
Abstract
Background
Chromosome deletions of the long arm of chromosome 4 in 4q syndrome are characterized by mild facial and digital dysmorphism, developmental delay, growth retardation, and skeletal and cardiac anomalies, which is regarded as an autism spectrum disorder. Moreover, some scarce reports indicate that patients with 4q interstitial deletion and 7p duplication may present symptoms associated with hearing loss.
Case presentation
A boy with a severe developmental delay not only post-natal but also intrauterine and several dysmorphic features including microcephaly, ocular hypertelorism, exophthalmos, low-set ears, single palmar flexion crease, and overlapping toes presented discontinued cyanosis and recurrent respiratory infections. MRI, BAEP, echocardiogram and bronchoscopy revealed that he had persistent falcine sinus with a thin corpus callosum, left auditory pathway disorder, patent foramen ovale (2 mm), and tracheobronchomalacia with the right superior bronchus arising from the lateral posterior wall of the right main bronchus. Finally, the patient died with severe pneumonia at 10 months. Array CGH revealed a 23.62 Mb deletion at chromosome 4q27, arr [hg19] 4q27-q31.21 (121, 148, 089–144, 769, 263) × 1, and a 0.85 Mb duplication at chromosome 7q36.1, arr [hg19] 7q36.1-q36.2 (152, 510, 685–153, 363,5 98) × 3. It is rare for 4q syndrome cases or 7q duplications previously reported to have a hearing disorder, pulmonary dysplasia, and pulmonary arterial hypertension.
Conclusions
The phenotype of our patient mainly reflects the effects of haploinsufficiency of FGF2, SPATA5, NAA15, SMAD1, HHIP genes combined with a microduplication of 7q36.1.
Similar content being viewed by others

Background
Chromosome deletion/duplication is associated with mental disorders and dysmorphism. 4q-syndrome is characterized by chromosomal deletion at the breakpoint 4q31 by Townes and colleagues firstly [1], and was subsequently extended to interstitial and terminal deletions of chromosome 4 [2,3,4,5] with an estimated incidence of roughly 1 in 10,000 live births [3, 4]. In the case of chromosomal duplications, the variable phenotypic expressions may vary due to different gene content as a result of the unbalanced rearrangement and the involvement of extrachromosomal material from other chromosomes [6]. The incidence of duplication in the long arm of chromosome 7 is much lower than 4q deletion, and most of the 7q duplicated cases showed unbalanced aberrations resulted from the inheritance of parental balanced chromosomal rearrangements [7,8,9,10,11].
Till now, there is no clinical report on patients with genetic abnormality associated with chromosome 4q deletion and 7q duplication. In this paper, we describe a patient with genetic abnormalities characterized with 23.62 Mb deletion on the long arm of chromosome 4 and a microduplication on the long arm of chromosome 7 presented with hearing impairment, severe developmental delay, and multisystem malformation.
Case presentation
The child was born as the third child and the first boy to non-consanguineous healthy parents, at completion of 36th weeks of gestation, with a reduced birth weight of 1350 g, length of 35 cm, and head circumference of 28 cm (less than -2SD), borne via cesarean delivery due to the reduction of amniotic fluid. The mother was 30 years old and the father was 36 years old at the time of delivery. Ultrasounds done during pregnancy was normal before the 30th week but indicated significant intrauterine growth retardation afterward. Dexamethasone was prenatally administered to promote lung maturation before delivery. The child suffered from irregular hypopnea immediately after birth and was relieved by positive pressure ventilation within 30 s. After 10 minutes, the child presented obvious respiratory distress syndrome with tachypnea, chest-wall retraction, and cyanosis, and was admitted to NICU for 41 days. However, on successful therapies such as nasal intermittent positive pressure ventilation, traumatic mechanical ventilation, and surfactant replacement therapy dyspnea was relieved. Ultrasonic cardiogram was performed during the first week revealed the patent ductus arteriosus and pulmonary arterial hypertension, for which the sildenafil therapy was initiated. Still, the cyanosis symptoms persisted when the child cried or screamed, and had shown complete hearing loss in the screening tests without congenital cytomegalovirus or syphilis infections. Chromosome analysis revealed 46 XY, 4q deletion.
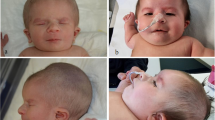
At the age of 3 months, the child was again hospitalized due to aspiration pneumonia and showed exophthalmos, single transverse palmar creases, overlapping toes, left inguinal hernia and severe dystrophy (Fig. 1), and also developed gastroesophageal reflux disease (GERD). MRI showed persistent falcine sinus with a thin corpus callosum (Fig. 2). Brainstem auditory evoked potential (BAEP) was tested at 4 months of age which revealed the left auditory pathway disorder showing no reaction to clicking sounds ranging from 30 to 120 dB. However, the right auditory pathway reaction was well with a BAEP threshold of 30 dB. Bronchoscopy showed tracheobronchomalacia (TBM) and the right superior bronchus arising from the lateral posterior wall of the right main bronchus. Ultrasonic cardiogram showed patent foramen ovale (2 mm) and normal pressure in the pulmonary artery. CT and CTA of the heart were performed which showed normal results. Metabolic diseases screened with serum amino acids and urine organic acids excluded congenital disorders, but all yielded normal results. During hospitalization, the child gained weight nearly 1 kg per month when fed on Alfaré, but was stopped due to family financial crisis and the weight growth speed reduced drastically to a low level of 5.3 kg at 8 months (less than -3SD). Finally, on reaching 10 months of age the child could not recover from cyanosis and died due to severe pneumonia.

Malformation of hands and feet. A single transverse palmar crease (a); The first toe overlapped with the second toe (b)

MRI image of head. Persistent falcine sinus and a thin corpus callosum confirmed by MRI
Chromosomal microarray analysis
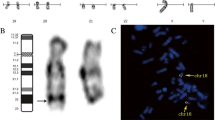
CMA (Chromosomal microarray analysis) was performed using SurePrint G3 customized array (Agilent Technologies, Santa Clara, CA, USA). Previously validated platform settings were consistently utilized for CNV detection and filtering. CNVs within the size range of 2–400 kb were detected via CMA and were further confirmed by manual inspection. It was revealed that there were 23.62 Mb deletion and 0.85 Mb microduplication at chromosome 4q27, arr[hg19] 4q27-q31.21 (121, 148, 089–144, 769, 263) × 1 (Fig. 3), and chromosome 7q36.1, arr[hg19] 7q36.1-q36.2 (152, 510, 685–153, 363, 598) × 3 (Fig. 4), respectively. Moreover, it was evident that within this deleted region there were 117 genes (64 listed in OMIM), and 10 genes listed in OMIM span over the duplicated region.

Red is the deletion at the long arm of chromosome 4

Blue is the duplication at the long arm of chromosome 7
Whole-exome sequencing
Genomic DNA samples were extracted from the patient’s peripheral blood using QIAamp® Blood Mini Kit (Qiagen, Hilden, Germany). DNA target regions were captured by hybridizing the genomic DNA sample library with the Agilent SureSelect Human All Exon V5 (Agilent, USA). The captured and amplified DNA samples were sequenced using Illumina NovaSeq6000 (Illumina, San Diego, CA, USA) with 150 base-paired end reads.
The bioinformatics analysis of the raw data according to ‘Standards and Guidelines for Validating Next-Generation Sequencing Bioinformatics Pipelines’ [12], include the following steps: Sequence Generation, Sequence Alignment, Variant Calling (SNV, INDEL, CNV), Variant Filtering, Variant Annotation, and Variant Prioritization. Indeed, we could find a deletion at chromosome 4q27 (chr:121302077–144,797,407) × 1, but could not get any other significant information from the results and analysis of WES to explain the real cause of hearing disorder of the child.
Discussion and conclusions
According to a literature search, there were no previous reports describing any case with deletion of the long arm of chromosome 4 and duplication of the long arm of chromosome 7. Even though the coordinates of the deletions have varied, our patient shared many clinical features with patients who had deletions in the similar region [13,14,15] (Table 1). All of them presented facial dysmorphism and developmental delay, which have reported that 99 and 94% of patients with 4q deletions presented these features respectively [4].
Unilateral hearing loss is more common than bilateral [20], and it was previously reported that more than one out of ten children initially diagnosed with unilateral hearing loss will progress to bilateral hearing loss [21,22,23]. Cochlear nerve deficiency is the most common type of malformation observed in the setting of congenital unilateral hearing loss [24,25,26]. Although there are reports of familial unilateral hearing loss [27,28,29,30], genetic mutations associated specifically with unilateral hearing loss have yet not been identified with certainty [31]. There are 4 reports of 4q deletions or 7q duplications with hearing impairment available in the literature. The four cases included a 8-year-old boy with deletion in 4q35.1q35.2 region [16], a male infant with deletion in 4q33q35 [17], a 3-year-old girl with duplication of 7q34q35 and deletion in 7q36 [18], and a girl with Complex rearrangement of 7q21.13-q22.1 [19], who were all having bilateral hearing loss with low-set ears (Table 1). Reviewing 141 cases in DECIPHER database showed that only a girl (DECIPHER ID: 293597) with mutations in SPATA5 (located in 4q28.1) and TSHR presented sensorineural hearing impairment.
SPATA5, also known as a spermatogenesis-associated factor (SPAF), was thought to express subcellular in the spermatogonia and spermatocytes, and was associated with mitochondrial function [32]. But the following studies of SPATA5 have suggested a role of the SPATA5 gene not only in neuronal development but also in spermatogenesis. It was dominantly cytosolic in cortical neurons [33,34,35,36]. The SPATA5 deficiency affects mitochondrial morphology and inhibits mitochondrial dynamics, delays neuronal development, and is also associated with decreased cellular ATP [36]. All the patients with SPATA5 variants reported in the literature so far have presented with developmental delay starting in early infancy, 77% presented sensorineural hearing loss, 73% suffered from gastrointestinal problems such as GERD and feeding problem, and 67% was revealed with abnormal brain MRI including hypoplasia of corpus callosum [36].
Furthermore, the deletion of fibroblast growth factor-2 (FGF2) might act an important role in our patient’s phenotype. FGF2 has a haploinsufficiency score (HI index) of 1.68% indicating a highly likely chance to exhibit haploinsufficiency. It plays an important role in the regulation of cell survival, cell division, angiogenesis, cell differentiation, and cell migration and reaches high concentrations in the brain and pituitary. Moreover, it encodes a kind of protein that is a member of the fibroblast growth factor (FGF) family which is not only implicated in limb development, wound healing and tumor growth [37], but also stimulates proliferation of neuronal precursor cells isolated from different regions of the developing central nervous system [38]. FGF signaling is critically required for the in vivo induction of the otic placode during embryonic inner ear development [39]. It is proved that FGF2 could induce the proliferation and survival of auditory neuroblasts in murine [40].
The NAA15 gene located at 4q31.1 involved in our patient’s deletion region has been proved to encode a component of the Nat A N-acetyltransferase complex, which is essential for normal cell function in humans, tethering the complex to the ribosome for posttranslational modification of proteins as they exit the ribosome [41]. Cheng et al. [42] proved that haploinsufficiency, patients with copy-number variation (CNV) deletions involving NAA15 and surrounding genes can lead to mild intellectual disability, mild dysmorphic features, motor delays, growth retardation through identifying and phenotypically characterizing 38 individuals with different likely gene disruption (LGD) variants in NAA15 that is followed by functional assays in yeast.
In addition to SPATA5, FGF2, and NAA15, the deficiency of SMAD1 may also play a role in the development of pulmonary hypertension [43,44,45],. and the HHIP possibly might have effected the lung malformation of our patient [46, 47].
In conclusion, we report a boy with a 23.62 Mb of 4q deletion and a 0.85 Mb of 7q duplication, suffered from severe developmental delay, and dysmorphic features similar to other patients of 4q deletion or 7q duplication. But his bronchial deformity, pulmonary arterial hypertension, especially unilateral hearing loss seems to be very unusual. The deletion of the region between 4q27-q31.21 and the duplication between 7q36.1-q36.2 have affected some genes leading to exhibit haploinsufficiency and resulted in these clinical symptoms. The deficiency of SPATA5 and FGF2 could give a possible explanation for the unilateral hearing loss. In the future, the molecular genetic techniques by combining transcriptomic and proteomic methods with array CGH, it would be possible to precisely examine this region to understand the complex genomic characterization leading to various pathophysiological abnormalities.
Availability of data and materials
The datasets (whole-exome sequencing, chromosomal microarray, and Sanger sequencing files) used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- BAEP:
-
Brainstem auditory evoked potential
- CGH:
-
Comparative genomic hybridization;
- CMA:
-
Chromosomal microarray analysis
- CT:
-
Computed tomography
- CTA:
-
Computed tomography angiography
- GERD:
-
Gastroesophageal reflux disease
- IUGR:
-
Intrauterine growth retardation
- MRI:
-
Magnetic resonance imaging
References
Townes PL, White M, Di Marzo SV. 4q- syndrome. Am J Dis Child. 1979;133(4):383–5.
Strehle EM. Dysmorphological and pharmacological studies in 4q- syndrome. Genet Couns. 2011;22(2):173–85.
Strehle EM, Ahmed OA, Hameed M, Russell A. The 4q-syndrome. Genet Couns. 2001;12(4):327–39.
Strehle EM, Bantock HM. The phenotype of patients with 4q-syndrome. Genet Couns. 2003;14(2):195–205.
Strehle EM, Middlemiss PM. Children with 4q-syndrome: the parents' perspective. Genet Couns. 2007;18(2):189–99.
Alfonsi M, Palka C, Morizio E, Gatta V, Franchi S, Guanciali Franchi P, Zori R, Calabrese G, Palka G, Chiarelli F. A new case of pure partial 7q duplication. Cytogenet Genome Res. 2012;136(1):1–5.
Verma RS, Conte RA, Pitter JH. Tandem duplication of the terminal band of the long arm of chromosome 7 (dir dup (7)(q36----qter)). J Med Genet. 1992;29(5):344–5.
Ndah BV, Stead JA, Brancazio LR, Hummel M, Wenger SL. Prenatal detection of trisomy for the entire long arm of chromosome 7. J Med Genet. 2000;37(7):551–3.
Megarbane A, Gosset P, Souraty N, Lapierre JM, Turleau C, Vekemans M, Loiselet J, Prieur M. Chromosome 7q22-q31 duplication: report of a new case and review. Am J Med Genet. 2000;95(2):164–8.
Robinet C, Douvier S, Khau Van Kien P, Favre B, Luquet I, Nadal N, Nivelon-Chevallier A, Mugneret F. Prenatal diagnosis of a partial trisomy 7q in two fetuses with bilateral ventriculomegaly. Prenat Diagn. 2000;20(11):936–8.
Zelante L, Croce AI, Grifa A, Notarangelo A, Calvano S. Interstitial "de novo" tandem duplication of 7(q31.1-q35): first reported case. Ann Genet-Paris. 2003;46(1):49–52.
Roy S, Coldren C, Karunamurthy A, Kip NS, Klee EW, Lincoln SE, Leon A, Pullambhatla M, Temple-Smolkin RL, Voelkerding KV, et al. Standards and guidelines for validating next-generation sequencing bioinformatics pipelines: a joint recommendation of the Association for Molecular Pathology and the College of American Pathologists. J Mol Diagn. 2018;20(1):4–27.
Strehle EM, Yu L, Rosenfeld JA, Donkervoort S, Zhou Y, Chen TJ, Martinez JE, Fan YS, Barbouth D, Zhu H, et al. Genotype-phenotype analysis of 4q deletion syndrome: proposal of a critical region. Am J Med Genet A. 2012;158A(9):2139–51.
Hickey SE, Biswas S, Thrush DL, Pyatt RE, Gastier-Foster JM, Astbury C, Atkin J. Multigeneration family with short stature, developmental delay, and dysmorphic features due to 4q27-q28.1 microdeletion. Eur J Med Genet. 2013;56(9):521–5.
Duga B, Czako M, Komlosi K, Hadzsiev K, Torok K, Sumegi K, Kisfali P, Kosztolanyi G, Melegh B. Deletion of 4q28.3–31.23 in the background of multiple malformations with pulmonary hypertension. Mol Cytogenet. 2014;7:36.
Vona B, Nanda I, Neuner C, Schroder J, Kalscheuer VM, Shehata-Dieler W, Haaf T. Terminal chromosome 4q deletion syndrome in an infant with hearing impairment and moderate syndromic features: review of literature. BMC Med Genet. 2014;15:72.
Calabrese G, Giannotti A, Mingarelli R, Di Gilio MC, Piemontese MR, Palka G. Two newborns with chromosome 4 imbalances: deletion 4q33-->q35 and ring r (4)(pterq35.2-qter). Clin Genet. 1997;51(4):264–7.
Pavone P, Ruggieri M, Lombardo I, Sudi J, Biancheri R, Castellano-Chiodo D, Rossi A, Incorpora G, Nowak NJ, Christian SL, et al. Microcephaly, sensorineural deafness and Currarino triad with duplication-deletion of distal 7q. Eur J Pediatr. 2010;169(4):475–81.
Bernardini L, Palka C, Ceccarini C, Capalbo A, Bottillo I, Mingarelli R, Novelli A, Dallapiccola B. Complex rearrangement of chromosomes 7q21.13-q22.1 confirms the ectrodactyly-deafness locus and suggests new candidate genes. Am J Med Genet A. 2008;146A(2):238–44.
Shargorodsky J, Curhan SG, Curhan GC, Eavey R. Change in prevalence of hearing loss in US adolescents. JAMA. 2010;304(7):772–8.
Haffey T, Fowler N, Anne S. Evaluation of unilateral sensorineural hearing loss in the pediatric patient. Int J Pediatr Otorhinolaryngol. 2013;77(6):955–8.
Uwiera TC, DeAlarcon A, Meinzen-Derr J, Cohen AP, Rasmussen B, Shott G, Greinwald J. Hearing loss progression and contralateral involvement in children with unilateral sensorineural hearing loss. Ann Otol Rhinol Laryngol. 2009;118(11):781–5.
Declau F, Boudewyns A, Van den Ende J, Peeters A, van den Heyning P. Etiologic and audiologic evaluations after universal neonatal hearing screening: analysis of 170 referred neonates. Pediatrics. 2008;121(6):1119–26.
Ito K, Ishimoto SI, Karino S. Isolated cochlear nerve hypoplasia with various internal auditory meatus deformities in children. Ann Otol Rhinol Laryngol. 2007;116(7):520–4.
Laury AM, Casey S, McKay S, Germiller JA. Etiology of unilateral neural hearing loss in children. Int J Pediatr Otorhinolaryngol. 2009;73(3):417–27.
Nakano A, Arimoto Y, Matsunaga T. Cochlear nerve deficiency and associated clinical features in patients with bilateral and unilateral hearing loss. Otol Neurotol. 2013;34(3):554–8.
Dikkers FG, Verheij JB, van Mechelen M. Hereditary congenital unilateral deafness: a new disorder? Ann Otol Rhinol Laryngol. 2005;114(4):332–7.
Patel N, Oghalai JS. Familial unilateral cochlear nerve aplasia. Otol Neurotol. 2006;27(3):443–4.
Dodson KM, Kamei T, Sismanis A, Nance WE. Familial unilateral deafness and delayed endolymphatic hydrops. Am J Med Genet A. 2007;143A(14):1661–5.
Dodson KM, Georgolios A, Barr N, Nguyen B, Sismanis A, Arnos KS, Norris VW, Chapman D, Nance WE, Pandya A. Etiology of unilateral hearing loss in a national hereditary deafness repository. Am J Otolaryngol. 2012;33(5):590–4.
Vila PM, Lieu JE. Asymmetric and unilateral hearing loss in children. Cell Tissue Res. 2015;361(1):271–8.
Liu Y, Black J, Kisiel N, Kulesz-Martin MF. SPAF, a new AAA-protein specific to early spermatogenesis and malignant conversion. Oncogene. 2000;19(12):1579–88.
Tanaka AJ, Cho MT, Millan F, Juusola J, Retterer K, Joshi C, Niyazov D, Garnica A, Gratz E, Deardorff M, et al. Mutations in SPATA5 are associated with microcephaly, intellectual disability, seizures, and hearing loss. Am J Hum Genet. 2015;97(3):457–64.
Kurata H, Terashima H, Nakashima M, Okazaki T, Matsumura W, Ohno K, Saito Y, Maegaki Y, Kubota M, Nanba E, et al. Characterization of SPATA5-related encephalopathy in early childhood. Clin Genet. 2016;90(5):437–44.
Buchert R, Nesbitt AI, Tawamie H, Krantz ID, Medne L, Helbig I, Matalon DR, Reis A, Santani A, Sticht H, et al. SPATA5 mutations cause a distinct autosomal recessive phenotype of intellectual disability, hypotonia and hearing loss. Orphanet J Rare Dis. 2016;11(1):130.
Puusepp S, Kovacs-Nagy R, Alhaddad B, Braunisch M, Hoffmann GF, Kotzaeridou U, Lichvarova L, Liiv M, Makowski C, Mandel M, et al. Compound heterozygous SPATA5 variants in four families and functional studies of SPATA5 deficiency. Eur J Hum Genet. 2018;26(3):407–19.
Ortega S, Ittmann M, Tsang SH, Ehrlich M, Basilico C. Neuronal defects and delayed wound healing in mice lacking fibroblast growth factor 2. Proc Natl Acad Sci U S A. 1998;95(10):5672–7.
Gage FH. Mammalian neural stem cells. Science. 2000;287(5457):1433–8.
Martin K, Groves AK. Competence of cranial ectoderm to respond to Fgf signaling suggests a two-step model of otic placode induction. Development. 2006;133(5):877–87.
Bruno M, Rizzo IM, Romero-Guevara R, Bernacchioni C, Cencetti F, Donati C, Bruni P. Sphingosine 1-phosphate signaling axis mediates fibroblast growth factor 2-induced proliferation and survival of murine auditory neuroblasts. Biochim Biophys Acta, Mol Cell Res. 2017;1864(5):814–24.
Stessman HA, Xiong B, Coe BP, Wang T, Hoekzema K, Fenckova M, Kvarnung M, Gerdts J, Trinh S, Cosemans N, et al. Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat Genet. 2017;49(4):515–26.
Cheng H, Dharmadhikari AV, Varland S, Ma N, Domingo D, Kleyner R, Rope AF, Yoon M, Stray-Pedersen A, Posey JE, et al. Truncating variants in NAA15 are associated with variable levels of intellectual disability, autism Spectrum disorder, and congenital anomalies. Am J Hum Genet. 2018;102(5):985–94.
Pannu J, Asano Y, Nakerakanti S, Smith E, Jablonska S, Blaszczyk M, ten Dijke P, Trojanowska M. Smad1 pathway is activated in systemic sclerosis fibroblasts and is targeted by imatinib mesylate. Arthritis Rheum. 2008;58(8):2528–37.
Nasim MT, Ogo T, Ahmed M, Randall R, Chowdhury HM, Snape KM, Bradshaw TY, Southgate L, Lee GJ, Jackson I, et al. Molecular genetic characterization of SMAD signaling molecules in pulmonary arterial hypertension. Hum Mutat. 2011;32(12):1385–9.
Han C, Hong KH, Kim YH, Kim MJ, Song C, Kim MJ, Kim SJ, Raizada MK, Oh SP. SMAD1 deficiency in either endothelial or smooth muscle cells can predispose mice to pulmonary hypertension. Hypertension. 2013;61(5):1044–52.
Li X, Howard TD, Moore WC, Ampleford EJ, Li H, Busse WW, Calhoun WJ, Castro M, Chung KF, Erzurum SC, et al. Importance of hedgehog interacting protein and other lung function genes in asthma. J Allergy Clin Immunol. 2011;127(6):1457–65.
Li X, Hawkins GA, Ampleford EJ, Moore WC, Li H, Hastie AT, Howard TD, Boushey HA, Busse WW, Calhoun WJ, et al. Genome-wide association study identifies TH1 pathway genes associated with lung function in asthmatic patients. J Allergy Clin Immunol. 2013;132(2):313–20 e315.
Acknowledgments
We are grateful for the diligent and thorough critical reading of our manuscript by Dr. Dai Lei, Prenatal Diagnosis Center, Xiangya Hospital, Central South University.
Funding
This study was supported by the National Natural Science Foundation of China (No. 81873851) and National Key Research and Development Project of China (No. 2016YFC1000307–17). The funding body had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
MW, XW and GZ acquired, analysis and interpreted the clinical data. JK carried out the molecular genetic testing. MW analyzed and interpreted the genetic testing and wrote the manuscript. XZ designed and organized this study, revised the manuscript critically for important intellectual content. All authors have read and approved the final version of the manuscript submitted by MW.
Authors’ information
All authors are from the Department of Pediatrics, XiangYa Hospital, Central South University, Changsha, China.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Committee on Ethics of Xiangya Hospital of Central South University (NO: 201908299) and was performed in accordance with the Declaration of Helsinki.
Consent for publication
Written informed consent was obtained from the patient’s parents for publication of the clinical and molecular data.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wu, M., Zheng, X., Wang, X. et al. 4q27 deletion and 7q36.1 microduplication in a patient with multiple malformations and hearing loss: a case report. BMC Med Genomics 13, 31 (2020). https://doi.org/10.1186/s12920-020-0697-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-020-0697-y