
Abstract
Coenzyme Q10 (CoQ10), a benzoquinone present in most organisms, plays an important role in the electron-transport chain, and its deficiency is associated with various neuropathies and muscular disorders. CoQ10 is the only lipid-soluble antioxidant found in humans, and for this, it is gaining popularity in the cosmetic and healthcare industries. To meet the growing demand for CoQ10, there has been considerable interest in ways to enhance its production, the most effective of which remains microbial fermentation. Previous attempts to increase CoQ10 production to an industrial scale have thus far conformed to the strategies used in typical metabolic engineering endeavors. However, the emergence of new tools in the expanding field of synthetic biology has provided a suite of possibilities that extend beyond the traditional modes of metabolic engineering. In this review, we cover the various strategies currently undertaken to upscale CoQ10 production, and discuss some of the potential novel areas for future research.
Similar content being viewed by others
Background
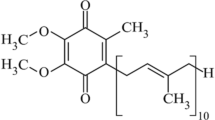
Coenzyme Q, commonly known as ubiquinone or CoQ, is a lipid-soluble, powerful antioxidant, and an essential cofactor in mitochondrial oxidative phosphorylation [1–3]. Coenzyme Q is species specific, with differences dictated by the number of isoprenyl units on the isoprenoid side chain. For example, 10 isoprenyl units are found in human and the fission yeast Schizosaccharomyces pombe but fewer units are found in other species (CoQ8 in Escherichia coli, CoQ9 in Arabidopsis thaliana, and CoQ6 in Saccharomyces cerevisiae) [1]. The isoprenoid side chain is responsible for the lipid-soluble nature of CoQ, whereas its antioxidant capacity derives from its quinone head, which can enable electron transfer (Fig. 1). Because of this electron-sequestering property, CoQ10 acts as an antioxidant at cellular membranes to counteract the oxidation of lipids or lipoproteins [4]. CoQ10 has roles in other physiological processes, including sulfide oxidation, regulating the mitochondrial permeability transition pore, and in the translocation of protons and Ca2+ across biological membranes [5, 6]. A detailed account of the various aspects of CoQ biosynthesis have been described at length elsewhere [1–6].

Chemical structure of coenzyme Q10. This molecule consists of a isoprenoid side chain composed of ten tandemly linked isoprenyl groups attached to a quinone head group
CoQ10 is the only lipid-soluble antioxidant produced by humans, and it localizes to almost every membrane, ranging from mitochondrial membranes to that of very low density lipoproteins (VLDL) [7]. This solubility means that CoQ can protect lipoproteins and lipids from peroxidation and oxidative damage [8]. CoQ10 also serves alongside other antioxidants, such as vitamins C and E, to combat free-radical damage arising from energetic mitochondrial reactions [9, 10]. Given its myriad functions and physiological importance, it is not surprising that CoQ deficiency can result in numerous diseases.
In model organisms, such as S. cerevisiae and S. pombe, CoQ deficiency is not lethal but results in growth defects on minimum medium, and a heightened sensitivity to oxidative stress [11–15]. In Caenorhabditis elegans, CoQ deficiency leads to GABA neuron degeneration, and in Drosophila melanogaster, it can cause mitochondrial stress and neuronal apoptosis [16, 17]. In humans, CoQ10 deficiency has been implicated in various diseases involving muscle and neural development, with the severity of the disease correlated with the acuteness of the CoQ10 shortfall [18]. These diseases may manifest in conditions such as central nervous system (CNS) dysfunction, myopathy or cardiomyopathy, among others [19–22]. The oxidative damage associated with impaired CoQ10 function has also been implicated in numerous clinical phenotypes [18, 23–25].
By virtue of its therapeutic relevance, CoQ10 is of particular importance in the biomedical and health supplement scene. Oral CoQ10 supplements are often prescribed alongside treatments for various diseases [26]. One example is its co-administration with HMG-CoA (3-hydroxy-3-methylglutaryl-coenzyme A) reductase (HMGR) inhibitors, widely used cholesterol-lowering drugs otherwise known as statins. HMGR catalyzes the formation of mevalonic acid, the precursor for cholesterol and CoQ10 biosyntheses [27]. Patients using statins show lower blood levels of CoQ10, and this justifies the need for CoQ10 supplementation to reduce the cardiomyopathy risk associated with statin use [27–30]. The presence of CoQ10 is however implicated in resistance to chemotherapeutic drugs, and this calls for caution in administering CoQ10 alongside certain agents [31, 32].
CoQ10 production decreases with aging [33], as does the antioxidant capability of the cell. Increased oxidative stress in aging cells may be ameliorated with dietary supplementation of CoQ10 [34]. Indeed, CoQ10 has garnered great popularity as an antioxidant in moisturizers, anti-wrinkle and anti-aging skin care treatments [35–37]. With the growing demand for skin care cosmetics and public awareness of the importance of antioxidants, we will likely see an increase in the demand for CoQ10 products on the market quite quickly [38]. Given that CoQ10 is endogenously synthesized, there should be fewer unwanted side effects from its therapeutic use as compared with other synthetic compounds, and this has been supported by tolerability studies for high CoQ10 doses [39]. Hence, attention has surged in the therapeutic use of CoQ10 in non-curable diseases challenging modern societies including Alzheimer’s, Huntington’s and Parkinson’s, and cardiovascular diseases [40–42].
Industrial production of CoQ10
The range of uses for CoQ10 across the pharmaceutical and cosmetics industries has meant that there is great commercial interest to scale up the production of CoQ10. Frederick Crane first isolated CoQ10 from a bovine heart source in the late 1950s [43]. Since then, industrial attempts to produce CoQ10 have centered on animal tissue extraction, semi-chemical synthesis, and microbial fermentation [44, 45].
The chemical synthesis of CoQ10 has typically involved solanesol as a starting substrate and the source of the isoprenoid tail, and this is carried out before it is combined with the quinone head [46]. However, as with most chemical processes, there are numerous costs associated with such high-energy catalysis reactions because of the need for expensive substrates and because of the significant chemical waste generated from its production [47–49]. The chemical synthesis of CoQ10 also lacks stereoselectivity, and this makes it difficult to separate optical isomers to obtain the all-trans biologically viable isomer [50].
Owing to these difficulties, microbial biosynthesis has become a preferred avenue of CoQ10 production. The cell-based catalysis of compounds does not require harsh catalytic conditions of heat and pressure that typify many chemical synthesis processes. Furthermore, the production costs tend to be lower, cheap growth media provides an appropriate substrate, and expensive co-substrates can be recycled [48, 51]. A living cellular system is also scalable, and the precision of the cellular catalytic machinery circumvents the problems of stereoselectivity [52, 53]. Furthermore, unlike with chemical processing, altered genetics does not significantly affect the operating costs, meaning that the efforts associated with constructing a high-titer-producing organism are worthwhile. Through microbial biosynthesis, metabolic engineering approaches can be used to increase the titer of CoQ10 and overcome some of the limiting steps along the biosynthetic pathway.
Metabolic engineering approaches initially used chemical mutagenesis-based selection and chemical engineering procedures that centered on manipulating substrate flux; however, the field has since expanded to include other strategies from a genetics standpoint [48, 54]. The process varies depending on promoter choice and strength, cassette copy number, and the localization or tethering of enzymes to scaffolds [55, 56]. The choice of cassette and promoter are typically host dependent, given that promoter strength and usability rely on a species-specific genetic environment and functionality. Furthermore, enzymes involved in the tail end of CoQ10 production are localized in the mitochondria, leading to models that propose the involvement of a membrane-bound complex containing multiple polypeptides of the CoQ10 biosynthesis enzymes [57].
Improving flux remains one of the most straightforward methods to increase yield [48, 58]. Typically, this involves finding and circumventing rate-limiting steps in biochemical pathways and then employing strong promoters to increase the expression of key pathway genes to direct biochemical flux. A parallel option entails knocking down the expression of genes in alternate pathways that branch off the pathway of interest, and this can be concomitantly administered, with care taken to ensure that these manipulations do not undermine cellular viability and robustness. Alleviating chemical bottlenecks that might hamper the production of the desired compound can also be achieved by including genes that reconstitute cofactors, such as NADPH and S-adenosyl methionine (SAM). These cofactors play essential roles in numerous biochemical pathways [54, 59]. Overall, it is clear that close scrutiny and careful optimization of biosynthetic pathways can optimize and direct the metabolic flux.
Biosynthesis of CoQ10
Entry points to CoQ10 biosynthesis
CoQ biosynthesis involves discrete synthetic stages: production of the aromatic group that forms the quinone head, production of the isoprene tail, attachment of the quinone head to the isoprene tail, and the subsequent steps that culminate in the formation of the final CoQ10 product [1, 60]. In yeast, mitochondria are responsible for CoQ synthesis. However, in humans, both mitochondria and Golgi apparatus are proposed sites for CoQ synthesis. The chemical precursors for both the quinone head and isoprene tail are organism specific. The quinone head is derived from the chorismate precursor in the shikimate pathway in prokaryotes but from tyrosine in higher eukaryotes (Fig. 2). The isoprene tail derives from MEP (2-C-methyl-d-erythritol 4-phosphate) in prokaryotes and plant plastids, which stems from glyceraldehyde 3-phosphate (G3P), whereas, in eukaryotes, the tail is produced from acetyl-CoA in the mevalonate pathway [2, 61]. These multiple entry points into the pathway could be exploited to optimize flux for yield improvement.

Biosynthesis of coenzyme Q10. Schematic showing the pathway of various metabolic precursors leading to the formation of the quinone head (PHB), the isoprene tail (decaprenyl diphosphate), and the final Coenzyme Q product. Reflected in red are the various enzymatic steps that are rate limiting. UbiC and UbiA are specific genes from E. coli, and Coq2 is from S. cerevisiae. Unlabelled arrows between chorismate and tyrosine and PHB; FPP and decaprenyl diphosphate; and decaprenyl-4-hydrobenzoic acid and coenzyme Q10 denote the presence of multiple steps that have been abbreviated
The engineering concept of ‘push and pull’ to divert metabolic flux implicates that both the inflow and outflow reactions must be increased synchronously, otherwise an accumulation of one product will limit the flux and cause an imbalance in the system. Therefore, it is crucial to understand the species-specific biosynthetic pathways that lead to CoQ10 production. There are several biosynthetic pathways of concern, each of which we will address separately.
Rate-limiting steps in biosynthesis of the isoprenoid chain
The first pathway provides the precursors for synthesizing the isoprene tail; if using a prokaryotic system, this is achieved through the MEP pathway. The MEP pathway starts with the interaction between G3P and pyruvate to form 1-deoxy-d-xylulose 5-phosphate (DXP) (Fig. 2), which is reported to be the major limiting step in the formation of the isoprene tail [62]. Indeed, efforts to increase the prokaryotic expression of carotenoids (which share the isoprenoid precursor pathway of MEP) have focused on improving the first catalytic step of DXP formation. Under such contexts, 1-deoxy-d-xylulose-5-phosphate synthase (DXS) and 1-deoxy-d-xylulose 5-phosphate reductoisomerase (DXR) are typically overexpressed to improve the catalytic formation of DXP and its subsequent conversion to MEP [60]. These reactions eventually yield isopentenyl diphosphate (IPP), which is used to initiate isoprene chain elongation in the isoprenoid pathway. Similar efforts can be co-opted for the production of CoQ [63].
Conversely, in the eukaryotic platform, the mevalonate pathway begins with acetyl-CoA and ends with the similar production of IPP (Fig. 2). Midway through the pathway is the catalysis of HMG-CoA to mevalonate by HMGR, the target of statins. Unlike with statins, however, which seek to reduce HMGR activity, here aiming to increase its activity instead, so as to increase flux to the IPP pathway. Indeed, the lower Km values of the downstream IPP pathway enzymes (farnesyl transferase and geranylgeranyl transferase) imply that the enzymatic reactions catalyzed (by enzymes including farnesyl and geranylgeranyl transferases) will reach saturation before that of HMG-CoA [7, 64]. This concept of exploiting HMGR for increased metabolic production is common; for example, a truncated HMGR lacking its inhibitory site can delay enzyme saturation [65, 66]. Regardless of the pathway source, downstream signaling leads to IPP and its isomer dimethylallyl diphosphate (DMAPP) (Fig. 2). IPP and DMAPP combine to form geranyl diphosphate (GPP), and this compound is sequentially lengthened by additional IPP moieties to form farnesyl diphosphate (FPP), geranylgeranyl diphosphate (GGPP), and the subsequent n-isoprene tail [61]. Depending on the host organism, components of the IPP pathway are also crucial branch points for several important compounds, which makes optimization of the isoprenoid pathway a lucrative endeavor (and one that has been done extensively in S. cerevisiae [59]). GPP can branch off and undergo reactions that lead to the formation of monoterpenoids; FPP, likewise, can form steroids and cholesterol; and GGPP can form carotenoids and retinoids before decaprenyl diphosphate [1]. Studies suggest that inhibiting these various branch points could direct metabolic flux from GPP towards decaprenyl diphosphate, as seen in FPP yields through the downregulation of squalene synthase [67].
CoQ10 production rates are thought to be limited by the availability of IPP, since the quinone head is produced from the relatively abundant chorismate or tyrosine [68, 69]. However, the tail length of CoQ, which contains varying numbers of IPP units, may also be rate-limiting. Although CoQ can be produced by multiple microbial platforms, each microbe synthesizes CoQ with a characteristic number of the IPP units. For example, S. cerevisiae and E. coli produce CoQ6 and CoQ8, respectively, whereas S. pombe and humans naturally produce CoQ10 [60]. Evidence shows that polyprenyl diphosphate synthase is the key determinant of IPP chain length, as this enzyme catalyzes polyisoprenoid tail extension [70]. In comparison, the polyprenyl diphosphate:4-HB transferase (UbiA/Coq2), which joins the tail and the quinone head, is promiscuous in terms of its isoprenoid chain length choice [71]. Therefore, any attempts to utilize a heterologous, non-native host to produce CoQ10 would need to optimize or replace the polyprenyl diphosphate synthase to achieve the appropriate tail length (10 isoprene subunits). Many groups have in fact approached this problem by introducing the decaprenyl diphosphate synthase (DPS) gene [72–74].
Rate-limiting steps in biosynthesis of the aromatic quinone group
Another likely avenue to increase metabolic flux is through the optimization of the aromatic quinone core. The precursor that contributes to the head group is 4-hydroxybenzoic acid (PHB or pHBA), which, in prokaryotes [60], forms from the condensation of phosphoenolpyruvate (PEP) and erythrose-4-phosphate, past shikimate, to chorismate and then PHB (Fig. 2) [68]. Chorismate is a branch point metabolite necessary in the formation of folate and aromatic amino acids (tyrosine and phenylalanine) [75]. Thus, it would be advantageous to increase the catalytic conversion of chorismate to PHB for both proper cell growth and metabolic flux [76].
Earlier work has also shown that CoQ production can be increased by the overexpression of chorismate pyruvate lyase (UbiC) in E. coli alongside the overexpression of several key catalytic enzymes that tend to limit CoQ production rates [77]. Similarly, an eightfold increase in CoQ10 was reported in the native producer Sporidiobolus johnsonii [78]. In other organisms, however, the source of PHB differs: mammals produce PHB from tyrosine, whereas yeast and plants use both chorismate and tyrosine (yeast) or a β-oxidation-like mechanism using p-hydroxycinnamic acid (plants) [60, 61]. In these cases, the exogenous addition of PHB can increase CoQ10 production; albeit, production rates are still reliant on the supply of IPP, which is rate-limiting [79, 80].
Rate-limiting steps in condensation of isoprenoid tail to the quinone group
In the final stages, polyprenyl-4-hydroxybenzoate transferase is required to combine the moieties to form the 4-hydroxy-3-polyprenylbenzoate precursor [60, 61, 81]. The isoprene group varies depending on the species, and the ring group undergoes a series of modifications (decarboxylation, hydroxylation and methylation) before the complete CoQ is synthesized. Flux is primarily determined by polyprenyl diphosphate transferase, and its overexpression in E. coli can generate a 3.4-fold increase in CoQ10 production [82]. Conversely, the overexpression of genes involved in ring modification leads to only a minor increase in CoQ10 content in E. coli and S. pombe, even if several genes are overexpressed together (in S. pombe) [83].
Overall, these findings suggest that the bottleneck in CoQ10 production still lies predominantly with IPP flux and is then limited by the quinone head formation and the required transfer steps [84].
Host platforms employed for CoQ10 production
CoQ10 is only native to a few organisms [2, 81] and it remains unknown whether human metabolic reactions can cope with a shorter CoQ [85, 86].
Traditionally, most efforts have focused on native CoQ10 producers, and screening for mutant strains that show higher CoQ10 yields. However, there is great potential in exploiting heterologous hosts armed with extensive toolbox like E. coli and S. cerevisiae into platforms for CoQ10 production. Here, we explore the benefits and disadvantages of both native and non-native producers.
Native producers of CoQ10
Native producers have an advantage over heterologous hosts, as they do not produce any unwanted CoQ species (CoQ8 or CoQ9), which vary by chain length that and are specific to the host. The additional costs required to extract and separate CoQ10 from other shorter-tailed CoQ products may shift the balance in favor of using native producers of the enzyme. Indeed, these other, shorter products will compete for the biochemical flux and affect the yield of the desired CoQ10 [60].
Several native producers of CoQ10 have been identified or optimized as candidates for CoQ10 production, including S. pombe, S. johnsonii, Rhodobacter sphaeroides and Agrobacterium tumefaciens [78, 83, 87, 88]. Several other organisms, including Pseudomonas, Paracoccus bacteria, Candida and Saitoella yeasts also produce CoQ10 natively but have not been sufficiently characterized as producing hosts, and many require the inclusion of expensive constituents in the growth media for proper function. Here, we will explore four of the most feasible native hosts for CoQ10 production: (1) S. pombe, (2) S. johnsonii, (3) R. sphaeroides and (4) A. tumefaciens.
Native producer: Schizosaccharomyces pombe
Schizosaccharomyces pombe (fission yeast) is a well-studied model organism with similar molecular pathway makeup and genetic mechanisms as those in humans [89, 90]. However, little effort has been made to develop S. pombe into a suitable framework for high-value compound production [91], and so efforts to increase CoQ10 in S. pombe have thus far been limited. In one study, genes encoding enzymes directly involved in CoQ10 biosynthesis (dps1 +–dlp1 +, ppt1 +, and coq3 +–coq9 +) and HMGR [83] were overexpressed. However, only overexpression of HMGR—and not the CoQ10 biosynthesis genes—led to a prominent 2.7-fold increase in CoQ10 yield (Table 1). It was posited that the lack of effect from the biosynthetic genes was because these enzymes are not rate-limiting.
More success has been attained in the production of ricinoleic acid, a fatty acid from castor oil in S. pombe [92], and it may be possible to hijack this system to co-produce both CoQ10 and fatty acids, with CoQ10 participating as a lipid-soluble antioxidant to protect polyunsaturated fatty acids (PUFA) against oxidative damage during storage. A similar approach has been explored in Yarrowia lipolytica, an oleaginous yeast, even though Y. lipolitica is a non-native producer of CoQ10, and this approach is currently undergoing approval for production [93]. The approach capitalizes on the same IPP pathway to produce carotenoids, and it has been suggested that this may lead to a reduction in flux and the generation of alterative products that will include CoQ10. Indeed, high CoQ10 selection based on mutant strains of Protomonas extorquens and R. sphaeroides are correlated with low carotenoid production [94].
Native producer: Sporidiobolus johnsonii
Sporidiobolus johnsonii was recently discovered as a natural producer of CoQ10 at 0.8–3.3 mg/g dry cell weight (DCW) (Table 1), which, in an unmodified strain, suggests a great potential as compared with the current top native (A. tumefaciens; 6.92–9.6 mg/g DCW) and heterologous (E. coli; 2.4 mg/g DCW; see below) producers [78, 95]. Efforts to use S. johnsonii as a production host at an industrial level have achieved 10 mg/g DCW; albeit, this yield involved exogenous PHB in the media [78]. Other mutagenesis attempts led to a mutant UF16 strain with 7.4 mg/g DCW [96].
Native producer: Rhodobacter sphaeroides
Rhodobacter sphaeroides is a photosynthetic bacterium [97] initially selected by screening mutant strains based on color change, which indicated a reduction in carotenoid production, and thus, by correlation, an increase in CoQ10 [94]. Promoter-based balancing of metabolic flux increased the production to 7.16–8.7 mg/g DCW [60, 98], and a recent study reported production as high as 12.96 mg/g DCW [87] (Table 1). However, other efforts to increase MEP pathway flux did not translate well into increased CoQ10 production, probably due to an accumulation of toxic intermediates [99]. R. sphaeroides, however, is reported to have limited growth rates, even when grown in optimal fermentation conditions [84]. This, coupled with other difficulties (such as requiring anaerobic and light conditions to produce higher CoQ10 titers) makes R. sphaeroides a less ideal host choice [100, 101].
Native producer: Agrobacterium tumefaciens
Agrobacterium tumefaciens is a Gram-negative bacterium that is widely used as a transmission vector tool for plant genetic modification [102]. Besides R. sphaeroides, it is one of the top producers of CoQ10 at 6.92–9.6 mg/g DCW [61, 83] (Table 1). Initial attempts to increase its production yield involved selecting cells based on their growth on inhibitory precursor analogues [103]. Later efforts involved targeting the overexpression of IPP pathway genes, especially DXS [60]. A. tumefaciens, however, produces unwanted exopolysaccharides, which increases the viscosity of the sample and affects CoQ extraction [88, 104].
Issues with native hosts
Native producers initially have higher CoQ10 yields as compared with non-native producers. However, few, if any, of the biosynthetic pathways leading to CoQ10 production have been optimized in these organisms, and the toolbox of promoters and genetic modules needed for effective tuning of native producers is lacking [84, 98]. Neither A. tumefaciens nor R. sphaeroides produce sufficient quantities of CoQ10 to meet current market demands, and this has led to higher prices of CoQ10 [38]. Furthermore, rather than optimizing the hosts, recent efforts in the field have been to develop toolkit pieces, such as promoter-regulated vectors [98, 99], or to determine ways to select for particular strains after mutagenesis [105]; only a few studies have attempted to harness metabolic engineering (to increase gene expression) or protein engineering [83, 87]. Other efforts garnered toward a more immediate solution have had to rely on the addition of precursors to increase yield, and this comes at a higher cost and therefore remains less feasible [106, 107].
Heterologous hosts
One method to circumvent the shortfalls seen with native producers is to use a heterologous platform that hosts high pliability towards genetic manipulation [108]. Heterologous systems are often avoided because of the production of unwanted CoQ species, the lengths of which are influenced by the chain length of the host organism and the nature of the heterologous polyprenyl diphosphate synthase; this is particularly complicated, as the synthases may function as either homo or hetero-dimers [109]. However, organisms that possess a large toolkit for host engineering are desirable, and their use holds promise to overcome some of the limitations seen with native hosts, assuming that these species can be appropriately optimized to produce CoQ with the correct chain length. In light of this, here, we explore two options—E. coli for prokaryotes and S. cerevisiae for eukaryotes [108]—as well as the utility of plants as heterologous hosts.
Heterologous host: Escherichia coli
The success in engineering E. coli to produce human insulin paved the way for a new frontier in metabolic engineering [110]. E. coli grows fast and is cheap to culture, and the large range of molecular tools, coupled with an extensive knowledge of its genetic, cellular and metabolic profiles, makes it a widely used production platform. Indeed, most compounds produced by metabolic engineering of E. coli command a good chance of success [108, 111, 112]. Hence, it is not surprising that strategies developed and optimized for the metabolite production in E. coli can be exploited for the production of CoQ10. However, E. coli natively produces CoQ8 not CoQ10 [77], and efforts to produce CoQ10 involved the addition of DPS from a native producer (A. tumefaciens or G. suboxydans) [113, 114]. Yet, despite producing CoQ10, the bacteria also produced CoQ products of variable tail lengths (CoQ8 and CoQ9) [115]. This was solved by knocking out the octaprenyl diphosphate synthase (IspB), which led to a minimal production of the other CoQ variants [116]. Other efforts used a DPS of greater stringency, and found that DPS from R. sphaeroides was more discerning in producing CoQ10 than DPS from A. tumefaciens [115].
Methods to improve the titer of CoQ10 in E. coli sought to increase the flux from the MEP pathway toward IPP [94, 116], while others reconstructed the complete mevalonate pathway to divert flux without encountering interference from negative regulators, such as HMGR by FPP in its native context [117, 118]. Although this reconstruction successfully increased CoQ10 yield, there was a metabolic bottleneck at the top end of the pathway involving mevalonate conversion (Fig. 2). When the lower part of the pathway was ectopically expressed, a twofold increase in yield was observed; yet, expression of the entire pathway led to only a 1.5-fold increase.
Several metabolomic studies in E. coli have investigated the rate-limiting steps in CoQ10 production [68, 119] by adding in the precursors exogenously to decouple the pathway away from cellular flux production. Not surprisingly, both the isoprenoid tail and aromatic quinone head are rate-limiting in E. coli [68, 120–122]. Yet, when these two precursors are no longer limiting, the downstream genes involved in ring modification (ubiB, ubiH and ubiG) becomes limiting [68]. In an effort to increase flux to the quinone precursor PHB, another study overexpressed chorismate pathway genes, including the gene encoding for 3-deoxy-d-arabinoheptulosonate 7-phosphate synthase, which initiates the first step in combining PEP with d-erythrose 4-phosphate [122] (Fig. 2). However, despite these efforts, CoQ10 levels in E. coli (0.45–3.63 mg/g DCW) still fall short of the levels produced by native producers (R. sphaeroides and A. tumefaciens) [99] (Table 2).
Heterologous host: Saccharomyces cerevisiae
Another popular host in metabolic engineering efforts is the budding yeast, Saccharomyces cerevisiae. As a model organism, the genome of S. cerevisiae has been extensively studied and modified, and there are many already optimized tools for efficient gene expression and genetic building blocks for promoters and other regulatory elements [123, 124]. S. cerevisiae has a fast growth cycle of about 90 min, and has a high cultivable density as compared with bacteria. The budding yeast can also perform homologous recombination and compartmentalize subcellular processes, making it an excellent host for metabolic engineering purposes. It is also a ‘Generally Recognized As Safe’ (GRAS) organism (United States Food and Drug Administration) (Table 2), and this reduces any potential complications that could arise from its use in the production of a health supplement or a nutritional product [56]. Most importantly, the IPP pathway has been extensively optimized in S. cerevisiae [59].
Unfortunately, similar to E. coli, S. cerevisiae natively produces CoQ6 not CoQ10 [1]. Early attempts to delete the COQ1 gene in S. cerevisiae and replace it with DPS from G. suboxydans under the COQ1 promoter reportedly yielded 12.3 µg/g DCW [85]. However, DPS tends to require a heterodimer formation for proper function and, when expressed, may instead form dimers with native polyprenyl diphosphate synthases to produce products of differing lengths [125] (Table 2). An alternative approach would be to examine the functionality of the DPS enzyme by fine-tuning is length-determining function. This would be advantageous on several levels, given that the DPS reaction is a limiting step in CoQ10 production. Indeed, polyprenyl diphosphate synthase belongs to the protein family of prenyl-synthases, many of which are involved in generating the polyisoprenoid chain components of commercially interesting compounds like alkaloids and monoterpenes [7, 68]. If successful, this will conceptually sidestep the aforementioned problem of homodimerization of overexpressed heterologous DPS. We thus propose that an understanding of the mechanism by which polyprenyl diphosphate synthase determines chain length may allow for the production of CoQ10 in S. cerevisiae without generating off-target products.
Heterologous host: plants
Another suggested strategy for CoQ10 production is the use of plant hosts for the ease of CoQ10 supplementation into the diet [38]. Such efforts are currently underway, in conjunction with other nutritional supplements, such as vitamin A (beta-carotene) in ‘golden rice’ (Oryza sativa), which can be likewise co-opted in the context of CoQ10 given that carotenoid production also employs the IPP pathway [38, 126]. However, the political hassle associated with the commercialization of ‘golden rice’ or other genetically modified foods is expected to be a counter-rationale for the biosynthesis of CoQ10 in plant hosts [127–130]. Furthermore, because CoQ10 is also prescribed for deficiency-associated diseases and as an ingredient in various cosmetics, it must be properly extracted. Plant production hosts also have further technical obstacles, such as difficulties in engineering and manipulating the plant host; the need for large plots of expensive, arable land; a dependency on harvesting time; and the risk of unpredictable climate conditions in sync with market demand. It is for these various reasons that plant hosts are not deemed economically viable for CoQ10 production. These challenges, along with the comparatively less effort in the scientific community to exploit plant hosts, has meant that microbial hosts are a better choice for CoQ10 production [131].
Potential future engineering approaches for CoQ10 production
There have been frequent attempts to engineer key enzymes within the CoQ10 pathway to increase the yield, including attempts to regulate IPP chain length. Recent interest in synthetic biology—which involves the fine-tuning of biosynthetic processes by controlling the genome and global organellar organization—promises to further revolutionize traditional bioengineering approaches. Several of the newly innovated methodologies will be discussed in the context of improving CoQ10 biosynthesis in the following sections.
Decaprenyl diphosphate synthase
In essence, there are two ways to induce a non-native heterologous host to make CoQ10: (1) Engineer the polyprenyl diphosphate synthase—which is solely responsible for chain length—to assume the function of DPS, or (2) introduce a DPS into the host and delete the native polyprenyl diphosphate synthase. The latter is based on earlier reports, where CoQ of differing tail lengths have been produced by heterologous hosts [2, 15, 71, 74]. Specifically, the introduction of ddsA and sdsA into E. coli from G. suboxydans and Rhodobacter capsulatus, respectively, can result in the formation of CoQ10 (and also CoQ9) [74, 113, 132]. PHB-polyprenyl diphosphate transferase (COQ2) lacks the specificity of polyprenyl diphosphate synthase, as it is able to transfer isoprenoid tails of varying length; e.g., E coli UbiA can utilize isoprenoid chains of 5–10 residues in length. Based on this promiscuity, the PHB:polyprenyl diphosphate transferase is expected not to be a limiting factor in engineering a non-native host for the production of CoQ10.
However, engineering a heterologous host via the introduction of exogenous DPS suffers from challenges that cannot, as yet, be explained. Even with the efforts of removing endogenous CoQ production by deleting the native polyprenyl diphosphate synthase gene, there remains a lack of stringency in these reactions. For example, when the DPS gene from G. suboxydans is expressed in E. coli, deletion of the native IspB gene only reduces the production of CoQ8 and CoQ9; even though it still predominantly produces CoQ10 [74, 113]. A further complication to the engineering effort lies with the complex formation of polyprenyl diphosphate synthases, which function as homodimers (IspB in E. coli, Coq1 protein in S. cerevisiae, and DdsA in G. suboxydans) or heterotetramers (Dps1-Dlp1 in S. pombe and HsPDSS1-HsPDSS2 in humans) [133, 134]. For instance, when heterologously expressed in E. coli, COQ1 from S. cerevisiae can replace IspB, an otherwise essential gene for the production of CoQ6 [70, 132]. However, when COQ1 from S. cerevisiae is expressed in Dlp1-deficient S. pombe, it rescues the dlp1 deletion by forming a heterodimer with Dps1 to produce CoQ10 [131]. Similarly, Dps1 or Dlp1 in S. pombe can complex with defective IspB mutants to restore functionality in E. coli [135]. In such cases, heterologous expression of DPS creates artificial interactions with the host DPS, calling for caution in considering CoQ10 production through host chassis engineering.
Polyprenyl diphosphate synthase residue functionality
Polyprenyl diphosphate synthases catalyze the formation of the polyprenoid tail by adding IPP units to an allylic diphosphate base [136]. These enzymes are categorized depending on the final carbon chain length of the synthesized product: class I for C10–20, class II for C30–35, class III for C40–50, and class IV for even longer products [115]. Class IV synthases also catalyze some cis-configuration double bonds, whereas the other classes all catalyze trans-configuration bonds. Synthases from class II and III categories should be chosen when studying tail length determination because these classes reflect both the final carbon chain length product and possess a similar stereo configuration of double bonds to that of DPS, with an average homology of 30–50% between the polyprenyl diphosphate synthases and DPS enzymes [113, 133].
There are seven conserved regions within trans-type prenyltransferases, two of which (domain II and VI) possess a DDXXD motif [71, 137]. These motifs are located in two helices that face each other, and are the binding sites for FPP (Helix D) and IPP (Helix H) with the aid of Mg2+ in substrate binding [136, 137] (Fig. 3a). The fifth residue before the DDXXD motif in domain II determines tail chain length. In GGPP and FPP (Thermoplasma), this residue is Tyr-89, a large bulky residue; in OPP (Thermotoga maritime) and IspB (E. coli), it is Ala-76 and Ala-79, respectively. These amino acid differences are associated with an inverse relationship between residue size and chain length [136]. Indeed, when Ala-76 and Ala-79 are changed to Tyr, the product chain length decreases from C40 to C20 [136]. In another study, this same substitution (in E. coli) in the absence of wild-type IspB, produces a non-functional protein, but one that is still able to heterodimerize with the wild-type protein to produce CoQ6 [71].

a Protein homology modeling of COQ1 (YBR003W) was performed using ModBase [159] and was viewed using Swiss PDB Viewer [160]. The template for modeling was based on the medium/long-chain length prenyl pyrophosphate synthase of Arabidopsis thaliana (3aq0A) with 42% sequence identity. Helix D and Helix H bind to the elongating isoprene chain and IPP, respectively, at the conserved DDXXD regions. Helix F contains Met-244 and Helix E contains Ser-231, which are thought to be the residues that regulate chain length elongation. The right figure represents the 180° view of that on the left and is superimposed with the structure of CoQ10. b Multiple sequence alignment of Q9X1M1_THEMA (T. maritime TM_1535), ISPB_ECOLI (E. coli IspB), COQ1_SCEREVISIAE (S. cerevisiae COQ1), DPS1_SPOMBE (S. pombe Dps1) and DPS1_HSAPIENS (Human PDSS1) using CLUSTAL W [161]. Helices D (grey), E (green), F (blue), and H (white) indicated in (a), are boxed in (b). Orange underline marks the DDXXD motif. Red asterisks indicate the positions of S. cerevisiae COQ1 Met-244, Ser-247 and Ser-231 residues. Met-244 corresponds to Leu-188 and Leu-231, and Ser-247 to Val-191 and Val-234 of S. pombe Dps1 and H. sapiens PDSS1, respectively. Labels of helices are marked with the same colors as those used for the helices in a
Elongation of the polyprenyl chain takes place in a ‘tunnel’ between helices H and D, where A76 (in T. maritime) lies at one end near the DDXXD motif and Phe-132 (Met-135 in E. coli) lies at the other end; this Phe residue is thought to serve as a cap-like residue [136, 138]. Mutating Phe-132 to Ala can increase the chain length from C40 to C50, which suggests a method to increase chain length synthesis by polyprenyl diphosphate synthases. This was confirmed by others, who, using a cis-type prenyltransferase, found that substituting leucine for alanine increased the chain length from C55 to C70 [139]. In addition to Met-135 in E. coli, another residue, Met-123, compositely serves to limit the elongation of the IPP chain; hence, Met-123 and Met-135 are proposed to contribute to a ‘double-floor’ (‘floor’ is synonymous with ‘barrier’), as opposed to the ‘single-floor’ created by Phe-132 in T. maritime [138].
Efforts to engineer the polyprenyl diphosphate synthases in a host that is highly malleable to genetic and metabolic engineering, such as S. cerevisiae, may provide a prospective avenue to increase the yield of CoQ10. A sequence alignment of polyprenyl diphosphate synthases from various organisms shows high similarity at the amino acid level in the helices that constitute the chain elongation tunnel (Fig. 3b). Such high conservation means that functional studies conducted with T. maritime polyprenyl diphosphate synthases could serve as a reference to guide engineering efforts in other species, such as COQ1 from S. cerevisiae.
Spatial metabolic organization with synthetic compartmentalization
Metabolic production of CoQ10 may also be increased by manipulating the spatial organization of the enzymes in the cell. This is particularly important when faced with potential off-target reactions or when the accumulation of products results in toxicity [140–142]; albeit, clinical studies indicate that toxicity from CoQ10 supplementation is not a huge concern [29].
Some of the more common ways to recruit the pathway into a localized complex involves the use of protein scaffolds or linkers to tether the pathway enzymes to proteins of interest [143–147]. This manipulation concentrates the substrate close to the enzyme, and may favor the forward metabolic flux, as an intermediary metabolite may be captured and shunted into the next step of the pathway. Conceptually, such spatial arrangement reduces the emergence of unwanted by-products, especially with more promiscuous enzymes.
When stoichiometric ratios of sequential reactions are of relevance, tethering also helps to modulate the ratio of enzyme to protein [56]. However, tethering may cause rigidity in the protein scaffold or direct enzyme fusions that could affect enzymatic function. However, these issues can be overcome with the use of a linker sequence, which provides increased flexibility to orientate the direction of the reaction and lower the risk of potential disruptions to enzyme folding.
In lieu of scaffold or linker systems, synthetic subcellular compartmentalization can also be used, whereby the enzyme complex is targeted to protein shells or organelles (Fig. 4). This would further reduce any unwanted side-effects or steric problems, which likely occur on protein scaffolding. The use of such synthetic compartmentalization may also sequester any toxic products produced by the reaction and preserve cell viability. One potential pathway for the ectopic induction of compartmentalization is through the use of bacterial microcompartments—proteinaceous organelles derived from prokaryotes [148–151]. These synthetic organelles possess selectively permeable surfaces comprising thousands of shell proteins and can sequester the enzymatic pathways by means of N-terminal targeting sequences to link the enzymes to the surface of the organelle. Carboxysomes are one example of a bacterial microcompartment that contains ribulose-1,5-bisphosphate carboxylase oxygenase (RuBisCO) for carbon-fixing activities [152–154]. In eukaryotes, protein-based compartments (which comprise ribonucleoprotein particles) known as ‘vaults’, can also be used; albeit, less is known about the structure and mode of formation of these compartments [155, 156]. Finally, it may be simpler to target the eukaryotic organelle pathways that already exist; for instance, one group sought to increase opioid production by altering the pathway of proteins to the endoplasmic reticulum (ER) by ER-tagging of the relevant enzymes. This modification increased the titer and specificity of the product of interest [157].

Spatial metabolic organization with synthetic compartmentalization. Diagrammatic representation of a synthetic proteinaceous or nanotube micro-compartmentalized organelle can be engineered in microbial cells [149–151]. The organelle consists of a scaffold on which the biosynthetic enzymes can be immobilized to direct the biochemical flux such that the substrate of an enzyme is the product of another juxtaposed enzyme. Toxic byproducts may conceptually be shunt into sub-compartments within the organelle and sequester therein to ensure optimal growth of the microbial host
In cases where modifications are made to the pre-combined quinone head and isoprene tail, the enzymes required are already localized in the mitochondria in a membrane-bound complex (eukaryotes) or on the cell membrane (prokaryotes); although, there is, as yet, no evidence for a complex in prokaryotes [11, 158]. However, the other pathways involved in generating the precursor head and which lack bio-orthogonal chemistry are still candidates for spatial organization; for example, the mevalonate pathway, which leads to the IPP precursor, could be one option. Indeed, SH3 ligands and domains are used to link HMG-CoA synthase with HMGR to prevent the accumulation of HMG-CoA and reduce its associated cytotoxicity [143].
Chorismate could be another option. As mentioned earlier, chorismate is a branch point metabolite and thus its recruitment could be spatially separated so as to prevent its conversion into off-target aromatic amino acids. This segregation would be advantageous, as this pathway is essential and cannot be completely disrupted. If a plant platform were to be used, attention would have to be given to the alternate and possibly competing products of GPP, FPP and GGPP. In non-native hosts, CoQ products will present with a range of tail lengths because of the use of the promiscuously inserted decaprenyl diphosphate synthase and its interactions with host polyprenyl diphosphate transferase. These are some possible candidate biosynthesis modules that may benefit from the manipulation of spatial organization and can be optimized in future experiments.
Conclusions
CoQ10 is a valuable and commercially important product that has yet to be produced to a level that can support market demands. This review gives an overview of the native and heterologous hosts reported thus far for the production of CoQ10. Currently Rhodobacter sphaeroides triumphed as the native host in producing 12.96 mg/g DCW of CoQ10. On the other hand, the most widely used workhorse for industrial production of valuable compounds—E. coli—only achieved 3.63 mg/g DCW as the most productive of the heterologous hosts by far. Thus, use of native hosts still remains as the best option for industrial scale production of CoQ10. However, with new tools and progress made in recent years with the advent of synthetic biology, CoQ10 production may stand a chance to be revolutionized. It will be exciting to expect future new technological breakthroughs in this field to take production to new levels either in native or heterologous producers.
Abbreviations
- CoQ:
-
coenzyme Q
- CoQ10 :
-
coenzyme Q with chain containing 10 isoprene subunits
- VLDL:
-
very low density lipoproteins
- CNS:
-
central nervous system
- PDSS1:
-
prenyl diphosphate synthase, subunit 1
- HMG-CoA:
-
3-hydroxy-3-methylglutaryl-coenzyme A
- SAM:
-
s-adenosyl methionine
- MEP:
-
2-C-methyl-d-erythritol 4-phosphate
- G3P:
-
glyceraldehyde 3-phosphate
- DXP:
-
1-deoxy-d-xylulose 5-phosphate
- DXS:
-
1-deoxy-d-xylulose-5-phosphate synthase
- DXR:
-
1-deoxy-d-xylulose 5-phosphate reductoisomerase
- IPP:
-
isopentenyl diphosphate
- HMGR:
-
HMG-CoA reductase
- DMAPP:
-
dimethylallyl diphosphate
- GPP:
-
geranyl disphosphate
- FPP:
-
farnesyl diphosphate
- GGPP:
-
geranylgeranyl diphosphate
- DPS:
-
decaprenyl diphosphate synthase
- PHB or pHBA:
-
4-hydroxybenzoic aid
- PEP:
-
phosphoenolpyruvate
- DCW:
-
dry cell weight
- GRAS:
-
generally recognized as safe
- RuBisCO:
-
ribulose-1,5-bisphosphate carboxylase/oxygenase
References
Kawamukai M. Biosynthesis of coenzyme Q in eukaryotes. Biosci Biotechnol Biochem. 2016;80:23–33.
Kawamukai M. Biosynthesis and bioproduction of coenzyme Q 10 by yeasts and other organisms. Biotechnol Appl Biochem. 2009;53:217–26.
Tran U, Clarke C. Endogenous synthesis of coenzyme Q in eukaryotes. Mitochondrion. 2007;7:S62–71.
Lankin VZ, Tikhaze AK, Kapel’ko VI, Shepel’kova GS, Shumaev KB, Panasenko OM, Konovalova GG, Belenkov YN. Mechanisms of oxidative modification of low density lipoproteins under conditions of oxidative and carbonyl stress. Biochemistry. 2007;72:1081–90.
Lagoutte E, Mimoun S, Andriamihaja M, Chaumontet C, Blachier F, Bouillaud F. Oxidation of hydrogen sulfide remains a priority in mammalian cells and causes reverse electron transfer in colonocytes. Biochim Biophys Acta. 2010;1797:1500–11.
Bogeski I, Gulaboski R, Kappl R, Mirceski V, Stefova M, Petreska J, Hoth M. Calcium binding and transport by coenzyme Q. J Am Chem Soc. 2011;133:9293–303.
Bentinger M, Tekle M, Dallner G. Coenzyme Q—biosynthesis and functions. Biochem Biophys Res Commun. 2010;396:74–9.
Lee B, Lin Y, Huang Y, Ko Y, Hsia S, Lin P. The relationship between coenzyme Q10, oxidative stress, and antioxidant enzymes activities and coronary artery disease. SciWorld J. 2012;2012:1–8.
Gvozdjáková A, Kucharská J, Dubravicky J, Mojto V, Singh RB. Coenzyme Q10, α-tocopherol, and oxidative stress could be important metabolic biomarkers of male infertility. Dis Markers. 2015;2015:827941.
Kobori Y, Ota S, Sato R, Yagi H, Soh S, Arai G, Okada H. Antioxidant cosupplementation therapy with vitamin C, vitamin E, and coenzyme Q10 in patients with oligoasthenozoospermia. Arch Ital Urol Androl. 2014;86:1–4.
Hayashi K, Ogiyama Y, Yokomi K, Nakagawa T, Kaino T, Kawamukai M. Functional conservation of coenzyme Q biosynthetic genes among yeasts, plants, and humans. PLoS ONE. 2014;9:e99038.
Allan C, Hill S, Morvaridi S, Saiki R, Johnson J, Liau W, Hirano K, Kawashima T, Ji Z, Loo J, Shepherd J, Clarke C. A conserved START domain coenzyme Q-binding polypeptide is required for efficient Q biosynthesis, respiratory electron transport, and antioxidant function in Saccharomyces cerevisiae. Biochim Biophys Acta. 2013;1831:776–91.
Barros M, Johnson A, Gin P, Marbois B, Clarke C, Tzagoloff A. The Saccharomyces cerevisiae COQ10 gene encodes a START domain protein required for function of coenzyme Q in respiration. J Biol Chem. 2005;280:42627–35.
Gin P, Hsu A, Rothman S, Jonassen T, Lee P, Tzagoloff A, Clarke C. The Saccharomyces cerevisiae COQ6 gene encodes a mitochondrial flavin-dependent monooxygenase required for coenzyme Q biosynthesis. J Biol Chem. 2003;278:25308–16.
Uchida N, Suzuki K, Saiki R, Kainou T, Tanaka K, Matsuda H, Kawamukai M. Phenotypes of fission yeast defective in ubiquinone production due to disruption of the gene for p-hydroxybenzoate polyprenyl diphosphate transferase. J Bacteriol. 2000;182:6933–9.
Earls L, Hacker M, Watson J, Miller D. Coenzyme Q protects Caenorhabditis elegans GABA neurons from calcium-dependent degeneration. Proc Natl Acad Sci USA. 2010;107:14460–5.
Grant J, Saldanha J, Gould A. A Drosophila model for primary coenzyme Q deficiency and dietary rescue in the developing nervous system. Dis Model Mech. 2010;3:799–806.
Quinzii C, DiMauro S, Hirano M. Human coenzyme Q10 deficiency. Neurochem Res. 2006;32:723–7.
Sharma A, Fonarow GC, Butler J, Ezekowitz JA, Felker GM. Coenzyme Q10 and heart failure: a state-of-the-art review. Circ Heart Fail. 2016;9:e002639.
Mancuso M, Orsucci D, Volpi L, Calsolaro V, Siciliano G, Spinelli A, Correale C, Szabo H, Montorsi M. Coenzyme Q10 in neuromuscular and neurodegenerative disorders. Curr Drug Targets. 2010;11:111–21.
Gempel K, Topaloglu H, Talim B, Schneiderat P, Schoser B, Hans V, Palmafy B, Kale G, Tokatli A, Quinzii C, Hirano M, Naini A, DiMauro S, Prokisch H, Lochmuller H, Horvath R. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain. 2007;130:2037–44.
Rötig A, Appelkvist E, Geromel V, Chretien D, Kadhom N, Edery P, Lebideau M, Dallner G, Munnich A, Ernster L, Rustin P. Quinone-responsive multiple respiratory-chain dysfunction due to widespread coenzyme Q10 deficiency. Lancet. 2000;356:391–5.
Jankowski J, Korzeniowska K, Cieślewicz A, Jabłecka A. Coenzyme Q10—a new player in the treatment of heart failure? Pharmacol Rep. 2016;68:1015–9.
Desbats MA, Morbidoni V, Silic-Benussi M, Doimo M, Ciminale V, Cassina M, Sacconi S, Hirano M, Basso G, Pierrel F, Navas P, Salviati L, Trevisson E. The COQ2 genotype predicts the severity of coenzyme Q10 deficiency. Hum Mol Genet. 2016. doi:10.1093/hmg/ddw257.
López L, Schuelke M, Quinzii C, Kanki T, Rodenburg R, Naini A, DiMauro S, Hirano M. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) Mutations. Am J Hum Genet. 2006;79:1125–9.
Cooper J, Korlipara L, Hart P, Bradley J, Schapira A. Coenzyme Q 10 and vitamin E deficiency in Friedreich’s ataxia: predictor of efficacy of vitamin E and coenzyme Q 10 therapy. Eur J Neurol. 2008;15:1371–9.
Langsjoen P, Langsjoen A. The clinical use of HMG CoA-reductase inhibitors and the associated depletion of coenzyme Q 10. A review of animal and human publications. BioFactors. 2003;18:101–11.
Braillon A. Coenzyme Q10 and statin-induced myopathy—II. Mayo Clinic Proc. 2015;90:420.
Potgieter M, Pretorius E, Pepper M. Primary and secondary coenzyme Q10 deficiency: the role of therapeutic supplementation. Nutr Rev. 2013;71:180–8.
Jula A, Marniemi J, Huupponen R, Virtanen A, Rastas M, Rönnemaa T. Effects of diet and simvastatin on serum lipids, insulin, and antioxidants in hypercholesterolemic men. JAMA. 2002;287:598.
Tay Z, Eng RJ, Sajiki K, Lim KK, Tang MY, Yanagida M, Chen ES. Cellular robustness conferred by genetic crosstalk underlies resistance against chemotherapeutic drug doxorubicin in fission yeast. PLoS ONE. 2013;8:e55041.
Nguyen TT, Chua JK, Seah KS, Koo SH, Yee JY, Yang EG, Lim KK, Pang SY, Yuen A, Zhang L, Ang WH, Dymock B, Lee EJ, Chen ES. Predicting chemotherapeutic drug combinations through gene network profiling. Sci Rep. 2016;6:18658.
Saini R. Coenzyme Q10: the essential nutrient. J Pharm Bioallied Sci. 2011;3:466.
Rafnsson S, Dilis V, Trichopoulou A. Antioxidant nutrients and age-related cognitive decline: a systematic review of population-based cohort studies. Eur J Nutr. 2013;52:1553–67.
Brugè F, Damiani E, Puglia C, Offerta A, Armeni T, Littarru G, Tiano L. Nanostructured lipid carriers loaded with CoQ10: effect on human dermal fibroblasts under normal and UVA-mediated oxidative conditions. Int J Pharm. 2013;455:348–56.
Rona C, Vailati F, Berardesca E. The cosmetic treatment of wrinkles. J Cosmet Dermat. 2004;3:26–34.
Chiu A, Kimball A. Topical vitamins, minerals and botanical ingredients as modulators of environmental and chronological skin damage. Br J Dermatol. 2003;149:681–91.
Parmar S, Jaiwal A, Dhankher O, Jaiwal P. Coenzyme Q10 production in plants: current status and future prospects. Crit Rev Biotechnol. 2013;35:152–64.
Ikematsu H, Nakamura K, Harashima S, Fujii K, Fukutomi N. Safety assessment of coenzyme Q10 (Kaneka Q10) in healthy subjects: a double-blind, randomized, placebo-controlled trial. Reg Toxicol Pharmacol. 2006;44:212–8.
McFarland R, Taylor R, Turnbull D. A neurological perspective on mitochondrial disease. Lancet Neurol. 2010;9:829–40.
Kumar A, Kaur H, Devi P, Mohan V. Role of coenzyme Q10 (CoQ10) in cardiac disease, hypertension and Meniere-like syndrome. Pharmacol Ther. 2009;124:259–68.
Anan R, Nakagawa M, Miyata M, Higuchi I, Nakao S, Suehara M, Osame M, Tanaka H. Cardiac involvement in mitochondrial diseases: a study on 17 patients with documented mitochondrial DNA defects. Circulation. 1995;91:955–61.
Crane FL, Lester RL, Widmer C, Hatefi Y. Studies on the electron transport system. XVIII. Isolation of coenzyme Q (Q275) from beef heart and beef heart mitochondria. Biochim Biophys Acta. 1959;32:73–9.
de Dieu Ndikubwimana J, Lee BH. Enhanced production techniques, properties and uses of coenzyme Q10. Biotechnol Lett. 2014;36:1917–26.
Kawamukai M. Biosynthesis, bioproduction and novel roles of ubiquinone. J Biosci Bioeng. 2002;94:511–51.
Mu F, Luo M, Fu Y, Zhang X, Yu P, Zu Y. Synthesis of the key intermediate of coenzyme Q10. Molecules. 2011;16:4097–103.
Sheldon R. Green and sustainable manufacture of chemicals from biomass: state of the art. Green Chem. 2014;16:950–63.
Murphy A. Metabolic engineering is key to a sustainable chemical industry. Nat Prod Rep. 2011;28:1406.
Wenda S, Illner S, Mell A, Kragl U. Industrial biotechnology—the future of green chemistry? Green Chem. 2011;13:3007.
Tian Y, Yue T, Yuan Y, Soma P, Williams P, Machado P, Fu H, Kratochvil R, Wei C, Lo Y. Tobacco biomass hydrolysate enhances coenzyme Q10 production using photosynthetic Rhodospirillum rubrum. Bioresour Technol. 2010;101:7877–81.
Du J, Shao Z, Zhao H. Engineering microbial factories for synthesis of value-added products. J Ind Microbiol Biotechnol. 2011;38:873–90.
Nielsen J, Fussenegger M, Keasling J, Lee SY, Liao JC, Prather K, Palsson B. Engineering synergy in biotechnology. Nat Chem Biol. 2014;10:319–22.
Pscheidt B, Glieder A. Yeast cell factories for fine chemical and API production. Microb Cell Fact. 2008;7:25.
Lee J, Na D, Park J, Lee J, Choi S, Lee S. Systems metabolic engineering of microorganisms for natural and non-natural chemicals. Nat Chem Biol. 2012;8:536–46.
Li M, Borodina I. Application of synthetic biology for production of chemicals in yeast Saccharomyces cerevisiae. FEMS Yeast Res. 2015. doi:10.1111/1567-1364.12213.
Siddiqui M, Thodey K, Trenchard I, Smolke C. Advancing secondary metabolite biosynthesis in yeast with synthetic biology tools. FEMS Yeast Res. 2012;12:144–70.
He C, Xie L, Allan C, Tran U, Clarke C. Coenzyme Q supplementation or over-expression of the yeast Coq8 putative kinase stabilizes multi-subunit Coq polypeptide complexes in yeast Coq null mutants. Biochim Biophys Acta. 2014;1841:630–44.
Jullesson D, David F, Pfleger B, Nielsen J. Impact of synthetic biology and metabolic engineering on industrial production of fine chemicals. Biotechnol Adv. 2015;33:1395.
Brown S, Clastre M, Courdavault V, O’Connor S. De novo production of the plant-derived alkaloid strictosidine in yeast. Proc Natl Acad Sci USA. 2015;112:3205–10.
Jeya M, Moon H, Lee J, Kim I, Lee J. Current state of coenzyme Q10 production and its applications. Appl Microbiol Biotechnol. 2009;85:1653–63.
Choi J, Ryu Y, Seo J. Biotechnological production and applications of coenzyme Q10. Appl Microbiol Biotechnol. 2005;68:9–15.
Zhou K, Zou R, Stephanopoulos G, Too H. Metabolite profiling Identified methylerythritol cyclodiphosphate efflux as a limiting step in microbial isoprenoid production. PLoS ONE. 2012;7:e47513.
Yuan L, Rouvière P, LaRossa R, Suh W. Chromosomal promoter replacement of the isoprenoid pathway for enhancing carotenoid production in E. coli. Metab Eng. 2006;8:79–90.
Brown M, Goldstein J. Multivalent feedback regulation of HMG CoA reductase, a control mechanism coordinating isoprenoid synthesis and cell growth. J Lipid Res. 1980;21:505–17.
Hey S, Powers S, Beale M, Hawkins N, Ward J, Halford N. Enhanced seed phytosterol accumulation through expression of a modified HMG-CoA reductase. Plant Biotechnol J. 2006;4:219–29.
Gardner R, Hampton R. A Highly Conserved signal controls degradation of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase in eukaryotes. J Biol Chem. 1999;274:31671–8.
Kampranis S, Makris A. Developing a yeast cell factory for the production of terpenoids. Comput Struct Biotechnol J. 2012;3:1–7.
Cluis C, Ekins A, Narcross L, Jiang H, Gold N, Burja A, Martin V. Identification of bottlenecks in Escherichia coli engineered for the production of CoQ10. Metab Eng. 2011;13:733–44.
Gin P, Clarke C. Genetic evidence for a multi-subunit complex in coenzyme Q biosynthesis in yeast and the role of the Coq1 hexaprenyl diphosphate synthase. J Biol Chem. 2004;280:2676–81.
Okada K, Suzuki K, Kamiya Y, Zhu X, Fujisaki S, Nishimura Y, Nishino T, Nakagawa T, Kawamukai M, Matsuda H. Polyprenyl diphosphate synthase essentially defines the length of the side chain of ubiquinone. Biochim Biophys Acta. 1996;1302:217–23.
Kainou T, Okada K, Suzuki K, Nakagawa T, Matsuda H, Kawamukai M. Dimer formation of octaprenyl-diphosphate synthase (IspB) is essential for chain length determination of ubiquinone. J Biol Chem. 2000;276:7876–83.
Martínez I, Méndez C, Berríos J, Altamirano C, Díaz-Barrera A. Batch production of coenzyme Q10 by recombinant Escherichia coli containing the decaprenyl diphosphate synthase gene from Sphingomonas baekryungensis. J Ind Microbiol Biotechnol. 2015;42:1283–9.
Huang M, Chen Y, Liu J. Chromosomal engineering of Escherichia coli for efficient production of coenzyme Q10. Chin J Chem Eng. 2014;22:559–69.
Okada K, Kainou T, Tanaka K, Nakagawa T, Matsuda H, Kawamukai M. Molecular cloning and mutational analysis of the ddsA gene encoding decaprenyl diphosphate synthase from Gluconobacter suboxydans. Eur J Biochem. 1998;255:52–9.
Nguyen T, Clarke C. Folate status of gut microbiome affects Caenorhabditis elegans lifespan. BMC Biol. 2012;10:66.
Huang J, Lin Y, Yuan Q, Yan Y. Production of tyrosine through phenylalanine hydroxylation bypasses the intrinsic feedback inhibition in Escherichia coli. J Ind Microbiol Biotechnol. 2015;42:655–9.
Zhu X, Yuasa M, Okada K, Suzuki K, Nakagawa T, Kawamukai M, Matsuda H. Production of ubiquinone in Escherichia coli by expression of various genes responsible for ubiquinone biosynthesis. J Perment Bioeng. 1995;79:493–5.
Dixson D, Boddy C, Doyle R. Reinvestigation of coenzyme Q10 isolation from Sporidiobolus johnsonii. Chem Biodivers. 2011;8:1033–51.
Viitanen P. Metabolic engineering of the chloroplast genome using the Echerichia coli ubiC gene reveals that chorismate Is a readily abundant plant precursor for p-hydroxybenzoic acid biosynthesis. Plant Physiol. 2004;136:4048–60.
Zahiri H, Yoon S, Keasling J, Lee S, Won Kim S, Yoon S, Shin Y. Coenzyme Q10 production in recombinant Escherichia coli strains engineered with a heterologous decaprenyl diphosphate synthase gene and foreign mevalonate pathway. Metab Eng. 2006;8:406–16.
Wang Y, Hekimi S. Molecular genetics of ubiquinone biosynthesis in animals. Crit Rev Biochem Mol Biol. 2013;48:69–88.
Zhang D, Shrestha B, Li Z, Tan T. Ubiquinone-10 production using Agrobacterium tumefaciens dps gene in Escherichia coli by coexpression system. Mol Biotechnol. 2007;35:1–14.
Moriyama D, Hosono K, Fujii M, Washida M, Nanba H, Kaino T, Kawamukai M. Production of CoQ 10 in fission yeast by expression of genes responsible for CoQ 10 biosynthesis. Biosci Biotechnol Biochem. 2015;79:1026–33.
Wang X, Chen J, Quinn P. Reprogramming microbial metabolic pathways. Dordrecht: Springer; 2012.
Okada K, Kainou T, Matsuda H, Kawamukai M. Biological significance of the side chain length of ubiquinone in Saccharomyces cerevisiae. FEBS Lett. 1998;431:241–4.
Hihi A, Kebir H, Hekimi S. Sensitivity of Caenorhabditis elegans clk-1 mutants to ubiquinone side-chain length reveals multiple uubiquinone-dependent processes. J Biol Chem. 2003;278:41013–8.
Lu W, Ye L, Lv X, Xie W, Gu J, Chen Z, Zhu Y, Li A, Yu H. Identification and elimination of metabolic bottlenecks in the quinone modification pathway for enhanced coenzyme Q10 production in Rhodobacter sphaeroides. Metab Eng. 2015;29:208–16.
Tokdar P, Wani A, Pratyush Kumar, Ranadive P, George S. Process and strain development for reduction of broth viscosity with improved yield in coenzyme Q10 fermentation by Agrobacterium tumefaciens ATCC 4452. Ferment Technol. 2013;2:110. doi:10.4172/2167-7972.1000110.
Hoffman C, Wood V, Fantes P. An ancient yeast for young geneticists: a primer on the Schizosaccharomyces pombe model system. Genetics. 2015;201:403–23.
Wood V, Gwilliam R, Rajandream MA, Lyne M, Lyne R, Stewart A, Sgouros J, Peat N, Hayles J, Baker S, et al. The genome sequence of Schizosaccharomyces pombe. Nature. 2002;415:871–90.
Fennessy D, Grallert A, Krapp A, Cokoja A, Bridge A, Petersen J, Patel A, Tallada V, Boke E, Hodgson B, Simanis V, Hagan I. Extending the Schizosaccharomyces pombe molecular genetic toolbox. PLoS ONE. 2014;9:e97683.
Yazawa H, Holic R, Kumagai H, Uemura H. Toxicity of ricinoleic acid production in fission yeast Schizosaccharomyces pombe is suppressed by the overexpression of plg7, a phospholipase A2 of a platelet-activating factor (PAF) family homolog. Appl Microbiol Biotechnol. 2013;97:8193–203.
Ye R. Coenzyme Q10 production in a recombinant oleaginous yeast. US Patent; 8,815,567 B2. 2014.
Cluis C, Burja A, Martin V. Current prospects for the production of coenzyme Q10 in microbes. Trends Biotechnol. 2007;25:514–21.
Doyle R, Dixson D. Coenzyme Q10 production using Sporidiobolus johnsonii. US Patent; 2011/0269196 A1. 2011.
Ranadive P, Mehta A, Chavan Y, Marx A, George S. Morphological and Molecular Differentiation of Sporidiobolus johnsonii ATCC 20490 and its coenzyme Q10 overproducing mutant strain UF16. Indian J Microbiol. 2014;54:343–57.
Cartron M, Olsen J, Sener M, Jackson P, Brindley A, Qian P, Dickman M, Leggett G, Schulten K, Hunter C. Integration of energy and electron transfer processes in the photosynthetic membrane of Rhodobacter sphaeroides. Biochim Biophys Acta. 2014;1837:e118.
Lu W, Shi Y, He S, Fei Y, Yu K, Yu H. Enhanced production of CoQ10 by constitutive overexpression of 3-demethyl ubiquinone-9 3-methyltransferase under tac promoter in Rhodobacter sphaeroides. Biochem Eng J. 2013;72:42–7.
Lu W, Ye L, Xu H, Xie W, Gu J, Yu H. Enhanced production of coenzyme Q 10 by self-regulating the engineered MEP pathway in Rhodobacter sphaeroides. Biotechnol Bioeng. 2013;111:761–9.
Kien N, Kong I, Lee M, Kim J. Coenzyme Q10 production in a 150-l reactor by a mutant strain of Rhodobacter sphaeroides. J Ind Microbiol Biotechnol. 2010;37:521–9.
Yen H, Chiu C. The influences of aerobic-dark and anaerobic-light cultivation on CoQ10 production by Rhodobacter sphaeroides in the submerged fermenter. Enzyme Microb Technol. 2007;41:600–4.
Henkel C, den Dulk-Ras A, Zhang X, Hooykaas P. Genome sequence of the octopine-type Agrobacterium tumefaciens strain Ach5. Genome Announcements. 2014;2:e00225-14.
Yuan Y, Tian Y, Yue T. Improvement of coenzyme Q10 production: mutagenesis induced by high hydrostatic pressure treatment and optimization of fermentation conditions. J Biomed Biotechnol. 2012;2012:1–8.
Ha S, Kim S, Seo J, Moon H, Lee K, Lee J. Controlling the sucrose concentration increases coenzyme Q10 production in fed-batch culture of Agrobacterium tumefaciens. Appl Microbiol Biotechnol. 2007;76:109–16.
Kim T, Yoo J, Kim S, Pan C, Kalia V, Kang Y, Lee J. Screening and characterization of an Agrobacterium tumefaciens mutant strain producing high level of coenzyme Q10. Process Biochem. 2015;50:33–9.
Qiu L. Coenzyme Q10 production by Sphingomonas sp. ZUTE03 with novel precursors isolated from tobacco waste in a two-phase conversion system. J Microbiol Biotechnol. 2011;21:494–502.
Yan N, Liu Y, Gong D, Du Y, Zhang H, Zhang Z. Solanesol: a review of its resources, derivatives, bioactivities, medicinal applications, and biosynthesis. Phytochem Rev. 2015;l14:403–17.
Kelwick R, MacDonald J, Webb A, Freemont P. Developments in the tools and methodologies of synthetic biology. Front Bioeng Biotechnol. 2014;2:60.
Trevisson E, DiMauro S, Navas P, Salviati L. Coenzyme Q deficiency in muscle. Curr Opin Neurol. 2011;24:449–56.
Williams DC, Van Frank RM, Muth WL, Burnett JF. Cytoplasmic inclusion bodies in Escherichia coli producing biosynthetic human insulin proteins. Science. 1982;215:687–9.
Vickers C, Klein-Marcuschamer D, Krömer J. Examining the feasibility of bulk commodity production in Escherichia coli. Biotechnol Lett. 2011;34:585–96.
Pasotti L, Politi N, Zucca S, Cusella De Angelis M, Magni P. Bottom-up engineering of biological systems through standard bricks: a modularity study on basic parts and devices. PLoS ONE. 2012;7:e39407.
Park Y, Kim S, Choi J, Lee W, Park K, Kawamukai M, Ryu Y, Seo J. Batch and fed-batch production of coenzyme Q10 in recombinant Escherichia coli containing the decaprenyl diphosphate synthase gene from Gluconobacter suboxydans. Appl Microbiol Biotechnol. 2005;67:192–6.
Huang M, Wang Y, Liu J, Mao Z. Multiple strategies for metabolic engineering of Escherichia coli for efficient production of coenzyme Q10. Chinese J Chem Eng. 2011;19:316–26.
Zahiri H, Noghabi K, Shin Y. Biochemical characterization of the decaprenyl diphosphate synthase of Rhodobacter sphaeroides for coenzyme Q10 production. Appl Microbiol Biotechnol. 2006;73:796–806.
Choi J, Ryu Y, Park Y, Seo J. Synergistic effects of chromosomal ispB deletion and dxs overexpression on coenzyme Q10 production in recombinant Escherichia coli expressing Agrobacterium tumefaciens dps gene. J Biotechnol. 2009;144:64–9.
Yang J, Xian M, Su S, Zhao G, Nie Q, Jiang X, Zheng Y, Liu W. Enhancing production of bio-isoprene using hybrid MVA pathway and isoprene synthase in E. coli. PLoS ONE. 2012;7:e33509.
Martin V, Pitera D, Withers S, Newman J, Keasling J. Engineering a mevalonate pathway in Escherichia coli for production of terpenoids. Nat Biotech. 2003;21:796–802.
Yoon S, Lee S, Das A, Ryu H, Jang H, Kim J, Oh D, Keasling J, Kim S. Combinatorial expression of bacterial whole mevalonate pathway for the production of β-carotene in E. coli. J Biotechnol. 2009;140:218–26.
Kim S, Keasling J. Metabolic engineering of the nonmevalonate isopentenyl diphosphate synthesis pathway in Escherichia coli enhances lycopene production. Biotechnol Bioeng. 2001;72:408–15.
Barker J, Frost J. Microbial synthesis of p-hydroxybenzoic acid from glucose. Biotechnol Bioeng. 2001;76:376–90.
Kim S, Kim M, Choi J, Kim S, Ryu Y, Seo J. Amplification of 1-deoxy-d-xyluose 5-phosphate (DXP) synthase level increases coenzyme Q10 production in recombinant Escherichia coli. Appl Microbiol Biotechnol. 2006;72:982–5.
Jensen MK, Keasling JD. Recent applications of synthetic biology tools for yeast metabolic engineering. FEMS Yeast Res. 2014. doi:10.1111/1567-1364.12185.
Philip J. Emerging policy issues in synthetic biology. Ind Biotechnol. 2014;10:256–8.
Zhang M, Luo J, Ogiyama Y, Saiki R, Kawamukai M. Heteromer formation of a long-chain prenyl diphosphate synthase from fission yeast Dps1 and budding yeast Coq1*. FEBS J. 2008;275:3653–68.
Paine J, Shipton C, Chaggar S, Howells R, Kennedy M, Vernon G, Wright S, Hinchliffe E, Adams J, Silverstone A, Drake R. Improving the nutritional value of golden rice through increased pro-vitamin A content. Nat Biotechnol. 2005;23:482–7.
Paarlberg R. The real threat to GM crops in poor countries: consumer and policy resistance to GM foods in rich countries. Food Policy. 2002;27:247–50.
Mayer J. The golden rice controversy: useless science or unfounded criticism? Bioscience. 2005;55:726.
Takahashi S, Ogiyama Y, Kusano H, Shimada H, Kawamukai M, Kadowaki K. Metabolic engineering of coenzyme Q by modification of isoprenoid side chain in plant. FEBS Lett. 2006;580:955–9.
Takahashi S, Ohtani T, Satoh H, Nakamura Y, Kawamukai M, Kadowaki K. Development of coenzyme Q10-enriched rice using sugary and shrunken mutants. Biosci Biotechnol Biochem. 2010;74:182–4.
Fischer R, Schillberg S, Hellwig S, Twyman R, Drossard J. GMP issues for recombinant plant-derived pharmaceutical proteins. Biotechnol Adv. 2012;30:434–9.
Okada K, Kamiya Y, Zhu X, Suzuki K, Tanaka K, Nakagawa T, Matsuda H, Kawamukai H. Cloning of the sdsA gene encoding solanesul diphosphate synthase from Rhodobacter capsulatus and its functional expression in Escherichia coli and Saccharomyces cerevisiae. J Bacteriol. 1997;179:5992–8.
Saiki R, Nagata A, Uchida N, Kainou T, Matsuda H, Kawamukai M. Fission yeast decaprenyl diphosphate synthase consists of Dps1 and the newly characterized Dlp1 protein in a novel heterotetrameric structure. Eur J Biochem. 2003;270:4113–21.
Saiki R, Nagata A, Kainou T, Matsuda H, Kawamukai M. Characterization of solenesyl and decaprenyl diphosphate synthases in mice and humans. FEBS J. 2005;272:5606–22.
Cui T, Kaino T, Kawamukai M. A subunit of decaprenyl diphosphate synthase stabilizes octaprenyl diphosphate synthase in Escherichia coli by forming a high-molecular weight complex. FEBS Lett. 2010;584:652–6.
Guo R. Crystal structure of octaprenyl pyrophosphate synthase from hyperthermophilic Thermotoga maritima and mechanism of product chain length determination. J Biol Chem. 2003;279:4903–12.
Chang K, Chen S, Kuo C, Chang C, Guo R, Yang J, Liang P. Roles of amino acids in the Escherichia coli octaprenyl diphosphate synthase active site probed by structure-guided site-directed mutagenesis. Biochemistry. 2012;51:3412–9.
Han X, Chen C, Kuo C, Huang C, Zheng Y, Ko T, Zhu Z, Feng X, Wang K, Oldfield E, Wang A, Liang P, Guo R, Ma Y. Crystal structures of ligand-bound octaprenyl pyrophosphate synthase from Escherichia coli reveal the catalytic and chain-length determining mechanisms. Proteins. 2014;83:37–45.
Chang S. Substrate binding mode and reaction mechanism of undecaprenyl pyrophosphate synthase deduced from crystallographic studies. Protein Sci. 2004;13:971–8.
Lee H, DeLoache W, Dueber J. Spatial organization of enzymes for metabolic engineering. Metab Eng. 2012;14:242–51.
Agapakis C, Boyle P, Silver P. Natural strategies for the spatial optimization of metabolism in synthetic biology. Nat Chem Biol. 2012;8:527–35.
Lassila J, Bernstein S, Kinney J, Axen S, Kerfeld C. Assembly of robust bacterial microcompartment shells using building blocks from an organelle of unknown function. J Mol Biol. 2014;426:2217–28.
Dueber J, Wu G, Malmirchegini G, Moon T, Petzold C, Ullal A, Prather K, Keasling J. Synthetic protein scaffolds provide modular control over metabolic flux. Nat Biotechnol. 2009;27:753–9.
Boyle P, Silver P. Parts plus pipes: synthetic biology approaches to metabolic engineering. Metab Eng. 2012;14:223–32.
Horn A, Sticht H. Synthetic protein scaffolds based on peptide motifs and cognate adaptor domains for improving metabolic productivity. Front Bioeng Biotechnol. 2015;3:191.
Yu K, Liu C, Kim B, Lee D. Synthetic fusion protein design and applications. Biotechnol Adv. 2015;33:155–64.
Zalatan J, Lee M, Almeida R, Gilbert L, Whitehead E, La Russa M, Tsai J, Weissman J, Dueber J, Qi L, Lim W. Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. Cell. 2015;160:339–50.
Sargent F, Davidson F, Kelly C, Binny R, Christodoulides N, Gibson D, Johansson E, Kozyrska K, Lado L, MacCallum J, Montague R, Ortmann B, Owen R, Coulthurst S, Dupuy L, Prescott A, Palmer T. A synthetic system for expression of components of a bacterial microcompartment. Microbiology. 2013;159:2427–36.
Pang A, Frank S, Brown I, Warren M, Pickersgill R. Structural insights into higher order assembly and function of the bacterial microcompartment protein PduA. J Biol Chem. 2014;289:22377–84.
Jakobson C, Kim E, Slininger M, Chien A, Tullman-Ercek D. Localization of proteins to the 1,2-propanediol utilization microcompartment by non-native signal sequences is mediated by a common hydrophobic motif. J Biol Chem. 2015;290:24519–33.
Pröschel M, Detsch R, Boccaccini A, Sonnewald U. Engineering of metabolic pathways by artificial enzyme channels. Front Bioeng Biotechnol. 2015;3:168.
Iancu C, Ding H, Morris D, Dias D, Gonzales A, Martino A, Jensen G. The structure of isolated Synechococcus strain WH8102 carboxysomes as revealed by electron cryotomography. J Mol Biol. 2007;372:764–73.
Yeates T, Kerfeld C, Heinhorst S, Cannon G, Shively J. Protein-based organelles in bacteria: carboxysomes and related microcompartments. Nat Rev Microbiol. 2008;6:681–91.
Kinney J, Axen S, Kerfeld C. Comparative analysis of carboxysome shell proteins. Photosynth Res. 2011;109:21–32.
Casañas A, Guerra P, Fita I, Verdaguer N. Vault particles: a new generation of delivery nanodevices. Curr Opin Biotechnol. 2012;23:972–7.
Mrazek J, Toso D, Ryazantsev S, Zhang X, Zhou Z, Fernandez B, Kickhoefer V, Rome L. Polyribosomes are molecular 3D nanoprinters that orchestrate the assembly of vault particles. ACS Nano. 2014;8:11552–9.
Thodey K, Galanie S, Smolke C. A microbial biomanufacturing platform for natural and semisynthetic opioids. Nat Chem Biol. 2014;10:837–44.
Aussel L, Pierrel F, Loiseau L, Lombard M, Fontecave M, Barras F. Biosynthesis and physiology of coenzyme Q in bacteria. Biochim Biophys Acta. 2014;1837:1004–11.
Pieper U, Webb B, Dong G, Schneidman-Duhovny D, Fan H, Kim S, Khuri N, Spill Y, Weinkam P, Hammel M, Tainer J, Nilges M, Sali A. ModBase, a database of annotated comparative protein structure models and associated resources. Nucleic Acids Res. 2014;42:D336–46.
Guex N, Peitsch M. SWISS-MODEL and the Swiss-Pdb viewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–23.
Larkin M, Blackshields G, Brown N, Chenna R, McGettigan P, McWilliam H, Valentin F, Wallace I, Wilm A, Lopez R, Thompson J, Gibson T, Higgins D. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–8.
Moriyama D, Kaino T, Yajima K, Yanai R, Ikenaka Y, Hasegawa J, Washida M, Nanba H, Kawamukai M. Cloning and characterization of decaprenyl diphosphate synthase from three different fungi. Appl Microbiol Biotechnol 2017;101(4):1559–71.
Yoo JH, Koo BS, Ha SJ, Lee HC, Lee JK, Kim SY, Cheong SR. Fermentation process for preparing coenzyme Q10 by the recombinant Agrobacterium tumefaciens. US patent 20080261282A1, 23 Oct 2008.
Authors’ contributions
SLQE, MK, and ESC wrote the manuscript; TST performed COQ1 modeling; ESC coordinated the work. All authors read and approved the final manuscript.
Acknowledgements
We apologize to colleagues whose work we have not cited due to space constraints. We thank Rebecca A. Jackson for critically editing this manuscript, and Pallavi Tripathi for discussion. E.S.C. thank Chi Bun Ching for inclusion as a member of NUS synthetic biology initiative program, and Matthew Wook Chang for support from NUS Synthetic Biology for Clinical and Technological Innovation (SynCTI) consortium.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All data supporting the conclusions of this article are included within the manuscript. The content of this manuscript has not been published or submitted for publication elsewhere.
Ethics approval
Data in this manuscript were not collected from humans or animals.
Funding
This work was supported by the DPRT NUS synthetic biology initiative grant (R-183-000-324-133) and a Singapore Ministry of Education Tier II Academic Research Fund (MOE2013-T2-1-112) awarded to E.S.C.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Lee, S.Q.E., Tan, T.S., Kawamukai, M. et al. Cellular factories for coenzyme Q10 production. Microb Cell Fact 16, 39 (2017). https://doi.org/10.1186/s12934-017-0646-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-017-0646-4