factor receptor (EGFR) drugs [62]. Furthermore, NF-κB plays a key role in EMT by regulating important molecules, such as MMP-9 [63]. It also protects cancer cells from apoptosis by induction of anti-apoptotic proteins, such as BCL2, BCLxL, XIAP, and so on [60, 63, 64]. NF-κB has interactions and cross talk with many intracellular molecules (e.g., NF-κB with estrogen receptor [ER] and progesterone receptor [PR] signaling pathways) [61, 65]. We are going to discuss the association between NF-κB and HGF/c-Met and EpCAM in the following sections.

C-Met can activate NF-κB in some cancer cells. By the way, indirect interactions can be assumed from experimental data. C-Met can activate STAT3 and PI3K/Akt, which are upstream and activators of NF-κB
Cross talk NF-κB and HGF/c-Met
Only few studies have shown that c-Met is upstream of NF-κB in BC [66], glioma [67], and renal cancer cells [68]. It can be mentioned that c-Met may activate NF-κB by activating other molecules and pathways. For example, PI3K/Akt and ERK/MAPK are downstream pathways of c-Met [69] and can activate NF-κB [12]. STAT3 activates NF-κB in BC cells [65], and c-Met promotes proliferation and migration by triggering STAT3 in these cells [62]. Hence, it can be assumed that c-Met activates NF-κB through STAT3, PI3K/Akt, and ERK/MAPK pathways. In other cancer cells, studies suggest more cross talk between NF-κB and c-Met. It was shown that blocking c-Met/HGF (C-Met ligand) interaction would suppress MMP-9 activity in lung cancer [70]. As you can see, there is no conclusive evidence of an interaction between NF-κB and c-Met in cancer cells, especially in BC cells; hence, more studies are needed to be done to clearly identify this interaction.
Cross talk NF-κB and EpCAM
The results showed that NF-κB-EpCAM was co-overexpressed in the nucleus of BC cells [71]. Downregulation of EpCAM was followed by downregulation of NF-κB in these cells [71]. As can be seen, the results from NF-κB-EpCAM and/or c-Met and NF-κB in BC cells are limited. The only way to find the possible interaction of NF-κB-EpCAM and c-Met-NF-κB in BC cells is to find a common molecule in the upstream or downstream of NF-κB-EpCAM and/or c-Met. Hence, the relation between NF-κB-c-Met or EpCAM may indirectly assume. For example, in BC cells, NF-κB regulates c-Myc expression [60, 72]; on the other hand, EpCAM-c-Myc is co-overexpressed in these cells [72]. Thus, it could be concluded that NF-κB-EpCAM has cross talk, which can regulate c-Myc expression. MMP-9 is another protein, which may explain the possible cross talk between NF-κB-EpCAM. MMP-9 is regulated by NF-κB [60] and promotes EpCAM activity in BC cells [73]. Hence, it can be concluded that NF-κB could regulate EpCAM through MMP-9. Expression of EpCAM is linked to COX2 expression [73], and COX2 is another target of NF-κB in BC cells [73]. The mentioned conclusions are possible here. As mentioned before, NF-κB is downstream of PI3K/Akt and ERK/MAPK pathways, and targeting NF-κB could be an option to block these pathways. PI3K/Akt and ERK/MAPK are downstream of several important receptors, such as EGFR, insulin-like growth factor (IGF-IR), and vascular endothelial growth factor receptor (VEGFR). Studies have shown that blocking NF-κB increased the sensitivity of cancer cells to therapy-induced apoptosis [63, 74]. Blocking NF-κB prevents tumor formation in mice or suppresses established tumors in these animal models [63]. Other results showed that inhibiting NF-κB signaling decreased progesterone-induced proliferation in BC cell lines [63]. Because of the lack of enough data on the interaction of NF-κB-EpCAM and c-Met-NF-κB in BC cells, there are only some possibilities that NF-κB-EpCAM and c-Met-NF-κB may have cross talk, and, to become clear, further studies are needed.
PI3K/Akt/mTOR signaling pathway and its importance in cancer
PI3K/Akt and mammalian target of rapamycin (mTOR; PAM) pathways are two signaling pathways that are necessary for many cellular activities, such as motility, proliferation, differentiation, metabolism, survival, and angiogenesis, in both normal physiology and development of cancer [75]. For as much as these pathways are closely connected to each other, they are often regarded as a unique pathway (PAM) [76, 77]. Phosphorylated tyrosines of receptor tyrosine kinases bind to PI3-kinase. This enzyme then phosphorylates the membrane lipid phosphatidylinositol-4,5-bisphosphate (PIP2) into phosphatidylinositol-3,4,5-trisphosphate (PIP3). Thus, PIP3 phosphorylates Akt, which is a serine/threonine kinase. Another crucial Akt activator is mTOR complex 2 (mTORC2). Activated Akt phosphorylates several substances, including mTORC1 and inhibitors (Tsc2 and PRAS40) to relieve the inhibition of mTORC1 kinase activity; thus, it stimulates protein synthesis, resulting in cell enlargement [78, 79]. Akt also inhibits apoptosis by deactivating pro-apoptotic protein Bcl-2 antagonist of cell death (BAD), as well as by promoting the breakdown of p53 as a pro-apoptotic protein via activation of E3 ubiquitin-protein ligase MDM2 [79].
PI3K/Akt/mTOR pathway has an integral role in cancer cells; for example, it has been estimated to be activated in 70% of all BCs [77, 80]. Akt phosphorylation, which demonstrates PAM activation, occurs in 50% to 70% of NSCLCs and 30% to 66% of head and neck squamous cell carcinomas (HNSCCs) [81, 82]. This pathway activation is also responsible for targeted therapy resistance in many cancer types [83].
EpCAM and PI3K/Akt/mTOR
Downregulation of PI3K/Akt/mTOR signaling pathway proteins occurs subsequent to EpCAM silencing and is associated with the decreased colony formation, proliferation, and cellular invasion in prostatic cancer cells [84]. EpCAM-regulated carcinogenesis was proved to be associated with PI3K/Akt/mTOR signaling pathway activation in animal experimentation on prostatic cancer [84]. Consequently, this implies that EpCAM expression is closely associated with activation of this pathway in tumorigenesis. Another study showed that the roles of the Akt signaling pathway in EpCAM were related to tumorigenesis. It revealed that nasopharyngeal carcinoma cell lines overexpressing EpCAM had a significant increase in the level of crucial molecules of this pathway, such as mTOR and activated Akt. Also, after treatment of this cell line with the Akt inhibitor, MK2206 or the mTOR inhibitor (rapamycin) reduced the expression of Vimentin and SLUG (as EMT biomarkers). Thus, EpCAM induced invasiveness. Also, it confirms that activation of the Akt/mTOR signaling pathway is necessary for EMT, resulted from EpCAM overexpression in nasopharyngeal carcinoma cells [85].
HGF/c-Met and PI3K/Akt/mTOR
PI3K/Akt/mTOR is also one of the pathways that controls c-Met regulated cell proliferation, survival, and migration [86]. Also, phosphorylated (activated) c-Met leads to phosphorylation of PI3K and RAS. Thus, it activates PI3K pathway indirectly later [79]. A new role for c-Met activated PI3K axes is involved in the constitution of lamellipodial protrusions that are crucial for transmigration cells through endothelial cells. This requires the mobilization of Rac1, p47phox, and localized reactive oxygen species (ROS) production [87]. It has also been shown that c-Met/PI3K/Akt signaling is responsible for resistance to photodynamic therapy and doxorubicin in carcinomas via enhancement of BCRP/ABCG2 expression [88]. Concurrent overexpression of c-Met and Akt leads to synergistic activity of these proto-oncogenes, resulting in rapid cancer expansion [89].
HGF/c-Met, EpCAM cross talk in PI3K/Akt/mTOR
It is well established that the c-Met/PI3K/Akt signaling axis is crucial to regulate tumor cell critical functions, such as proliferation and survival. It is well established that c-Met uses the PAM axis to regulate tumor cells’ critical functions, such as proliferation and survival; on the other hand, some studies have proven that EpCAM uses the same axis for carcinogenesis. Although different outcomes are expected regarding which antigen has triggered the pathway, it could be suggested that these two TAAs interact through this signaling axis (Fig. 2).
Wnt signaling pathway and its importance in cancer
Three different Wnt axes have been characterized, i.e., Ca, planar cell polarity (PCP), and canonical β-catenin/T cell factor 1 (TCF-1) pathways. The two latter branches are considered to antagonize each other. Once the canonical axis initiates, before nuclear translocation, β-catenin protein aggregates in the cytoplasm; then, at the level of the genome, it binds to the specific transcription factors to trigger the expression of specific genes [90]. Manifold localization of β-catenin in the nucleus and cytosol is identified in human BC frequently [91]. In the PCP axis, JUN-N-terminal kinase and GTPases are involved in regulating c-Jun-dependent transcription and cell migration. The Ca axis is associated with activation of phospholipase C or GMP-specific phosphodiesterase, followed by the release of intracellular Ca into the cytosol to activate downstream signaling proteins consisting of CREB and NFAT transcription factors mainly to regulate migration [90, 92]. One of the most critical signaling axes in embryonic development is the Wnt pathway, which regulates cell self-renewal, differentiation, proliferation, and migration [90]. This axis is generally silent until cell regeneration is needed in stem cells [93]. Hyperactive Wnt signaling can lead to aberrant cell proliferation and has been signified in the pathogenesis of BC, CRC, melanoma, and leukemia [94]. It is activated in more than 50% of BCs and is associated with decreased overall survival [95, 96]. Of note, the significance of the Wnt axis role in triggering, progression, or maintenance of distinct BC subtypes is controversial [96].
EPCAM and Wnt pathway
Several different studies have indicated that EpCAM causes tumorigenesis using the Wnt axis. EpCAM signaling is activated when EpCAM is cleaved into EpEX and intracellular domain (EpICD) by specific enzymes. This cleavage only occurs when a temporary proliferation is required. EpEX sheds outside the cell, and EPICD is released to the cytoplasm. Four and one-half LIM domain protein 2 (FHL2) is a cytosolic protein, which is an interaction partner for EpICD and a co-activator of β-catenin [97, 98]. In a study by Yamashita et al. [99], EpCAM was found as a novel Wnt axis target gene, which could be considered as a biomarker for this pathway activation. They found a positive correlation between the EpCAM expression level and Wnt signaling genes, such as BAMBI and DKK. Also, it was discovered that activation of this pathway enriched the population of EpCAM+ cells [100]. In another study, it was detected that EpCAM overexpression in BC cell lines regulated Wnt axis components. The mRNA level of negative regulators of Wnt signaling (SFRP1 and TCF7L2) decreased in EpCAM-expressing cell lines after EpCAM overexpression; it means that Wnt signaling increased [101]. Transcription of EpCAM target genes, including c-Myc cyclins and TCF1, is activated as a result of translocation of β-catenin bound to FHL2 and EpICD into the nucleus [97, 98].
HGF/c-Met and Wnt pathway
C-Met/HGF signaling has been shown to be an upstream regulator for the Wnt pathway [86, 102]. C-Met expression is controlled by the β-catenin signaling and downregulated by inhibition of Wnt signaling using Frzb and DNLRP5, two Wnt antagonists [103, 104]. C-Met and α3β1 integrin response to HGF and laminin, respectively, to regulate cell survival via the Wnt cascade [105]. Frizzled-8 (FZD8) as a Wnt receptor is known to be necessary for the interaction between the Wnt/β-catenin axis and c-Met since it is upregulated through the ERK/c-Fos cascade by c-Met in cancer stem-like cells [106]. Moreover, Wnt signaling is responsible for the development of acquired anti-c-Met therapy resistance since its components are upregulated in resistant cells [107].
HGF/c-Met and EpCAM cross talking in the Wnt pathway
Several lines of evidence suggest that EpCAM and c-Met as signal transducers contribute to Wnt downstream effectors to initiate and develop cancer. It provides a logical explanation for the development of dual targeting drugs such as MM-131 a bispecific anti-MET/EpCAM mAb [108] and also further drug discoveries targeting concurrently all three c-Met, EpCAm and Wnt pathway simultaneously to overcome both innate and acquired resistance to tyrosin kinase inhibitors (TKIs).
RAS/MAPK signaling pathway and its importance in cancer
Following the autophosphorylation of receptor tyrosine kinases, such as c-Met or many other receptor types, the activated receptor binds to the adaptor protein growth-factor-receptor-bound protein 2 (GRB2). GRB2 is then bound to the nucleotide exchange factor Son of sevenless (SOS); thus, phosphorylation of RTK recruits SOS to the plasma membrane, where RAS is also localized [109]. GTP-bound RAS (activated form) binds to RAF proteins (c-RAF, BRAF, and ARAF) and induces conformational alternations to dimerize RAF, which is activated and turns on the kinase cascade [110]. As mentioned above, RAS can also activate PI3K, triggering the PI3K/Akt/mTOR axis [111]. Activated RAF phosphorylates and activates mitogen-activated protein kinase kinases 1 and 2 (MEK1 and MEK2); these kinases are able to catalyze the phosphorylation of MAPKs, ERK1, and ERK2, thus rank-ordering the MAPK cascade from RAS, RAF, MEK, and finally to ERK [112, 113]. Eventually, the expression of immediate-early genes, c-fos, and c-Myc is upregulated [113]. Ultimately, ERK activates transcription factors involved in fundamental cellular processes, such as proliferation, apoptosis, differentiation, migration, adhesion, and cell polarity [114]. RAS/MAPK is one of the dominant cancer-initiating pathways and has a well-known impression in tumorigenesis by enhancing the survival and metastasis of cancer cells [115]. It is noteworthy that activating point mutations across any of the components of the pathway have been known as either trigger of cancer (mutations of RAF and RAS family genes) or as poor prognosis indicator (mutations of ERK and MEK) [114, 116]. Mutations in RAS genes, which make this enzyme continually active, are frequently detected in many cancer types; however, specific patterns exist between the cancer type and mutation frequencies associated with each RAS gene [113]. Mutations in RAF and RAS genes are considered the most common ones associated with chemotherapy and targeted therapy resistance [115].
EPCAM and RAS/MAPK
It has been discovered that synergistic activation of this pathway by several factors (such as FGF, SCF, or HGF) stimulates the activation of the MAPK pathway [117]. In addition to RTKs and G-protein-coupled receptors, EpCAM has been considered as the RAS/MAPK cascade regulator. Gao et al. found that knockdown of EpCAM by small interfering RNA (siRNA) significantly reduced the expression and phosphorylation of RAS/MAPK components, p-ERK, p-RAF, and RAS in BC cells and repressed their malignant behavior [118]. In accordance with the latter report, phosphorylated ERK was decreased in EpCAM CRISPR/Cas9-mediated knockout cells, suggesting EpCAM augments integrin-mediated signaling [119]. Sankpal et al. defined a double-negative feedback loop between EpCAM and the activated MAPK pathway. They examined signaling pathway activity and EpCAM expression in a panel of 31 epithelial cancer cell lines; a strong inverse correlation between MAPK/ERK cascade activity and EpCAM expression was observed. To confirm their results, they used TGF-β1/TNF-α or EGF to induce MAPK/ERK activity; decreased EpCAM expression was subsequently observed [120].
HGF/c-Met and the RAS/MAPK signaling pathway
As mentioned above, the RAS/MAPK pathway is also considered a downstream effector of HGF/c-Met signals, among other signal transduction pathways [86, 121]. Stimulated c-Met activates RAS to induce the expression of c-fos and c-Jun via the MAPK cascade in order to mediate cell scattering, invasion, and mobility [122]. The MAPK pathway is also responsible for HGF induced cell morphogenesis [123]. HGF induces the expression of ETS proto-oncogene 1, transcription factor (ETS1) by activation of ERK1. Activated ERK1 phosphorylates the threonine 38 residue of ETS1 to induce transcriptional activation [124]. Hypoxia-induced VEGF expression is known to be regulated by HGF/c-Met signaling via the MAPK axis [121]. Co-expression of Akt/c-Met stimulates the activation of the MAPK pathway as a result of the synergistic activity [125].
C-Met and EpCAM cross talk in the RAS/MAPK pathway
The role of the MAPK cascade in the transduction of c-Met signaling into the nucleus is almost well defined; however, since research studies conducted on the cross talk between EpCAM and the RAS/RAF/MEK/ERK cascade have been limited to recent years and even sometimes are conflicting, it is almost impossible to define a conclusive opinion regarding the cross talk between c-Met and EpCAM through this pathway (Fig. 3).

Cross talk between EpCAM, c-Met, and Wnt-β-catenin signaling pathways. Full-length EpCAM is cleaved, releasing EpCAM’s ectodomain (EpEX). Following the cleavage step, EpCAM’s cytoplasmic tail (EpICD) is released and associates with FHL-2 and β-catenin and translocates to the nucleus, upregulating transcription of EpCAM target genes via LEF consensus sites. C-Met activates MAP kinase (RAF/MEK/ERK) and PI3K/Akt signaling to induce cell proliferation and survival in cancer cells. C-Met and Wnt-β-catenin signaling pathways mainly cooperate in regulating EMT. C-Met contributes to nuclear translocation of β-catenin by its tyrosine phosphorylation or inhibition of the β-catenin degradation complex by Akt that phosphorylates glycogen synthase kinase-3β (GSK3β). This might, in turn, result in increased availability of non-bound β-catenin that may be stabilized by association with EpICD
Resistance to targeted therapy in EpCAM and c-Met
Drug resistance
Failure in cancer therapy can happen via two general causes. The first one is intrinsic in cancer cells; for example, cell membrane transporter proteins are important in drug resistance because they alter drug transport. ABC transporters (ATP-binding cassette) specially P-gp (P-glycoprotein) play an important role in clinical drug resistance by many of mechanisms for example high level expression by gene rearrangement. The other is the gradual genetic and epigenetic abnormalities that occur in cancer cells [126]. Tumors such as renal cancer, hepatocellular carcinoma, and malignant melanoma display intrinsic resistance [127]. In BC, it was seen that cancer therapy resulted in a stem cell-like phenotype in non-stem tumor cells [128]. In some cases, it was demonstrated that chemotherapy could potentially raise the levels of circulating endothelial progenitor cells (EPCs) that cause tumor growth and metastasis [129]. Researches have shown a positive correlation between chemotherapeutic resistance and the number of spontaneous genetic mutations [130]. Mutation in several genes confers resistance to cancer therapy. For example, in ovarian cancer, a mutation in the p53 gene was seen in platinum chemotherapy [131] and, in BC treated with anticancer drugs, reduced response to chemotherapy; most cases had a mutation in p53 [132]. Also, in head and neck cancer, polymorphism in p53 influenced clinical response to cisplatin-based chemoradiotherapy [133].
In metastatic colorectal cancer, mutations of K-Ras have an adverse effect on response to anti-EGFR antibody and cetuximab [134]; also, in NSCLC, mutations in K-Ras affect resistance to treatment with gefitinib or erlotinib drugs [135]. Gene amplification is the other way that confers resistance to cancer therapy. For example, an increasing copy number of the dihydrofolate reductase (DHFR) gene was seen in a patient that received methotrexate [136]. In breast cancer, a correlation between the copy number of the HER2 gene and sensitivity to Herceptin was seen [137]. Moreover, gene amplification is a way that in chronic myelogenous leukemia (CML) lead to over expression of the Bcr-Abl protein as a mechanism of acquired imatinib resistance [137]. The use of cetuximab in patients with colon cancer results in EGFR gene amplification and treatment resistance [134]. Another mechanism, which is a problem in treatment, is gene rearrangement; for example, in leukemia, rearrangements of the MDR-1 gene cause drug refractory [138]. Epigenetic changes are another obstacle in cancer therapy; for example, in patients with gliomas, methylation of the promoter of the DNA-repair enzyme O6-methylguanine-DNA methyltransferase (MGMT) is a problem in cancer therapy with alkylating agents [139] (Table 2).
Roles of EpCAM in drug resistance
Most studies have analyzed the relationship between high EpCAM expression in cancer cells and permanent proliferation signals, as well as overexpression of various targeted genes, including c-Myc and cyclins [140]. EpCAM has been identified as a cancer stem cell marker in solid tumors and has a significant correlation with all the characteristics of cancer stem cells, EMT, and metastasis [123]. Besides, EpCAM overexpression is related to Wnt/β-catenin pathway activation in cancers, which is a potential causal pathway for cytoplasmic and nuclear accumulation of β-catenin, indicating proliferation and reduced apoptosis in cancer cells [141]. EpCAM is applied as a suitable target in immunotherapy approaches, such as antibody-based approaches for in vitro/in vivo and ongoing clinical trials. Recent results have indicated that humanized anti-EpCAM antibodies are successful in both preclinical and early clinical studies and selectively targeted EpCAM-positive cell lines with a greater potency without any relapse [142]. However, despite these favorable achievements, anti-EpCAM monoclonal antibodies have not shown as much promise as initially indicated, and resistance to mAb has become a major obstacle recently. Several studies have shown that anti-EpCAM antibodies limit therapeutic efficacies as the results of showing dose- and target-dependent in metastatic cancers, short serum half-life, altered EpCAM expression pattern, and differential cleavage and localization of EpICD in cancer cells (Fig. 3) [143, 144]. Thus, larger clinical trials are necessary to approve novel antibodies in the treatment of specific tumors.
Role of c-Met in drug resistance
A variety of preclinical data have demonstrated that amplification of c-Met signaling occurs in many malignancies as a result of MET gene mutation or overexpression. Resistance to targeted therapeutics developed by the activation of c-Met kinase has appeared as a considerable mechanism of resistance in multiple types of cancers [145]. As depicted in Fig. 2, in c-Met signaling, overexpression of the c-Met receptor and HGF results in sustained PI3K/Akt/mTOR and MAPK signaling activation through Gab1 and mutation of MET kinase domain, which are associated with therapy resistance [87]. Additionally, c-Met signaling can also mediate aberrant localization and phosphorylation of β-catenin in cancer cells. Phosphorylated β-catenin translocates to the nucleus, binds to TCF/LEF transcription factors, and promotes the expression of many target genes, including those involved in cell proliferation, anti-apoptosis, invasion, and angiogenesis (Fig. 3) [146]. Overexpression of HGF and/or c-Met can contribute to therapy resistance in radiotherapy, chemotherapy, and targeted therapies (including EGFR inhibitors in NSCLC and CRC) [109, 147]. Since patients who developed EGFR mutant lung adenocarcinomas with acquired resistance to EGFR inhibitor drugs (gefitinib or erlotinib) exhibit increased copy numbers of MET, c-Met-HGF inhibitors are used for lung cancer treatment alone or in combination with other drugs [148]. Targeting c-Met has been investigated in anti-EGFR resistant cells because c-Met is a key player in anti-EGFR resistance [148]. The results showed that c-Met inhibitors could overcome anti-EGFR resistance and suppress resistant cell proliferation [148]. The important role of c-Met in anti-EGFR resistance promoted researchers to investigate the effects of EGFR and c-Met inhibitors [149]. The findings showed that using bispecific Ab (anti-EGFR and anti-c-Met) could suppress growth in anti-EGFR resistant cells more efficiently compared to anti-EGFR agents, such as gefitinib, erlotinib, and so on [149]. As can be seen, the results of different studies demonstrate the efficacy of c-Met targeting; in the following, we will discuss the advantages of targeting c-Met and EpCAM.
Targeting c-Met and EpCAM: a way to overcome drug resistance
It has been well established that aberrantly expressed EpCAM has a direct impact on the cell cycle, proliferation, and upregulation of the oncogenic transcription factors c-Myc and cyclin A/E [150]. As a matter of fact, the intracellular part of EpCAM (EpICD) is crucial and sufficient to upregulate proto-oncogenes activation [151]. Moreover, c-Myc alterations contribute to acquired resistance to c-Met inhibitors in different MET-overexpressed cancers [152]. It is conceivable that targeting c-Met, along with other aberrant proto-oncogenes, can modulate c-Myc expression and cell survival in EpCAM-positive and overexpressed c-Met-resistant cancer cells [102, 153]. In addition, several studies have shown co-overexpression of EpCAM and c-Met pathways and their significant interaction in developing resistance to currently targeted therapies in cancer tissues. Similar to EpCAM, c-Met also has a putative role in the acquisition of the stem cell status and EMT in cancer that causes tumor cell dissociation and metastasis [154]. Understanding different resistance mechanisms to currently-existing inhibitors can provide new insights into combinatorial targeting of the driver oncoprotein and further downstream effectors to avoid or postpone the therapy resistance. For instance, MM-131, a bispecific anti-MET/EpCAM mAb, has been developed to inhibit cell proliferation and migration in c-Met positive/HGF-positive tumors, which also overexpresses EpCAM [109]. Thus, concurrent administrations of novel agents against c-Met and EpCAM are recommended to implement optimal efficacy in current treatments.
Undoubtedly, reviewing existing evidence and mechanisms regarding therapy resistance confirms the structural and functional intricacy of aberrantly expressed EpCAM and c-Met, which makes them function as proto-oncogenes in several malignancies. Nevertheless, it is still needed to be elucidated whether these two oncogenic signaling pathways possess any initiating roles in the development of cancer or they are merely contributing molecules in the propagation of tumors or development of resistance to existing therapeutic agents. As we discussed in this review, the upregulation of EpCAM and c-Met takes place in many human carcinomas and is in close relation to the development and propagation of cancers; thus, suppressing overexpression of EpCAM and c-Met may represent a potential potent therapeutic approach. In addition, their cross talks with other oncogenic pathways result in amplification of their underlying signaling cascades and resistance development to different targeted therapeutic agents.
Novel therapy targeting for c-Met and EPCAM
C-Met and novel therapy
Anti-c-Met agents can be classified into four types: selective c-Met tyrosine kinase inhibitors (TKIs), non-selective c-Met inhibitors, monoclonal antibodies against HGF/c-Met, and microRNAs (miRNAs). Selective c-Met inhibitors have a very high selectivity (more than ten thousand times) for c-Met compared to other kinases [155]. However, this selectivity does not mean that other tyrosine kinases are not inhibited at all. Volitinib (Savolitinib), SAR125844, tepotinib (EMD1214063), capmatinib (INCB28060), AMG337, Indo5, tivantinib, and PHA665752 are of such selective c-Met inhibitors [156,157,158,159,160], which showed anti-tumor effects in various cancers with high MET expression, including gastric and hepatocellular carcinoma, papillary renal carcinoma, and NSCLC [158, 161]. Volitinib, SAR125844, and PHA665752 have anti-proliferative and pro-apoptotic effects by inhibition of RAS/MAPK and PI3K/Akt signaling pathways; meanwhile, tepotinib inhibits c-Met signaling in both HGF-dependent and independent ways [156].
One way to improve the response to TKIs in patients with lower c-Met overexpression is to expand the target kinases [162]. Non-selective c-Met inhibitors include crizotinib (PF-02341066), foretinib (XL-880), cabozantinib (XL-184), glesatinib (MGCD265), BMS-777607, and MK2461, which in addition to c-Met, can inhibit other tyrosine kinases, such as ALK, RON, VEGFR2, KIT, TIE2, PDGFR, VEGFR1, VEGFR3, RET, and FLT-3 [163,164,165], through binding to the ATP binding site on tyrosine kinases. The considerable effects of non-selective TKIs, especially crizotinib, led to the US Food and Drug Administration (FDA) approval of crizotinib in ALK+ cancers [166]. VEGFR inhibitors are also capable of inhibiting angiogenesis along with TKIs benefits. The combination of non-selective TKIs with chemotherapy and checkpoint inhibitors has been investigated in various trials with promising results [162]. The point that should be considered about non-selective TKIs is that the anti-tumor function of these inhibitors might be higher than that of selective c-Met inhibitors due to inhibition of multiple kinases. Meanwhile, the toxicity of non-selective TKIs is higher than selective c-Met inhibitors, limiting their prescribed dosage [167]. In some cases, high toxicity and damage of non-selective TKIs to various organs might be comparable to those caused by the tumor itself. Therefore, it is better to use specific TKIs based on the overexpressed tyrosine kinase in each cancer type [165].
Various monoclonal antibodies have been developed to block c-Met/HGF signaling pathways, some of which competitively bind to c-Met, and the others are served as traps for c-Met, preventing ligand attachment and dimerization of c-Met [168]. Onartuzumab (MetMAb), SAIT301, ABT-700 (h224G11), ARGX-111, and DN30 are specific antibodies against c-Met, preventing the binding of c-Met to HGF [169,170,171]. SAIT301 also internalizes and destroys c-Met after binding to it [171]. The mechanism of ARGX-111 is through stimulating antibody-dependent cellular cytotoxicity (ADCC) in cells with excessive c-Met expression [172]. DN30 is involved in decreasing the c-Met signaling pathway through a variety of ways, including c-Met destruction, reducing c-Met expression, c-Met proteolytic detachment, and shedding c-Met from the cell surface, providing traps for HGF [162, 170]. Thus, DN30 with dual effects on c-Met and HGF separately can inhibit the c-Met/HGF pathway [173].
Bispecific antibodies with different specifications are an interesting option in targeted therapy. Emibetuzumab (LY2875358) against c-Met and HGF and MP0250 against HGF and VEGF are two bispecific antibodies that prevent c-Met attachment to the ligand, increase the internalization, destroy c-Met molecules, and thereby reduce the signaling of the c-Met pathway [149, 174, 175]. Moreover, LY3164530 and JNJ-61186372 bispecific antibodies bind to c-Met and EGFR and have shown good results in inhibiting tumor growth [176, 177].
HGF targeting antibodies, including rilotumumab (AMG-102), ficlatuzumab, TAK-701, and YYB-101, bind to HGF and prevent c-Met attachment, leading to the induction of synergistic anti-tumor responses combined with other TKIs and EGFR inhibitors [162, 172, 178, 179]. Clinical trials have examined the safety and efficacy of these antibodies in cancers and have shown that combinational therapy with other treatments has improved patients’ responses and their survival [169, 178, 179].
Nowadays, one of the new methods of targeted therapies is based on miRNAs. The expression of miRNAs in cancerous tissue is different from normal tissue, leading to cancer progression, tumor growth, invasion, and metastasis [180]. Some of these miRNAs target c-Met by increasing or decreasing its signaling and play a role in tumor progression or suppression. For example, miR-93 activates c-Met and PI3K/Akt signaling pathways and leads to tumor progression and invasion [181]. However, miR-101, miR-206, and miR-26a target c-Met/HGF and inhibit tumor growth and metastasis [182,183,184]. Therefore, the use of either miRNA antagonists, agonists, or miRNA mimics that inhibit c-Met signaling is a potential and promising treatment for cancer [165].
Taken together, c-Met could be targeted using selective and non-selective inhibitors, monoclonal or bispecific antibodies, and miRNAs to inhibit tumor progression. Selective inhibitors have more specificity and low adverse events compared to non-selective ones. The clinical trials on c-Met inhibitors are still at the beginning of the way and require further evaluation.
EpCAM and novel therapy
EpCAM (CD326) is found only in the basolateral membrane of epithelial tissue while overexpressed throughout the cancerous tissue membrane, making this molecule a good candidate for targeting in the treatment of cancer [185]. Tight junctions hamper the targeting of EpCAM in normal tissues and prevent unspecific tissue damage [186]. Due to the loose adhesion of EpCAM compared to other CAMs, increased expression of EpCAM is associated with invasion, metastasis, and poor prognosis in many cancers, including basal-like and luminal B BCs [185, 187, 188].
The first anti-EpCAM monoclonal antibody was a mouse antibody developed in 1979 called edrecolomab [189]. Edrecolomab causes cytotoxicity for the tumor through cell-dependent mechanisms, such as ADCC and complement-dependent cytotoxicity (CDC) [190, 191]. Despite its benefits in early clinical trials, the trials were halted due to limited efficiency and several side effects of the murine and chimeric types [142, 192, 193]. Studies have suggested that the inefficiency of edrecolomab is due to its low affinity to EpCAM. Two high-affinity antibodies, 3622W94 and ING-1, were developed but bound to all EpCAM-expressing cells, leading to side effects such as acute pancreatitis in clinical trials [142]. Adecatumumab, which has a moderate affinity to EpCAM, can target tumor cells with high EPCAM expression and reduce aggression and metastasis in patients with fewer side effects [194]. In addition to ADCC and CDC, one of the therapeutic mechanisms of adecatumumab is through inhibition of tumor cell metabolism [142]. There are specific conditions needed for an efficient ADCC or CDC, including enough number of antibodies attached to the target cell, a sufficient number of effector immune cells (such as natural killer [NK] cells, monocytes, and granulocytes), and the absence of ADCC suppressor cells (such as regulatory T cells [Tregs] and myeloid-derived suppressor cells [MDSCs]) in the tumor microenvironment. Moreover, the internalization of EpCAM and bounded antibodies pose challenges to ADCC/CDC. Therefore, it seems that other mechanisms should be considered in designing anti-EpCAM antibodies [143].
The next generation of EpCAM-targeting antibodies is based on closing immune cells to tumor cells. Catumaxomab is a bispecific antibody against EpCAM and CD3, which brings T cells closer to EpCAM+ cells. Besides, the fragment crystallizable (Fc) of the antibody can bind to Fc receptors (FcRs) on the surface of immune cells and induce ADCC, making it a three-functional antibody (Triomab) [195, 196]. Fc also binds to FcRs on the surface of DCs and connects them to T cells to activate T cells [197]. In clinical trials, catumaxomab showed promising efficacy by increasing anti-tumor cytokines and had low toxicity that led to its approval [195, 196].
Another bispecific molecule is the bispecific T cell engagers (BiTEs), consisting of two single-chain variable fragments with different specificities [198]. Solitomab or MT-110 is a BiTE against EPCAM and CD3, which bridges between tumor cells and T cells, leading to lysis of tumor cells [196, 198]. The advantages of this molecule are smaller size and greater penetration into solid tumors compared to conventional antibodies [143]. Another way to activate immune cells, including T and NK cells, is to use IL-2 linked to an anti-EpCAM antibody. Tucotuzumab celmoleukin (EMD 273066 or huKS-IL2) is an immunocytokine consisting of tucotuzumab (anti-EpCAM antibody) and IL-2 (celmoleukin) [199]. The combination of huKS-IL2 with cyclophosphamide prevents Treg proliferation and increases anti-tumor effects [144]. An important point to consider when using T cell-involving antibodies is that the tumor microenvironment is immunosuppressive, and the infiltrated T cells are mostly exhausted. Therefore, simultaneous targeting of immunosuppressive agents, such as IDO and immune checkpoints (including PD-1 and CTLA-4), improves anti-tumor response [186, 196, 198, 200].
A common way to kill EpCAM+ tumor cells is to use immunotoxins. This means that EpCAM-specific antibodies are conjugated with cytotoxic agents, such as doxorubicin, paclitaxel, calicheamicin, topoisomerase I inhibitors, alpha amanitin, and indolinobenzodiazepine pseudodimers (IGNs), to specifically kill the EpCAM+ tumor cells. Antibodies enter the cell to release the toxin in the cytoplasm; thus, the extent of antigen expression, antibody affinity, and the ratio of toxic agents conjugated with each antibody is effective in its performance [201, 202]. Some of these substances have direct cytotoxic effects, and some others, such as fungal derivative alpha amanitin, induce apoptosis in tumor cells [203]. Oportuzumab monatox (VB4-845) and citatuzumab bogatox (VB6-845) are fusion proteins comprised of an anti-EPCAM single-chain variable fragment (ScFv) or Fab bound to pseudomonas endotoxin A or non-immunogenic toxin of Bougainvillea spectabilis. These fusion proteins enter EpCAM+ cells, and the conjugated toxin inhibits protein synthesis leading to cell apoptosis. Studies have reported tolerability and acceptable side effects of toxin-conjugated fusion proteins, as well as great anti-tumor effects in bladder, head and neck cancers, and epithelial tumors, which has led them to the next stages of clinical trials [204,205,206].
One of the problems with using antibodies is their large size; thus, researchers are trying to increase the tumor permeability by replacing antibodies with smaller peptides and aptamers [143, 207]. Using specific peptides instead of antibodies could solve this limitation (large size) and enhance the tumor permeability. Two protein families are mostly used in targeted therapy: (1) designed ankyrin repeat proteins (DARPins), a type of non-immunoglobulin-engineered protein derived from the ankyrin protein and (2) macrocyclic peptides produced by the random non-standard peptides integrated discovery (RaPID) system. These two protein families have high specificity and affinity to their targets, as well as high resistance to proteases. The EpCAM-specific types of these peptides have shown promising anti-tumor results and are being further investigated [208, 209].
Aptamers, also known as chemical antibodies, are single-stranded oligonucleotides that shape a three-dimensional structure and bind to their targets just like antibodies [210]. Aptamers have different advantages over antibodies, including much smaller size (making them more appropriate to penetrate solid tumors), less immunogenicity (reducing resistance to treatment), the capability to manipulate their length and physicochemical properties (making them possible to change the half-life, pH sensitivity, flexibility, and conjugation capacity), fast and simple production procedure, negligible inter-batch variation, and better temperature resistance [210,211,212,213,214]. Xiang et al. showed that EpCAM-specific aptamers had better biodistribution and had up to four times high tumor penetration, and therefore better performance than anti-EpCAM antibodies [215]. Similar to antibodies, one of the applications of EpCAM-specific aptamers is to deliver cytotoxic molecules, such as doxorubicin and neocarzinostatin, to target cells [216]. The interesting point about the binding of drugs to aptamers is that by changing the length of the aptamer and its physicochemical features, the number of conjugating molecules can be increased without any particular effect on the affinity and specificity of the aptamer [211, 216]. The application of aptamers also faces challenges that are mainly related to their small size and nucleotide nature. These challenges include degradation by cellular nucleases, high renal clearance, and low penetration in biological membranes due to their negative charge. These challenges can be met by nucleotide modifications and choosing conjugates with appropriate molecular weight and positive charge [213, 214].
Moreover, EpCAM is associated with some miRNAs and genes involved in tumor progression [217]. The high levels of miR-130, miR-181, miR-17-92, and miR-92b have been associated with EpCAM overexpression in hepatocellular carcinoma [218, 219]. Further, miR-181 and miR-130 are overexpressed in EpCAM+ cells than in EpCAM cells and cause stemness features in EpCAM+ tumor cells. Hence, these miRNA families are associated with cancer stem cells in EpCAM+ tumors, and their inhibition reduces tumor cell proliferation.
To sum up, there are several generations of anti-EpCAM antibodies with different effector mechanisms, including ADCC, CDC, T-cell engaging, providing activatory cytokines for immune cells, and delivering toxins to the tumor cells. Although low-affinity mAbs have low efficacy, high-affinity mAbs might demonstrate more adverse events due to binding to all EpCAM+ cells. Researchers are trying to overcome the low-penetration issues by using ScFvs, BiTEs, aptamers, DARPins, RaPID system, and miRNAs instead of full-sized antibodies. The clinical trials on combining EpCAM targeting with other immunotherapies (such as checkpoint blockade) are ongoing.
Conclusion and prospective
As mentioned before, tumors, stroma, and the immune system communicate with each other and contribute to tumor resistance. Furthermore, tumor cells are capable of escape from the immune system. TAAs on tumor cells deceive the immune system, leading to inappropriate and inadequate responses. EpCAM and c-Met are not only considered two prominent tumor-associated antigens but also expressed on healthy tissues. However, their overexpression on tumor cells results in over-activation of certain intercellular signaling pathways of proliferation, migration, and invasion of tumor cells. Subsequent the HGF coupling with c-Met as a tyrosine kinas receptor, angiogenesis could be initiated through migration and invasion to adjusting tissues. EpCAM is also associated with NF-κB signaling pathway activation leading to IL-8 production, which is one of the key mediators in angiogenesis. Hence, targeting both of them simultaneously might be a promising approach to hinder therapeutic resistance by blocking all potential alternative routes. Indeed, the investigation of cross talk between mentioned molecules and tumor stroma may shed light on finding new targets and could provide beneficial information for the rational design of a novel therapeutic approach in cancer.
Availability of data and materials
Not applicable.
Abbreviations
- NF-κB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- STAT3:
-
Signal transducer and activator of transcription 3
- c-MET:
-
Mesenchymal epithelial transition factor (MET) or hepatocyte growth factor receptor (HGFR)
- EpCAM:
-
Epithelial cell adhesion molecule
- COX2:
-
Cyclooxygenase-2
- MMP9:
-
Matrix metallopeptidase 9
- IKK:
-
IκB kinase
- PI3K:
-
Phosphoinositide 3-kinase
- AKT:
-
Protein kinase B or (PKB)
- ERK:
-
Extracellular signal-regulated kinase
- MAPK:
-
Mitogen-activated protein kinase
- EGFR:
-
Epidermal growth factor receptor tyrosine kinase
- EMT:
-
Epithelial–mesenchymal transition
- BCL2:
-
B-cell lymphoma 2
- BCLxL:
-
B-cell lymphoma-extra large
- XIAP:
-
X-linked inhibitor of apoptosis protein
- ER:
-
Estrogen receptor
- PR:
-
Progesterone receptor
- IGFIR:
-
Insulin-like growth factor
- VEGFR:
-
Vascular endothelial growth factor receptor
- c-Myc:
-
Myelocytomatosis
- IKB:
-
Inhibitor of kB
References
Papaccio F, Della Corte CM, Viscardi G, Di Liello R, Esposito G, Sparano F, et al. HGF/MET and the immune system: relevance for cancer immunotherapy. Int J Mol Sci. 2018;19(11):3595.
Jedeszko C, Victor BC, Podgorski I, Sloane BF. Fibroblast hepatocyte growth factor promotes invasion of human mammary ductal carcinoma in situ. Can Res. 2009;69(23):9148–55.
Phan LM, Fuentes-Mattei E, Wu W, Velazquez-Torres G, Sircar K, Wood CG, et al. Hepatocyte growth factor/cMET pathway activation enhances cancer hallmarks in adrenocortical carcinoma. Can Res. 2015;75(19):4131–42.
Trusolino L, Comoglio PM. Scatter-factor and semaphorin receptors: cell signalling for invasive growth. Nat Rev Cancer. 2002;2(4):289–300.
Schmidt L, Duh F-M, Chen F, Kishida T, Glenn G, Choyke P, et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet. 1997;16(1):68–73.
Di Renzo MF, Olivero M, Martone T, Maffe A, Maggiora P, De Stefani A, et al. Somatic mutations of the MET oncogene are selected during metastatic spread of human HNSC carcinomas. Oncogene. 2000;19(12):1547–55.
Park S, Choi Y-L, Sung CO, An J, Seo J, Ahn M-J, et al. High MET copy number and MET overexpression: poor outcome in non-small cell lung cancer patients. 2012.
Morgillo F, Della Corte CM, Fasano M, Ciardiello F. Mechanisms of resistance to EGFR-targeted drugs: lung cancer. ESMO Open. 2016;1(3): e000060.
Huang L, Yang Y, Yang F, Liu S, Zhu Z, Lei Z, et al. Functions of EpCAM in physiological processes and diseases. Int J Mol Med. 2018;42(4):1771–85.
Schmelzer E, Zhang L, Bruce A, Wauthier E, Ludlow J, Yao H-L, et al. Human hepatic stem cells from fetal and postnatal donors. J Exp Med. 2007;204(8):1973–87.
Kamimoto K, Kaneko K, Kok CY-Y, Okada H, Miyajima A, Itoh T. Heterogeneity and stochastic growth regulation of biliary epithelial cells dictate dynamic epithelial tissue remodeling. Elife. 2016;5: e15034.
Zheng X, Fan X, Fu B, Zheng M, Zhang A, Zhong K, et al. EpCAM inhibition sensitizes chemoresistant leukemia to immune surveillance. Can Res. 2017;77(2):482–93.
Baeuerle P, Gires O. EpCAM (CD326) finding its role in cancer. Br J Cancer. 2007;96(3):417–23.
Ding X, Ji J, Jiang J, Cai Q, Wang C, Shi M, et al. HGF-mediated crosstalk between cancer-associated fibroblasts and MET-unamplified gastric cancer cells activates coordinated tumorigenesis and metastasis. Cell Death Dis. 2018;9(9):1–16.
Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16(9):582.
Matsuo Y, Ochi N, Sawai H, Yasuda A, Takahashi H, Funahashi H, et al. CXCL8/IL-8 and CXCL12/SDF-1α co-operatively promote invasiveness and angiogenesis in pancreatic cancer. Int J Cancer. 2009;124(4):853–61.
Wang X, Zhou Q, Yu Z, Wu X, Chen X, Li J, et al. Cancer-associated fibroblast-derived Lumican promotes gastric cancer progression via the integrin β1-FAK signaling pathway. Int J Cancer. 2017;141(5):998–1010.
Spina A, De Pasquale V, Cerulo G, Cocchiaro P, Della Morte R, Avallone L, et al. HGF/c-MET axis in tumor microenvironment and metastasis formation. Biomedicines. 2015;3(1):71–88.
Matsumoto K, Date K, Ohmichi H, Nakamura T. Hepatocyte growth factor in lung morphogenesis and tumor invasion: role as a mediator in epithelium-mesenchyme and tumor-stroma interactions. Cancer Chemother Pharmacol. 1996;38(1):S42–7.
Yamaura K, Ito K-I, Tsukioka K, Wada Y, Makiuchi A, Sakaguchi M, et al. Suppression of acute and chronic rejection by hepatocyte growth factor in a murine model of cardiac transplantation: induction of tolerance and prevention of cardiac allograft vasculopathy. Circulation. 2004;110(12):1650–7.
Adams DH, Harvath L, Bottaro DP, Interrante R, Catalano G, Tanaka Y, et al. Hepatocyte growth factor and macrophage inflammatory protein 1 beta: structurally distinct cytokines that induce rapid cytoskeletal changes and subset-preferential migration in T cells. Proc Natl Acad Sci. 1994;91(15):7144–8.
Kurz SM, Diebold SS, Hieronymus T, Gust TC, Bartunek P, Sachs M, et al. The impact of c-met/scatter factor receptor on dendritic cell migration. Eur J Immunol. 2002;32(7):1832–8.
Mizuno S, Kurosawa T, Matsumoto K, Mizuno-Horikawa Y, Okamoto M, Nakamura T. Hepatocyte growth factor prevents renal fibrosis and dysfunction in a mouse model of chronic renal disease. J Clin Investig. 1998;101(9):1827–34.
Kuruvilla A, Shah R, Hochwald G, Liggitt H, Palladino M, Thorbecke G. Protective effect of transforming growth factor beta 1 on experimental autoimmune diseases in mice. Proc Natl Acad Sci. 1991;88(7):2918–21.
Ilangumaran S, Villalobos-Hernandez A, Bobbala D, Ramanathan S. The hepatocyte growth factor (HGF)–MET receptor tyrosine kinase signaling pathway: diverse roles in modulating immune cell functions. Cytokine. 2016;82:125–39.
Schag K, Schmidt SM, Müller MR, Weinschenk T, Appel S, Weck MM, et al. Identification of C-met oncogene as a broadly expressed tumor-associated antigen recognized by cytotoxic T-lymphocytes. Clin Cancer Res. 2004;10(11):3658–66.
Benkhoucha M, Santiago-Raber M-L, Schneiter G, Chofflon M, Funakoshi H, Nakamura T, et al. Hepatocyte growth factor inhibits CNS autoimmunity by inducing tolerogenic dendritic cells and CD25+ Foxp3+ regulatory T cells. Proc Natl Acad Sci. 2010;107(14):6424–9.
Okunishi K, Dohi M, Nakagome K, Tanaka R, Mizuno S, Matsumoto K, et al. A novel role of hepatocyte growth factor as an immune regulator through suppressing dendritic cell function. J Immunol. 2005;175(7):4745–53.
Finisguerra V, Di Conza G, Di Matteo M, Serneels J, Costa S, Thompson AR, et al. MET is required for the recruitment of anti-tumoural neutrophils. Nature. 2015;522(7556):349–53.
Raman D, Baugher PJ, Thu YM, Richmond A. Role of chemokines in tumor growth. Cancer Lett. 2007;256(2):137–65.
Tanaka T, Narazaki M, Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol. 2014;6(10): a016295.
Barzaman K, Samadi M, Moradi-Kalbolandi S, Majidzadeh-A K, Salehi M, Jalili N, et al. Development of a recombinant anti-VEGFR2-EPCAM bispecific antibody to improve antiangiogenic efficiency. Exp Cell Res. 2021;405: 112685.
Huang R-P. Cytokines in cancer drug resistance: cues to new therapeutic strategies. Biochim Biophys Acta Rev Cancer. 2016;1865(2):255–65.
Mitra A, Yan J, Xia X, Zhou S, Chen J, Mishra L, et al. IL6-mediated inflammatory loop reprograms normal to epithelial-mesenchymal transition+ metastatic cancer stem cells in preneoplastic liver of transforming growth factor beta–deficient β2-spectrin+/− mice. Hepatology. 2017;65(4):1222–36.
Puchalski T, Prabhakar U, Jiao Q, Berns B, Davis HM. Pharmacokinetic and pharmacodynamic modeling of an anti-interleukin-6 chimeric monoclonal antibody (siltuximab) in patients with metastatic renal cell carcinoma. Clin Cancer Res. 2010;16(5):1652–61.
Guo Y, Xu F, Lu T, Duan Z, Zhang Z. Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat Rev. 2012;38(7):904–10.
Li P, Shan J-X, Chen X-H, Zhang D, Su L-P, Huang X-Y, et al. Epigenetic silencing of microRNA-149 in cancer-associated fibroblasts mediates prostaglandin E2/interleukin-6 signaling in the tumor microenvironment. Cell Res. 2015;25(5):588–603.
To Y, Dohi M, Matsumoto K, Tanaka R, Sato A, Nakagome K, et al. A two-way interaction between hepatocyte growth factor and interleukin-6 in tissue invasion of lung cancer cell line. Am J Respir Cell Mol Biol. 2002;27(2):220–6.
Wróblewski M, Szewczyk-Golec K, Hołyńska-Iwan I, Wróblewska J, Woźniak A. Characteristics of selected adipokines in ascites and blood of ovarian cancer patients. Cancers. 2021;13(18):4702.
Urbantat RM, Blank A, Kremenetskaia I, Vajkoczy P, Acker G, Brandenburg S. The CXCL2/IL8/CXCR2 pathway is relevant for brain tumor malignancy and endothelial cell function. Int J Mol Sci. 2021;22(5):2634.
Bar-Eli M. Role of interleukin-8 in tumor growth and metastasis of human melanoma. Pathobiology. 1999;67(1):12–8.
Huang S, DeGuzman A, Bucana CD, Fidler IJ. Level of interleukin-8 expression by metastatic human melanoma cells directly correlates with constitutive NF-κB activity. Cytokines Cell Mol Ther. 2000;6(1):9–17.
de Cássia NN, Mizukami A, Caliári-Oliveira C, Cominal JG, Rocha JLM, Covas DT, et al. Priming approaches to improve the efficacy of mesenchymal stromal cell-based therapies. Stem Cell Res Ther. 2019;10(1):1–21.
Boissinot M, Vilaine M, Hermouet S. The hepatocyte growth factor (HGF)/met axis: a neglected target in the treatment of chronic myeloproliferative neoplasms? Cancers. 2014;6(3):1631–69.
Boissinot M, Cleyrat C, Vilaine M, Jacques Y, Corre I, Hermouet S. Anti-inflammatory cytokines hepatocyte growth factor and interleukin-11 are over-expressed in Polycythemia vera and contribute to the growth of clonal erythroblasts independently of JAK2 V617F. Oncogene. 2011;30(8):990–1001.
Kapka-Skrzypczak L, Popek S, Sawicki K, Drop B, Czajka M, Jodłowska-Jędrych B, et al. IL-6 prevents CXCL8-induced stimulation of EpCAM expression in ovarian cancer cells. Mol Med Rep. 2019;19(3):2317–22.
Chaudry M, Sales K, Ruf P, Lindhofer H, Winslet M. EpCAM an immunotherapeutic target for gastrointestinal malignancy: current experience and future challenges. Br J Cancer. 2007;96(7):1013–9.
Armeanu-Ebinger S, Hoh A, Wenz J, Fuchs J. Targeting EpCAM (CD326) for immunotherapy in hepatoblastoma. Oncoimmunology. 2013;2(1): e22620.
Ziegler A, Heidenreich R, Braumüller H, Wolburg H, Weidemann S, Mocikat R, et al. EpCAM, a human tumor-associated antigen promotes Th2 development and tumor immune evasion. Blood J Am Soc Hematol. 2009;113(15):3494–502.
Powell DJ, Dudley ME, Hogan KA, Wunderlich JR, Rosenberg SA. Adoptive transfer of vaccine-induced peripheral blood mononuclear cells to patients with metastatic melanoma following lymphodepletion. J Immunol. 2006;177(9):6527–39.
Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10(9):909–15.
Freund A, Jolivel V, Durand S, Kersual N, Chalbos D, Chavey C, et al. Mechanisms underlying differential expression of interleukin-8 in breast cancer cells. Oncogene. 2004;23(36):6105–14.
Sankpal NV, Fleming TP, Gillanders WE. EpCAM modulates NF-κB signaling and interleukin-8 expression in breast cancer. Mol Cancer Res. 2013;11(4):418–26.
Martin TA. Interleukin-8 and angiogenesis. In: Growth factors and their receptors in cancer metastasis. Dordrecht: Springer; 2001. p. 51–65.
Neiva KG, Warner KA, Campos MS, Zhang Z, Moren J, Danciu TE, et al. Endothelial cell-derived interleukin-6 regulates tumor growth. BMC Cancer. 2014;14(1):99.
Huang S-P, Wu M-S, Shun C-T, Wang H-P, Lin M-T, Kuo M-L, et al. Interleukin-6 increases vascular endothelial growth factor and angiogenesis in gastric carcinoma. J Biomed Sci. 2004;11(4):517–27.
Basseres D, Baldwin A. Nuclear factor-κ B and inhibitor of κ B kinase pathways in oncogenic initiation and progression. Oncogene. 2006;25(51):6817–30.
Biswas DK, Shi Q, Baily S, Strickland I, Ghosh S, Pardee AB, et al. NF-κB activation in human breast cancer specimens and its role in cell proliferation and apoptosis. Proc Natl Acad Sci. 2004;101(27):10137–42.
Cao Y, Karin M. NF-κB in mammary gland development and breast cancer. J Mammary Gland Biol Neoplasia. 2003;8(2):215–23.
Aggarwal BB, Shishodia S, Takada Y, Banerjee S, Newman RA, Bueso-Ramos CE, et al. Curcumin suppresses the paclitaxel-induced nuclear factor-κB pathway in breast cancer cells and inhibits lung metastasis of human breast cancer in nude mice. Clin Cancer Res. 2005;11(20):7490–8.
Agrawal AK, Pielka E, Lipinski A, Jelen M, Kielan W, Agrawal S. Clinical validation of nuclear factor kappa B expression in invasive breast cancer. Tumor Biol. 2018;40(1):1010428317750929.
Chen M-K, Du Y, Sun L, Hsu JL, Wang Y-H, Gao Y, et al. H2O2 induces nuclear transport of the receptor tyrosine kinase c-MET in breast cancer cells via a membrane-bound retrograde trafficking mechanism. J Biol Chem. 2019;294(21):8516–28.
Park YH. The nuclear factor-kappa B pathway and response to treatment in breast cancer. Pharmacogenomics. 2017;18(18):1697–709.
Graveel CR, Tolbert D, Woude GFV. MET: a critical player in tumorigenesis and therapeutic target. Cold Spring Harb Perspect Biol. 2013;5(7): a009209.
Fan P, Tyagi AK, Agboke FA, Mathur R, Pokharel N, Jordan VC. Modulation of nuclear factor-kappa B activation by the endoplasmic reticulum stress sensor PERK to mediate estrogen-induced apoptosis in breast cancer cells. Cell Death Discov. 2018;4(1):1–14.
Tacchini L, De Ponti C, Matteucci E, Follis R, Desiderio M. Hepatocyte growth factor-activated NF-κB regulates HIF-1 activity and ODC expression, implicated in survival, differently in different carcinoma cell lines. Carcinogenesis. 2004;25(11):2089–100.
Cheng F, Guo D. MET in glioma: signaling pathways and targeted therapies. J Exp Clin Cancer Res. 2019;38(1):270.
Golovine K, Makhov P, Naito S, Raiyani H, Tomaszewski J, Mehrazin R, et al. Piperlongumine and its analogs down-regulate expression of c-Met in renal cell carcinoma. Cancer Biol Ther. 2015;16(5):743–9.
Ho-Yen CM, Jones JL, Kermorgant S. The clinical and functional significance of c-Met in breast cancer: a review. Breast Cancer Res. 2015;17(1):52.
Parr C, Ali AY. Boswellia frereana suppresses HGF-mediated breast cancer cell invasion and migration through inhibition of c-Met signalling. J Transl Med. 2018;16(1):1–12.
Zhang D, Yang L, Liu X, Gao J, Liu T, Yan Q, et al. Hypoxia modulates stem cell properties and induces EMT through N-glycosylation of EpCAM in breast cancer cells. J Cell Physiol. 2020;235(4):3626–33.
Ahmadi Shadmehri A, Namvar F. A review on green synthesis, cytotoxicity mechanism and antibacterial activity of Zno-NPs. Int J Res Appl Basic Med Sci. 2020;6(1):23–31.
Gao S, Sun Y, Liu X, Zhang D, Yang X. EpCAM and COX-2 expression are positively correlated in human breast cancer. Mol Med Rep. 2017;15(6):3755–60.
Sarink D, Schock H, Johnson T, Chang-Claude J, Overvad K, Olsen A, et al. Receptor activator of nuclear factor kB ligand, osteoprotegerin, and risk of death following a breast cancer diagnosis: results from the EPIC cohort. BMC Cancer. 2018;18(1):1010.
Karar J, Maity A. PI3K/AKT/mTOR pathway in angiogenesis. Front Mol Neurosci. 2011;4:51.
Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR signaling in cancer. Front Oncol. 2014;4:64.
Lee JJ, Loh K, Yap Y-S. PI3K/Akt/mTOR inhibitors in breast cancer. Cancer Biol Med. 2015;12(4):342.
Lauring J, Park BH, Wolff AC. The phosphoinositide-3-kinase-Akt-mTOR pathway as a therapeutic target in breast cancer. J Natl Compr Cancer Netw. 2013;11(6):670–8.
Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. 2010;11(12):834–48.
Li X, Wu C, Chen N, Gu H, Yen A, Cao L, et al. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget. 2016;7(22):33440.
Cooper WA, Lam DC, O’Toole SA, Minna JD. Molecular biology of lung cancer. J Thorac Dis. 2013;5(Suppl 5):S479.
Lui VW, Hedberg ML, Li H, Vangara BS, Pendleton K, Zeng Y, et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov. 2013;3(7):761–9.
LoRusso PM. Inhibition of the PI3K/AKT/mTOR pathway in solid tumors. J Clin Oncol. 2016;34(31):3803.
Ni J, Cozzi P, Hao J, Beretov J, Chang L, Duan W, et al. Epithelial cell adhesion molecule (EpCAM) is associated with prostate cancer metastasis and chemo/radioresistance via the PI3K/Akt/mTOR signaling pathway. Int J Biochem Cell Biol. 2013;45(12):2736–48.
Ni J, Cozzi P, Beretov J, Duan W, Bucci J, Graham P, et al. Epithelial cell adhesion molecule (EpCAM) is involved in prostate cancer chemotherapy/radiotherapy response in vivo. BMC Cancer. 2018;18(1):1–12.
Wang M-H, Sun R, Zhou X-M, Zhang M-Y, Lu J-B, Yang Y, et al. Epithelial cell adhesion molecule overexpression regulates epithelial–mesenchymal transition, stemness and metastasis of nasopharyngeal carcinoma cells via the PTEN/AKT/mTOR pathway. Cell Death Dis. 2018;9(1):1–16.
Zhang Y, Xia M, Jin K, Wang S, Wei H, Fan C, et al. Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol Cancer. 2018;17(1):45.
Usatyuk PV, Fu P, Mohan V, Epshtein Y, Jacobson JR, Gomez-Cambronero J, et al. Role of c-Met/phosphatidylinositol 3-kinase (PI3k)/Akt signaling in hepatocyte growth factor (HGF)-mediated lamellipodia formation, reactive oxygen species (ROS) generation, and motility of lung endothelial cells. J Biol Chem. 2014;289(19):13476–91.
Jung K-A, Choi B-H, Kwak M-K. The c-MET/PI3K signaling is associated with cancer resistance to doxorubicin and photodynamic therapy by elevating BCRP/ABCG2 expression. Mol Pharmacol. 2015;87(3):465–76.
Hu J, Che L, Li L, Pilo MG, Cigliano A, Ribback S, et al. Co-activation of AKT and c-Met triggers rapid hepatocellular carcinoma development via the mTORC1/FASN pathway in mice. Sci Rep. 2016;6:20484.
Shaw HV, Koval A, Katanaev V. Targeting the Wnt signalling pathway in cancer: prospects and perils. Swiss Med Wkly. 2019;149: w20129.
Suzuki H, Toyota M, Caraway H, Gabrielson E, Ohmura T, Fujikane T, et al. Frequent epigenetic inactivation of Wnt antagonist genes in breast cancer. Br J Cancer. 2008;98(6):1147–56.
Pohl S-G, Brook N, Agostino M, Arfuso F, Kumar AP, Dharmarajan A. Wnt signaling in triple-negative breast cancer. Oncogenesis. 2017;6(4): e310.
Sun Q, Fu Q, Li S, Li J, Liu S, Wang Z, et al. Emetine exhibits anticancer activity in breast cancer cells as an antagonist of Wnt/β-catenin signaling. Oncol Rep. 2019;42(5):1735–44.
Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. 2017;36(11):1461–73.
Lin S-Y, Xia W, Wang JC, Kwong KY, Spohn B, Wen Y, et al. β-catenin, a novel prognostic marker for breast cancer: its roles in cyclin D1 expression and cancer progression. Proc Natl Acad Sci. 2000;97(8):4262–6.
van Schie EH, van Amerongen R. Aberrant WNT/CTNNB1 signaling as a therapeutic target in human breast cancer: weighing the evidence. Front Cell Dev Biol. 2020;8:25.
Vasanthakumar S, Sasikala P, Padma M, Balachandar V, Venkatesh B, Ganesan S. EpCAM as a novel therapeutic target for hepatocellular carcinoma. J Oncol Sci. 2017;3(2):71–6.
Maetzel D, Denzel S, Mack B, Canis M, Went P, Benk M, et al. Nuclear signalling by tumour-associated antigen EpCAM. Nat Cell Biol. 2009;11(2):162–71.
Yamashita T, Budhu A, Forgues M, Wang XW. Activation of hepatic stem cell marker EpCAM by Wnt-β-catenin signaling in hepatocellular carcinoma. Can Res. 2007;67(22):10831–9.
Terris B, Cavard C, Perret C. EpCAM, a new marker for cancer stem cells in hepatocellular carcinoma. J Hepatol. 2010;52(2):280–1.
Gostner JM, Fong D, Wrulich OA, Lehne F, Zitt M, Hermann M, et al. Effects of EpCAM overexpression on human breast cancer cell lines. BMC Cancer. 2011;11(1):45.
Lee Y, Lee J-K, Ahn SH, Lee J, Nam D-H. WNT signaling in glioblastoma and therapeutic opportunities. Lab Invest. 2016;96(2):137–50.
Boon EM, van der Neut R, van de Wetering M, Clevers H, Pals ST. Wnt signaling regulates expression of the receptor tyrosine kinase met in colorectal cancer. Can Res. 2002;62(18):5126–8.
Guo Y, Xie J, Rubin E, Tang Y-X, Lin F, Zi X, et al. Frzb, a secreted Wnt antagonist, decreases growth and invasiveness of fibrosarcoma cells associated with inhibition of Met signaling. Can Res. 2008;68(9):3350–60.
Liu Y, Chattopadhyay N, Qin S, Szekeres C, Vasylyeva T, Mahoney ZX, et al. Coordinate integrin and c-Met signaling regulate Wnt gene expression during epithelial morphogenesis. Development. 2009;136(5):843–53.
Sun S, Liu S, Duan SZ, Zhang L, Zhou H, Hu Y, et al. Targeting the c-Met/FZD8 signaling axis eliminates patient-derived cancer stem-like cells in head and neck squamous carcinomas. Can Res. 2014;74(24):7546–59.
Fong JT, Jacobs RJ, Moravec DN, Uppada SB, Botting GM, Nlend M, et al. Alternative signaling pathways as potential therapeutic targets for overcoming EGFR and c-Met inhibitor resistance in non-small cell lung cancer. PLoS ONE. 2013;8(11): e78398.
Casaletto JB, Geddie ML, Abu-Yousif AO, Masson K, Fulgham A, Boudot A, et al. MM-131, a bispecific anti-Met/EpCAM mAb, inhibits HGF-dependent and HGF-independent Met signaling through concurrent binding to EpCAM. Proc Natl Acad Sci. 2019;116(15):7533–42.
Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3(1):11–22.
Ritt DA, Abreu-Blanco MT, Bindu L, Durrant DE, Zhou M, Specht SI, et al. Inhibition of Ras/Raf/MEK/ERK pathway signaling by a stress-induced phospho-regulatory circuit. Mol Cell. 2016;64(5):875–87.
Regad T. Targeting RTK signaling pathways in cancer. Cancers. 2015;7(3):1758–84.
Degirmenci U, Wang M, Hu J. Targeting aberrant RAS/RAF/MEK/ERK signaling for cancer therapy. Cells. 2020;9(1):198.
Dunn KL, Espino PS, Drobic B, He S, Davie JR. The Ras-MAPK signal transduction pathway, cancer and chromatin remodeling. Biochem Cell Biol. 2005;83(1):1–14.
Santarpia L, Lippman SM, El-Naggar AK. Targeting the MAPK–RAS–RAF signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16(1):103–19.
Deng Y-N, Xia Z, Zhang P, Ejaz S, Liang S. Transcription factor RREB1: from target genes towards biological functions. Int J Biol Sci. 2020;16(8):1463.
Prior IA, Hood FE, Hartley JL. The frequency of Ras mutations in cancer statistics. Cancer Res. 2020;80(14):2969–74.
Wellbrock C, Arozarena I. The complexity of the ERK/MAP-kinase pathway and the treatment of melanoma skin cancer. Front Cell Dev Biol. 2016;4:33.
Gao J, Liu X, Yang F, Liu T, Yan Q, Yang X. By inhibiting Ras/Raf/ERK and MMP-9, knockdown of EpCAM inhibits breast cancer cell growth and metastasis. Oncotarget. 2015;6(29):27187.
Yang J, Isaji T, Zhang G, Qi F, Duan C, Fukuda T, et al. EpCAM associates with integrin and regulates cell adhesion in cancer cells. Biochem Biophys Res Commun. 2020;522(4):903–9.
Sankpal NV, Fleming TP, Sharma PK, Wiedner HJ, Gillanders WE. A double-negative feedback loop between EpCAM and ERK contributes to the regulation of epithelial–mesenchymal transition in cancer. Oncogene. 2017;36(26):3706–17.
Matsumura A, Kubota T, Taiyoh H, Fujiwara H, Okamoto K, Ichikawa D, et al. HGF regulates VEGF expression via the c-Met receptor downstream pathways, PI3K/Akt, MAPK and STAT3, in CT26 murine cells. Int J Oncol. 2013;42(2):535–42.
Abounader R, Ranganathan S, Kim B, Nichols C, Laterra J. Signaling pathways in the induction of c-met receptor expression by its ligand scatter factor/hepatocyte growth factor in human glioblastoma. J Neurochem. 2001;76(5):1497–508.
Tulasne D, Bori T, Watson SP. Regulation of RAS in human platelets: evidence that activation of RAS is not sufficient to lead to ERK1-2 phosphorylation. Eur J Biochem. 2002;269(5):1511–7.
Paumelle R, Tulashe D, Kherrouche Z, Plaza S, Leroy C, Reveneau S, et al. Hepatocyte growth factor/scatter factor activates the ETS1 transcription factor by a RAS-RAF-MEK-ERK signaling pathway. Oncogene. 2002;21(15):2309–19.
Pluen A, Boucher Y, Ramanujan S, McKee TD, Gohongi T, Di Tomaso E, et al. Role of tumor–host interactions in interstitial diffusion of macromolecules: cranial vs. subcutaneous tumors. Proc Natl Acad Sci. 2001;98(8):4628–33.
Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53(1):615–27.
Pavlopoulou A, Oktay Y, Vougas K, Louka M, Vorgias CE, Georgakilas AG. Determinants of resistance to chemotherapy and ionizing radiation in breast cancer stem cells. Cancer Lett. 2016;380(2):485–93.
Shaked Y, Henke E, Roodhart JM, Mancuso P, Langenberg MH, Colleoni M, et al. Rapid chemotherapy-induced acute endothelial progenitor cell mobilization: implications for antiangiogenic drugs as chemosensitizing agents. Cancer Cell. 2008;14(3):263–73.
Goldie J. A mathematical model for relating the drug sensitivity of tumor to their spontaneous mutation rate. Cancer Treat Rep. 1979;63:172–84.
Concin N, Hofstetter G, Berger A, Gehmacher A, Reimer D, Watrowski R, et al. Clinical relevance of dominant-negative p73 isoforms for responsiveness to chemotherapy and survival in ovarian cancer: evidence for a crucial p53–p73 cross-talk in vivo. Clin Cancer Res. 2005;11(23):8372–83.
Anelli A, Brentani R, Gadelha A, De Albuquerque AA, Soares F. Correlation of p53 status with outcome of neoadjuvant chemotherapy using paclitaxel and doxorubicin in stage IIIB breast cancer. Ann Oncol. 2003;14(3):428–32.
Gasco M, Bergamaschi D, Sullivan A, Hiller L, Trigiante G, Merlano M, et al. p53 polymorphism predicts outcome in advanced head and neck cancer. Ann Oncol. 2003;14:222–31.
Lievre A, Bachet J-B, Le Corre D, Boige V, Landi B, Emile J-F, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Can Res. 2006;66(8):3992–5.
Aviel-Ronen S, Blackhall FH, Shepherd FA, Tsao M-S. K-ras mutations in non-small-cell lung carcinoma: a review. Clin Lung Cancer. 2006;8(1):30–8.
Bertino J, Carman M, Weiner H, Cashmore A, Moroson B, Srimatkandada S, et al. Gene amplification and altered enzymes as mechanisms for the development of drug resistance. Cancer Treat Rep. 1983;67(10):901–4.
Carlson RW, Moench SJ, Hammond MEH, Perez EA, Burstein HJ, Allred DC, et al. HER2 testing in breast cancer: NCCN Task Force report and recommendations. J Natl Compr Cancer Netw. 2006;4(S3):S-1-S-22.
Huff LM, Lee J-S, Robey RW, Fojo T. Characterization of gene rearrangements leading to activation of MDR-1. J Biol Chem. 2006;281(48):36501–9.
Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000;343(19):1350–4.
Keller L, Werner S, Pantel K. Biology and clinical relevance of EpCAM. Cell Stress. 2019;3(6):165.
Zhou F, Qi Y, Xu H, Wang Q, Gao X, Guo H. Expression of EpCAM and Wnt/β-catenin in human colon cancer. Genet Mol Res. 2015;14(2):4485–94.
Münz M, Murr A, Kvesic M, Rau D, Mangold S, Pflanz S, et al. Side-by-side analysis of five clinically tested anti-EpCAM monoclonal antibodies. Cancer Cell Int. 2010;10(1):44.
Macdonald J, Henri J, Roy K, Hays E, Bauer M, Veedu RN, et al. EpCAM immunotherapy versus specific targeted delivery of drugs. Cancers. 2018;10(1):19.
Eyvazi S, Farajnia S, Dastmalchi S, Kanipour F, Zarredar H, Bandehpour M. Antibody based EpCAM targeted therapy of cancer, review and update. Curr Cancer Drug Targets. 2018;18(9):857–68.
Ko B, He T, Gadgeel S, Halmos B. MET/HGF pathway activation as a paradigm of resistance to targeted therapies. Ann Transl Med. 2017;5(1):4.
Kim KH, Seol HJ, Kim EH, Rheey J, Jin HJ, Lee Y, et al. Wnt/β-catenin signaling is a key downstream mediator of MET signaling in glioblastoma stem cells. Neuro Oncol. 2013;15(2):161–71.
Moradi-Kalbolandi S, Hosseinzade A, Salehi M, Merikhian P, Farahmand L. Monoclonal antibody-based therapeutics, targeting the epidermal growth factor receptor family: from herceptin to Pan HER. J Pharm Pharmacol. 2018;70(7):841–54.
Tong M, Gao M, Xu Y, Fu L, Li Y, Bao X, et al. SHR-A1403, a novel c-mesenchymal–epithelial transition factor (c-Met) antibody-drug conjugate, overcomes AZD9291 resistance in non-small cell lung cancer cells overexpressing c-Met. Cancer Sci. 2019;110(11):3584.
Patnaik A, Gordon M, Tsai F, Papadopoulous K, Rasco D, Beeram SM, et al. A phase I study of LY3164530, a bispecific antibody targeting MET and EGFR, in patients with advanced or metastatic cancer. Cancer Chemother Pharmacol. 2018;82(3):407–18.
Münz M, Kieu C, Mack B, Schmitt B, Zeidler R, Gires O. The carcinoma-associated antigen EpCAM upregulates c-myc and induces cell proliferation. Oncogene. 2004;23(34):5748–58.
Park SY, Bae JS, Cha EJ, Chu HH, Sohn JS, Moon WS. Nuclear EpICD expression and its role in hepatocellular carcinoma. Oncol Rep. 2016;36(1):197–204.
Shen A, Wang L, Huang M, Sun J, Chen Y, Shen Y-Y, et al. c-Myc alterations confer therapeutic response and acquired resistance to c-Met inhibitors in MET-addicted cancers. Can Res. 2015;75(21):4548–59.
Sadeghi S, Hojati Z, Tabatabaeian H. Cooverexpression of EpCAM and c-myc genes in malignant breast tumours. J Genet. 2017;96(1):109–18.
Jeon H-M, Lee J. MET: roles in epithelial–mesenchymal transition and cancer stemness. Ann Transl Med. 2017;5(1):5.
Whang YM, Jung SP, Kim M-K, Chang IH, Park SI. Targeting the hepatocyte growth factor and c-Met signaling axis in bone metastases. Int J Mol Sci. 2019;20(2):384.
Bladt F, Faden B, Friese-Hamim M, Knuehl C, Wilm C, Fittschen C, et al. EMD 1214063 and EMD 1204831 constitute a new class of potent and highly selective c-Met inhibitors. Clin Cancer Res. 2013;19(11):2941–51.
Egile C, Kenigsberg M, Delaisi C, Bégassat F, Do-Vale V, Mestadier J, et al. The selective intravenous inhibitor of the MET tyrosine kinase SAR125844 inhibits tumor growth in MET-amplified cancer. Mol Cancer Ther. 2015;14(2):384–94.
Gavine PR, Ren Y, Han L, Lv J, Fan S, Zhang W, et al. Volitinib, a potent and highly selective c-Met inhibitor, effectively blocks c-Met signaling and growth in c-MET amplified gastric cancer patient-derived tumor xenograft models. Mol Oncol. 2015;9(1):323–33.
Luo T, Zhang S-G, Zhu L-F, Zhang F-X, Li W, Zhao K, et al. A selective c-Met and Trks inhibitor Indo5 suppresses hepatocellular carcinoma growth. J Exp Clin Cancer Res. 2019;38(1):130.
Du Z, Caenepeel S, Shen Y, Rex K, Zhang Y, He Y, et al. Preclinical evaluation of AMG 337, a highly selective small molecule MET inhibitor, in hepatocellular carcinoma. Mol Cancer Ther. 2016;15(6):1227–37.
Schuller AG, Barry ER, Jones RD, Henry RE, Frigault MM, Beran G, et al. The MET inhibitor AZD6094 (savolitinib, HMPL-504) induces regression in papillary renal cell carcinoma patient-derived xenograft models. Clin Cancer Res. 2015;21(12):2811–9.
Miranda O, Farooqui M, Siegfried JM. Status of agents targeting the HGF/c-Met axis in lung cancer. Cancers. 2018;10(9):280.
Engstrom LD, Aranda R, Lee M, Tovar EA, Essenburg CJ, Madaj Z, et al. Glesatinib exhibits antitumor activity in lung cancer models and patients harboring MET exon 14 mutations and overcomes mutation-mediated resistance to type I MET inhibitors in nonclinical models. Clin Cancer Res. 2017;23(21):6661–72.
Leighl NB, Tsao M-S, Liu G, Tu D, Ho C, Shepherd FA, et al. A phase I study of foretinib plus erlotinib in patients with previously treated advanced non-small cell lung cancer: Canadian cancer trials group IND. 196. Oncotarget. 2017;8(41):69651.
Wang H, Rao B, Lou J, Li J, Liu Z, Li A, et al. The function of the HGF/c-Met axis in hepatocellular carcinoma. Front Cell Dev Biol. 2020;8:55.
Kwak EL, Bang Y-J, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363(18):1693–703.
Bouattour M, Raymond E, Qin S, Cheng AL, Stammberger U, Locatelli G, et al. Recent developments of c-Met as a therapeutic target in hepatocellular carcinoma. Hepatology. 2018;67(3):1132–49.
Michieli P, Mazzone M, Basilico C, Cavassa S, Sottile A, Naldini L, et al. Targeting the tumor and its microenvironment by a dual-function decoy Met receptor. Cancer Cell. 2004;6(1):61–73.
Aftimos PG, Barthelemy P, Rolfo CD, Hanssens V, De Jonge N, Silence K, et al. A phase I, first-in-human study of argx-111, a monoclonal antibody targeting c-met in patients with solid tumors. Am Soc Clin Oncol. 2015. https://doi.org/10.1200/jco.2015.33.15_suppl.2580.
Petrelli A, Circosta P, Granziero L, Mazzone M, Pisacane A, Fenoglio S, et al. Ab-induced ectodomain shedding mediates hepatocyte growth factor receptor down-regulation and hampers biological activity. Proc Natl Acad Sci. 2006;103(13):5090–5.
Lee B, Kang S, Kim K, Song Y, Cheong K, Cha H, et al. Met degradation by SAIT301, a Met monoclonal antibody, reduces the invasion and migration of nasopharyngeal cancer cells via inhibition of EGR-1 expression. Cell Death Dis. 2014;5(4): e1159.
Kim K, Hur Y, Ryu E-K, Rhim J-H, Choi CY, Baek C-M, et al. A neutralizable epitope is induced on HGF upon its interaction with its receptor cMet. Biochem Biophys Res Commun. 2007;354(1):115–21.
Cignetto S, Modica C, Chiriaco C, Fontani L, Milla P, Michieli P, et al. Dual constant domain-Fab: a novel strategy to improve half-life and potency of a Met therapeutic antibody. Mol Oncol. 2016;10(6):938–48.
Fiedler U, Ekawardhani S, Cornelius A, Gilboy P, Bakker TR, Dolado I, et al. MP0250, a VEGF and HGF neutralizing DARPin® molecule shows high anti-tumor efficacy in mouse xenograft and patient-derived tumor models. Oncotarget. 2017;8(58):98371.
Liu L, Zeng W, Wortinger MA, Yan SB, Cornwell P, Peek VL, et al. LY2875358, a neutralizing and internalizing anti-MET bivalent antibody, inhibits HGF-dependent and HGF-independent MET activation and tumor growth. Clin Cancer Res. 2014;20(23):6059–70.
Jarantow SW, Bushey BS, Pardinas JR, Boakye K, Lacy ER, Sanders R, et al. Impact of cell-surface antigen expression on target engagement and function of an epidermal growth factor receptor× c-MET bispecific antibody. J Biol Chem. 2015;290(41):24689–704.
Emdal KB, Dittmann A, Reddy RJ, Lescarbeau RS, Moores SL, Laquerre S, et al. Characterization of in vivo resistance to osimertinib and JNJ-61186372, an EGFR/Met bispecific antibody, reveals unique and consensus mechanisms of resistance. Mol Cancer Ther. 2017;16(11):2572–85.
Tarhini AA, Rafique I, Floros T, Tran P, Gooding WE, Villaruz LC, et al. Phase 1/2 study of rilotumumab (AMG 102), a hepatocyte growth factor inhibitor, and erlotinib in patients with advanced non-small cell lung cancer. Cancer. 2017;123(15):2936–44.
Iveson T, Donehower RC, Davidenko I, Tjulandin S, Deptala A, Harrison M, et al. Rilotumumab in combination with epirubicin, cisplatin, and capecitabine as first-line treatment for gastric or oesophagogastric junction adenocarcinoma: an open-label, dose de-escalation phase 1b study and a double-blind, randomised phase 2 study. Lancet Oncol. 2014;15(9):1007–18.
Yousefi H, Maheronnaghsh M, Molaei F, Mashouri L, Aref AR, Momeny M, et al. Long noncoding RNAs and exosomal lncRNAs: classification, and mechanisms in breast cancer metastasis and drug resistance. Oncogene. 2019;39(5):953–74.
Ji C, Liu H, Yin Q, Li H, Gao H. miR-93 enhances hepatocellular carcinoma invasion and metastasis by EMT via targeting PDCD4. Biotech Lett. 2017;39(11):1621–9.
Yang X, Zhang XF, Lu X, Jia HL, Liang L, Dong QZ, et al. MicroRNA-26a suppresses angiogenesis in human hepatocellular carcinoma by targeting hepatocyte growth factor-cMet pathway. Hepatology. 2014;59(5):1874–85.
Wu H, Tao J, Li X, Zhang T, Zhao L, Wang Y, et al. MicroRNA-206 prevents the pathogenesis of hepatocellular carcinoma by modulating expression of met proto-oncogene and cyclin-dependent kinase 6 in mice. Hepatology. 2017;66(6):1952–67.
Liu Y, Tan J, Ou S, Chen J, Chen L. MicroRNA-101-3p suppresses proliferation and migration in hepatocellular carcinoma by targeting the HGF/c-Met pathway. Invest New Drugs. 2020;38(1):60–9.
van der Gun BT, Melchers LJ, Ruiters MH, de Leij LF, McLaughlin PM, Rots MG. EpCAM in carcinogenesis: the good, the bad or the ugly. Carcinogenesis. 2010;31(11):1913–21.
Amann M, Friedrich M, Lutterbuese P, Vieser E, Lorenczewski G, Petersen L, et al. Therapeutic window of an EpCAM/CD3-specific BiTE antibody in mice is determined by a subpopulation of EpCAM-expressing lymphocytes that is absent in humans. Cancer Immunol Immunother. 2009;58(1):95.
Hamzehlou S, Momeny M, Zandi Z, Kashani B, Yousefi H, Dehpour AR, et al. Anti-tumor activity of neratinib, a pan-HER inhibitor, in gastric adenocarcinoma cells. Eur J Pharmacol. 2019;863: 172705.
Gholami MD, Falak R, Heidari S, Khoshmirsafa M, Kazemi MH, Zarnani A-H, et al. A truncated snail1 transcription factor alters the expression of essential EMT markers and suppresses tumor cell migration in a human lung cancer cell line. Recent Pat Anti-Cancer Drug Discov. 2019;14(2):158–69.
Shaw DR, Khazaeli M, Sun L, Ghrayeb J, Daddona PE, McKinney S, et al. Characterization of a mouse/human chimeric monoclonal antibody (17-1A) to a colon cancer tumor-associated antigen. J Immunol. 1987;138(12):4534–8.
Göitlinger HG, Funke I, Johnson JP, Gokel JM, Riethmüller G. The epithelial cell surface antigen 17-1A, a target for antibody-mediated tumor therapy: its biochemical nature, tissue distribution and recognition by different monoclonal antibodies. Int J Cancer. 1986;38(1):47–53.
Adkins JC, Spencer CM. Edrecolomab (monoclonal antibody 17-1A). Drugs. 1998;56(4):619–26.
Fields AL, Keller A, Schwartzberg L, Bernard S, Kardinal C, Cohen A, et al. Adjuvant therapy with the monoclonal antibody edrecolomab plus fluorouracil-based therapy does not improve overall survival of patients with stage III colon cancer. J Clin Oncol. 2009;27(12):1941–7.
Riethmüller G, Gruber R, Schneider-Gädicke E, Schlimok G, Witte J, Raab R, et al. Randomised trial of monoclonal antibody for adjuvant therapy of resected Dukes’ C colorectal carcinoma. Lancet. 1994;343(8907):1177–83.
Schmidt M, Scheulen ME, Dittrich C, Obrist P, Marschner N, Dirix L, et al. An open-label, randomized phase II study of adecatumumab, a fully human anti-EpCAM antibody, as monotherapy in patients with metastatic breast cancer. Ann Oncol. 2010;21(2):275–82.
Seimetz D, Lindhofer H, Bokemeyer C. Development and approval of the trifunctional antibody catumaxomab (anti-EpCAM× anti-CD3) as a targeted cancer immunotherapy. Cancer Treat Rev. 2010;36(6):458–67.
Yuraszeck T, Kasichayanula S, Benjamin J. Translation and clinical development of bispecific T-cell engaging antibodies for cancer treatment. Clin Pharmacol Ther. 2017;101(5):634–45.
Zeidler R, Reisbach G, Wollenberg B, Lang S, Chaubal S, Schmitt B, et al. Simultaneous activation of T cells and accessory cells by a new class of intact bispecific antibody results in efficient tumor cell killing. J Immunol. 1999;163(3):1246–52.
Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol. 2015;93(3):290–6.
Connor JP, Cristea MC, Lewis NL, Lewis LD, Komarnitsky PB, Mattiacci MR, et al. A phase 1b study of humanized KS-interleukin-2 (huKS-IL2) immunocytokine with cyclophosphamide in patients with EpCAM-positive advanced solid tumors. BMC Cancer. 2013;13(1):20.
Hajifathali A, Parkhideh S, Kazemi MH, Chegeni R, Roshandel E, Gholizadeh M. Immune checkpoints in hematologic malignancies: what made the immune cells and clinicians exhausted! J Cell Physiol. 2020;235(12):9080–97.
Hamilton GS. Antibody-drug conjugates for cancer therapy: the technological and regulatory challenges of developing drug-biologic hybrids. Biologicals. 2015;43(5):318–32.
Sievers EL, Senter PD. Antibody-drug conjugates in cancer therapy. Annu Rev Med. 2013;64:15–29.
Moldenhauer G, Salnikov AV, Lüttgau S, Herr I, Anderl J, Faulstich H. Therapeutic potential of amanitin-conjugated anti-epithelial cell adhesion molecule monoclonal antibody against pancreatic carcinoma. J Natl Cancer Inst. 2012;104(8):622–34.
Entwistle J, Kowalski M, Brown J, Cizeau J, MacDonald GC. The preclinical and clinical evaluation of VB6-845: an immunotoxin with a de-immunized payload for the systemic treatment of solid tumors. In: Antibody-drug conjugates and immunotoxins. New York: Springer; 2013. p. 349–67.
Cizeau J, Grenkow DM, Brown JG, Entwistle J, MacDonald GC. Engineering and biological characterization of VB6-845, an anti-EpCAM immunotoxin containing a T-cell epitope-depleted variant of the plant toxin bouganin. J Immunother. 2009;32(6):574–84.
Kowalski M, Entwistle J, Cizeau J, Niforos D, Loewen S, Chapman W, et al. A phase I study of an intravesically administered immunotoxin targeting EpCAM for the treatment of nonmuscle-invasive bladder cancer in BCGrefractory and BCG-intolerant patients. Drug Des Dev Ther. 2010;4:313.
Samadi M, Majidzadeh-A K, Salehi M, Jalili N, Noorinejad Z, Mosayebzadeh M, et al. Engineered hypoxia-responding Escherichia coli carrying cardiac peptide genes, suppresses tumor growth, angiogenesis and metastasis in vivo. J Biol Eng. 2021;15(1):1–15.
Iwasaki K, Goto Y, Katoh T, Yamashita T, Kaneko S, Suga H. A fluorescent imaging probe based on a macrocyclic scaffold that binds to cellular EpCAM. J Mol Evol. 2015;81(5–6):210–7.
Leenheer D, ten Dijke P, Hipolito CJ. A current perspective on applications of macrocyclic-peptide-based high-affinity ligands. Pept Sci. 2016;106(6):889–900.
Zhou J, Rossi J. Aptamers as targeted therapeutics: current potential and challenges. Nat Rev Drug Discov. 2017;16(3):181.
de Almeida CE, Alves LN, Rocha HF, Cabral-Neto JB, Missailidis S. Aptamer delivery of siRNA, radiopharmaceutics and chemotherapy agents in cancer. Int J Pharm. 2017;525(2):334–42.
Xiang D, Shigdar S, Qiao G, Wang T, Kouzani AZ, Zhou S-F, et al. Nucleic acid aptamer-guided cancer therapeutics and diagnostics: the next generation of cancer medicine. Theranostics. 2015;5(1):23.
Shigdar S, Ward AC, De A, Yang CJ, Wei M, Duan W. Clinical applications of aptamers and nucleic acid therapeutics in haematological malignancies. Br J Haematol. 2011;155(1):3–13.
Shigdar S, Macdonald J, O’Connor M, Wang T, Xiang D, Qiao L, et al. Aptamers as theranostic agents: modifications, serum stability and functionalisation. Sensors. 2013;13(10):13624–37.
Xiang D, Zheng C, Zhou S-F, Qiao S, Tran PH-L, Pu C, et al. Superior performance of aptamer in tumor penetration over antibody: implication of aptamer-based theranostics in solid tumors. Theranostics. 2015;5(10):1083.
Xiang D, Shigdar S, Bean AG, Bruce M, Yang W, Mathesh M, et al. Transforming doxorubicin into a cancer stem cell killer via EpCAM aptamer-mediated delivery. Theranostics. 2017;7(17):4071.
Wiemer EA. The role of microRNAs in cancer: no small matter. Eur J Cancer. 2007;43(10):1529–44.
Beta M, Khetan V, Chatterjee N, Suganeswari G, Rishi P, Biswas J, et al. EpCAM knockdown alters microRNA expression in retinoblastoma-functional implication of EpCAM regulated miRNA in tumor progression. PLoS ONE. 2014;9(12): e114800.
Qian N-S, Liu W-H, Lv W-P, Xiang X, Su M, Raut V, et al. Upregulated microRNA-92b regulates the differentiation and proliferation of EpCAM-positive fetal liver cells by targeting C/EBPß. PLoS ONE. 2013;8(8): e68004.
Abbas AK, Lichtman AH, Pillai S. Cellular and molecular immunology. 7th ed. Philadelphia: Elsevier Health Sciences; 2012.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
Study concepts: LF; study design: KB; data acquisition: LF; drawing figure(s): RV, AH, PM; quality control of data and algorithms: LF; manuscript preparation: KB, RV, MHK, AH, PM, AH, HD; manuscript editing: KB, RV; manuscript review: LF. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Barzaman, K., Vafaei, R., Samadi, M. et al. Anti-cancer therapeutic strategies based on HGF/MET, EpCAM, and tumor-stromal cross talk. Cancer Cell Int 22, 259 (2022). https://doi.org/10.1186/s12935-022-02658-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-022-02658-z