
Abstract
Background
Rearrangements of unstable DNA sequences may alter the structural integrity or the copy number of dose-sensitive genes, resulting in copy number variations. They may lead more frequently to deletions, in addition to duplications and/or inversions, which are the underlying pathogenic mechanism of a group of conditions known as genomic disorders (or also contiguous gene syndromes). Interstitial deletions of the short arm of chromosome 1 are rare, and only about 30 patients have been reported. Their clinical features are variable, in respect of the extent of the deleted region. They include global developmental delay, central nervous system (CNS) malformations, craniosynostosis, dysmorphic face, ocular defects, cleft palate, urinary tract anomalies and hand/foot abnormalities.
Case presentation
Hereby, we report on an Italian female newborn with craniosynostosis, facial dysmorphisms including bilateral microphthalmia and coloboma, cleft palate, and a severe global developmental and growth delay, associated to a 1p31.3p22.2 deletion of 20.7 Mb. This was inherited from the healthy mother, who was carrier of a smaller (2.6 Mb) deletion included within the centromeric region (1p22.3p22.2) of the same rearrangement, in addition to a translocation between chromosomes 1p and 4q. The deleted region of the proband contains about ninety genes. We focus on the genotype–phenotype correlations.
Conclusions
The results of the present study further confirm that microdeletions at 1p31.3 constitute a contiguous gene syndrome. It is hard to establish whether the critical rearrangement of such syndrome may involve the centromeric band p22.3p22.2, or more likely do not, also in light of the genomic profile of the healthy mother of our patient. Neonatologists and pediatricians should take into consideration 1p31 microdeletion in cases of developmental and growth delay associated to craniosynostosis, peculiar facial dysmorphisms, cleft palate and hand/foot abnormalities. The present report provides new data about 1p31 microdeletion syndrome, in view of a better characterization of its genomic and phenotypic profile.
Similar content being viewed by others


Background
Rearrangements of unstable DNA sequences may alter the structural integrity or the copy number of dose-sensitive genes, resulting in copy number variations (CNVs). CNVs mainly occur in some genomic regions flanked by highly homologous sequences, defined Segmental Duplications (SDs) or Low Copy Repeats (LCRs), characterized by high similarity, and at greater risk of mismatch (homologous non-allelic recombination, NAHR) [1]. This may cause more frequently deletions, in addition to duplications and/or inversions, which represent the underlying pathogenic mechanism of a group of conditions known as genomic disorders (or also contiguous gene syndromes) [2]. In recent years, the increasing availability and clinical application of high resolution array comparative genomic hybridization (a-CGH) allowed the identification of a growing number of microdeletions and neurogenetic syndromes [2,3,4]. Interstitial deletions of the short arm of chromosome 1 are rare, with only about 30 patients reported [1]. They present with variable clinical manifestations, in respect of the extent of the deleted region, including global developmental delay, central nervous system (CNS) malformations, dysmorphic features, urinary tract anomalies, as well as craniosynostosis, ocular defects, cleft palate and hand/foot abnormalities [5]. Hereby, we report on an Italian female newborn with craniosynostosis, facial dysmorphisms including bilateral microphthalmia and coloboma, cleft palate and a severe global developmental and growth delay, associated to a 1p31.3-p22.2 deletion. Her clinical and genomic findings were compatible with a 1p31 microdeletion syndrome diagnosis. The 20.7 Mb rearrangement was inherited from the healthy mother, who was carrier of a much smaller (2.6 Mb) deletion included within the centromeric region (1p22.3-p22.2) of the same genomic abnormality of the daughter, in addition to a translocation between chromosomes 1p and 4q. The deleted region of the proband contains about ninety genes, but we focused on those which may play a role in determining her phenotype, in an attempt to suggest possible genotype–phenotype correlations also in light of the maternal genomic profile.
Case presentation
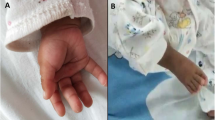
A female newborn, first child of healthy and nonconsanguineous parents, was born at term by spontaneous delivery. Family history disclosed two relatives, on the maternal side (brothers of maternal grandmother and great-grandfather), affected with cleft palate. Pregnancy was marked by fetal growth restriction, which arose during the third trimester. Apgar scores were 6, 8 and 9, at 1, 5 and 10 min respectively. At birth, anthropometric measures were as follows: weight 2090 g (3rd centile, -1.91 standard deviation, SD), length 44 cm (3rd centile, -1.88 SD) and occipitofrontal circumference (OFC) 33 cm (43th centile, -0.18 SD). Due to difficulties in adapting to extrauterine life requiring non-invasive ventilatory support (continuous positive airway pressure administration), she was transferred to the Neonatal Intensive Care Unit. At admission, physical examination showed high forehead, frontal bossing, brachycephaly with flattened occiput, round face, hypotelorism, bilateral microphthalmia, convergent strabismus, epicanthal folds, narrow and down slanting palpebral fissures, broad and depressed nasal root, bulbous tip, anteverted nares, prominent columella, long and hypoplastic philtrum, thin lips with “M” shaped mouth, cleft palate and microretrognathia. Cupped and small ears (with low-set of the left one) with thick helix, and incisions of the upper gingival mucosa completed her craniofacial profile (Fig. 1a/b/c/d). Pectus excavatum and widely spaced nipples were also observed. Bilateral adducted thumbs, calcaneus-valgus talipes and crowded toes (broad first, proximal position of the second and clinodactyly of the fourth and fifth ones) with nail dysplasia, outlined the abnormalities of hands and foot (Fig. 2a/b/c). A severe generalized hypotonia, in addition to poor reactivity, spontaneous motor activity and suction, as well as impaired swallowing and decreased archaic reflexes, marked her neurological features. After birth she suffered from mild respiratory distress, which needed oxygen support until the 15th day of life. Laboratory analyses including complete blood count, serum electrolytes, liver and kidney function tests, and hormonal levels (thyroid stimulating hormone, TSH; basal growth hormone, GH, performed during the first week of life) showed normal results. Head ultrasound (US) revealed isolated moderate widening of the III ventricle, along with kinked corpus callosum. Abdominal US showed hypertrophy of the left hepatic segments, and dysmorphic gallbladder. US heart evaluation showed pulmonary hypertension (pressure gradient around 40–50 mmHg) and patent foramen ovale. Ophthalmological evaluation disclosed bilateral coloboma of iris and optic nerve, extending to the posterior pole, arteriovenous shunts of the retinal vessels, as well as bilateral esotropia.

a and b Patient’s front view at birth and age 4 months: high forehead, frontal bossing, round face, hypotelorism, bilateral microphtalmia, convergent strabismus, epicanthal folds, narrow and down slanting palpebral fissures, broad and depressed nasal root, bulbous tip, anteverted nares, prominent columella, long and hypoplastic philtrum, thin lips with “M” shaped mouth. c and d Lateral view at birth and age 4 months: brachicephaly with flattened occiput, cupped, small and low-set left ear with thick helix, microretrognathia

a Adducted thumb. b and c Bilateral calcaneovalgus talipes, crowded toes with nail dysplasia, broad first, proximal position of the second and clinodactyly of the fourth and fifth ones
aCGH analysis (100–150 Kb resolution, genome assembly GRCh37.p13) identified a 1p31.3-p22.2 deletion of 20.7 Mb and indicated the positions 67,721,572 and 88,415,438 as the breakpoints of the rearrangement. The deleted region involved about ninety genes, including IL23R, RPE65, LRRC7, SRSF11, ANKRD13C, CTH, NEGR1, TTNI3K, LHX8, ACADM, PIGK, ZZZ3, USP33, NEXN, FUBP1, ADGRL2, LPAR3, BCL10, CCN1, CLCA3P, and SH3GLB1, which are mainly those with higher scores of probability of loss-of-function (LoF) intolerance (pLI) (Fig. 3). Fluoresence in situ hydridization (FISH) was then performed in both parents, showing normal results in the father and a translocation between the short arm of chromosome 1 and the subtelomeric region of the long arm of chromosome 4, t(4;1)(q35;p31.1p31.1), in the healthy mother, identifying thus the cause of the genetic abnormality of the proband. Moreover, the aCGH analysis performed in the latter (65 Kb resolution, genome assembly GRCh37.p13) showed a 1p22.3-p22.2 deletion of 2.6 Mb, enclosed within the same rearrangement of the daughter and with overlapping centromeric breakpoint (from positions 85,869,876 to 88,477,895) (Fig. 3).

Gene content in relation to the size of deletions between the patient and her mother
The absence of some specific signs (syndactyly of hands and/or feet, ocular proptosis, ankylosis/synostosis of limb bones and cervical vertebrae), and the type of craniosynostosis (brachycephaly instead of acrocephaly for early fusion of coronal and sagittal sutures) made the present patient’s clinical picture poorly compatible with the main syndromic craniosynostoses, like Apert, Crouzon and Pfeiffer syndromes. Among them, Pfeiffer syndrome (PS) may have the highest phenotypic overlap with our newborn, owing to craniosynostosis and wide first toes (present also in our patient). However, the absence of enlarged and medially deviated thumbs (which were conversely adducted), midfacial hypoplasia and ocular proptosis (which are all distinctive and prevalent features of PS) in our patient, who instead presented bilateral microphthalmia and coloboma, did not direct the diagnostic suspicion towards PS. Furthermore, the absence of macrocephaly and brachydactyly made the diagnostic hypothesis not very consistent also with Muenke syndrome. Then, based both on the phenotype and the genetic findings, it was not considered necessary to proceed with target next generation sequencing (NGS) analysis of craniosynostosis-related genes (FGFR1, FGFR2, FGFR3, TWIST, EFNB1 and TCF12).
The following clinical evolution was characterized by persistent severe generalized hypotonia, developmental delay and feeding difficulties, which needed enteral nutrition by nasogastric tube during the whole hospital stay. She was discharged at 2 months and 20 days of age in good general conditions, despite poor weight and length growth, and included in a multidisciplinary follow-up. Hearing screening through transient-evoked otoacoustic emissions (TEOAEs) revealed abnormal results. In order to ascertain and characterize the hearing loss, an audiological assessment was started. It included serial auditory brainstem response (ABR) evaluations at 2, 4 and 6 months of age, which disclosed, and then confirmed, a bilateral moderate sensorineural hypoacusis requiring hearing-aid, applied at 6 months. Moreover, at the age of 4 months, a three-dimensional reconstruction computed tomography (3D-CT) of the cranium showed bilateral coronal craniosynostyosis, due to reduced representation for fusion of both such sutures. No further alterations of the other sutures were observed, while reduction of the antero-posterior diameter (APD, 106 mm measured on a plan passing through the lateral ventricles), and increase of the transversal one (TD, 114 mm measured on a plan passing through the two temporal bones), with Cephalic Index (CI = TD/APD × 100) 107, compatible with severe brachycephaly (normal CI 75–90, mild alteration 91–93, moderate 94–97, severe > 97; brachycephaly if CI > 90%, dolichocephaly if CI < 76%) [6], were documented. Furthermore, head US ruled out hydrocephalus. She currently is 1 year and 26 days old, and her anthropometric measures, according to World Health Organization growth chart for neonatal and infant close monitoring [7], are: weight 6650 g (< 1st centile, -2.67 SD), length 65 cm (< 1st centile, -2.68 SD) and OFC 43.2 cm (8th centile, -1.42 SD). She has a severe global developmental delay, with a central type axial and lower limbs hypotonia, and absent lateral parachute reaction on both sides. She cannot sit unsupported, and is not able to speak two syllables. She can turn the head, follow and reach an object (red cube and suspended red ring) in the midline with both her hands. Blood examination, heart and abdominal US show no further abnormalities.
Discussion and conclusions
Although chromosome 1 is the largest chromosome in the human genome, only about 30 patients with interstitial microdeletions of its short arm were reported [1, 8,9,10]. Few of them [11,12,13] were described in the current microarray era and, then, the centromeric and telomeric breakpoints delimiting the exact rearranged region and the precise number of genes involved in the microdeletions were not clearly defined. Clinical features of these subjects include developmental delay, seizures, CNS malformations, macrocephaly (40% of cases), elongated or rounded face with prominent nose, micro/retrognathia, half-opened mouth, short neck, congenital heart malformations, hernia, hand/foot malformations, renal anomalies, abnormal external genitalia, joint hyperlaxity and cutis laxa [1, 5]. We identified a 20.7 Mb deletion at chromosome 1p31.3-p22.2, in an Italian female newborn with craniosynostosis (brachycephaly for premature fusion of both coronal sutures), bilateral microphthalmia and coloboma in addition to other facial dysmorphic features, cleft secondary palate, hands and foot abnormalities and severe developmental and growth delay. Unlike to the few reported cases, our patient harbors a 1p31 microdeletion spanning towards the centromere to reach the p22.2 band (less frequently reported than the involvement of the telomeric region p32) [13], and did not show either renal malformations or hormonal (TSH and GH) defects (Table 1).
Indeed, the interstitial deletions previously described were mainly smaller than ours and involved the chromosomal region towards the telomere including some genes which are spared in present newborn. Among these patients, the one described by Rivera-Pedroza et al.[5] is more similar from a genetic as well as clinical point of view. This female newborn carried a 1p31.1p31.3 deletion of 18.6 Mb, and presented with a phenotype considerably overlapping with that of our proband, and including cloverleaf skull, round face, hypotelorism, severe exophthalmos with the absence of eyelids, ectopia lentis, sclerocornea, prominent nose, cleft palate, half-opened mouth, microretrognathia, low-set ears, cutis laxa, hand/foot abnormalities, congenital heart disease and developmental delay. Compared to our newborn, she additionally had bilateral renal hypoplasia, abnormal external genitalia, hernia, as well as seizures, obstructive hydrocephalus, small posterior cranial fossa and intracerebral hemorrhage.
The deleted region of the present newborn contains about ninety OMIM genes. We paid attention to about 20 of them, which are mainly those considered LoF intolerant according to their haploinsufficiency score (pLI score): IL23R, RPE65, LRRC7, SRSF11, ANKRD13C, CTH, NEGR1, TTNI3K, LHX8, ACADM, PIGK, ZZZ3, USP33, NEXN, FUBP1, ADGRL2, LPAR3, BCL10, CCN1, CLCA3P, and SH3GLB1 (Fig. 3).
Among these, IL23R (interleukin 23 receptor) may have a potential association with craniosynostosis, as already suggested by previous genetic and population analyses [5]. Thus, although partially deleted, IL23R may be related to the craniosynostosis also in our patient. LHX8 (LIM homeobox 8) encodes a transcriptional regulator of the family LIM-homeobox, which is expressed in the first branchial arch and the basal forebrain. Its haploinsufficiency was found in patients with cleft palate in addition to microcephaly and severe learning difficulties, carrying smaller chromosome 1 deletions, and it is deleted also in our proposita. Moreover, this gene is highly expressed in connective tissue, skin and its appendages (tooth formation). Therefore, it is hard to establish a clear relation between its defect and the cutaneous abnormalities observed in 1p31 microdeletion patients, as well as the nail dysplasia observed in our newborn. RPE65 (retinoid isomerohydrolase RPE65) encodes for a protein which is a component of the vitamin A visual cycle of the retina. It is a member of the carotenoid cleavage oxygenase superfamily, and performs the essential enzymatic isomerization step in the synthesis of 11-cis retinal. Its mutations are associated with early-onset severe blinding disorders such as Leber congenital amaurosis, and it may be linked to the ocular abnormalities (microphthalmia, coloboma, retinal vessels malformations) present in our patient. LRRC7 (leucine rich repeat containing 7), GADD45A (growth arrest and DNA damage inducible alpha), and NEGR1 (neuronal growth regulator 1) are candidate genes for the developmental delay, and the psychiatric and language disorders reported in 1p31 microdeletion patients [11, 14, 15], and two of them (LRRC7 and NEGR1) are deleted in the proband (who actually presents with a severe neuromotor and language delay). Conversely, NFIA (nuclear factor IA) gene, which was associated to CNS malformations (corpus callosum and cerebellar anomalies, ventriculomegaly, hydrocephalus, tethered spinal cord, type I Chiari malformation) craniofacial abnormalities (metopic synostosis, facial dysmorphisms), developmental delay and genitourinary tract defects [16,17,18,19,20,21,22], is spared in the proposita, who actually shows only minimal morphological CNS abnormalities (isolated moderate widening of the III ventricle and kinked corpus callosum) with no genitourinary abnormalities. These findings agree with the possible involvement of such gene in renal development, but it seems unlikely its role in causing the CNS defects of present patient, although indirect mechanisms (i.e. disruption of regulatory elements) may not be excluded. LEPR (leptin receptor) and JAK1 (Janus kinase 1) haploinsufficiency were associated to abnormal pituitary development and obesity [23]. Also these genes are not included in the deletion of our newborn, who does not show to date either hormonal (TSH and GH) abnormalities or other clinical signs of endocrine dysfunction, including excess weight.
Then, we identified and analyzed, according to literature data, the genes which may be responsible for the clinical findings of our newborn. It is hard to establish whether the other genes in the deleted centromeric region p22.3-p22.2 (i.e., LPAR3, BCL10, CCN1, SH3GLB1, HS2ST1) may also play a causative role for the phenotype of present patient, and be then considered within the critical rearrangement of 1p31 microdeletion syndrome, or more likely do not, also in light of the genomic profile (deletion of such p22.3-p22.2 genes) of the healthy mother of the proband (Fig. 3). The results of present study further confirm that microdeletions at 1p31.3 constitute a contiguous gene syndrome, whose genotype–phenotype correlations are still only partially elucidated, due to variability both in phenotype expressivity and deletion size typical of genomic disorders [24,25,26]. Further characterization of this genomic region, analysis of other patients, and functional studies will provide more insights both on the number and type of critical genes and their impact on this contiguous gene syndrome.
1p31 microdeletion syndrome must be distinguished from FGFR3-related craniosynostoses, from which it differs for associated manifestations and underlying pathogenic (genetic and molecular) mechanisms. Specifically, some suggestive clinical signs (syndactyly of hands and/or feet, ocular proptosis, ankylosis/synostosis of limb bones and cervical vertebrae), and the type of craniosynostosis (acrocephaly for early fusion of coronal and sagittal sutures, rather than brachicephaly) may help in the differential diagnosis, since they are more frequently observed in the main syndromic craniosynostoses, like Apert, Crouzon, and Pfeiffer syndromes. Neonatologists and pediatricians should take into consideration interstitial deletions of chromosome 1 in cases of developmental and growth delay associated to craniosynostosis, peculiar facial dysmorphisms, cleft palate and hand/foot abnormalities [27]. The present report provides new data about 1p31 microdeletion syndrome, in view of a better characterization of its genomic and phenotypic profile.
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- 3D-CT:
-
Three-dimensional reconstruction computed tomography
- a-CGH:
-
Array comparative genomic hybridization
- ABR:
-
Auditory brainstem response
- APD:
-
Antero-posterior diameter
- CI:
-
Cephalic index
- CNS:
-
Central nervous system
- CNVs:
-
Copy number variations
- FISH:
-
Fluoresence in situ hydridization
- GH:
-
Growth hormone
- LoF:
-
Loss of function
- OFC:
-
Occipitofrontal circumference
- OMIM:
-
Online Mendelian Inheritance in Man
- pLI:
-
Probability of loss-of-function intolerance
- PS:
-
Pfeiffer syndrome
- SD:
-
Standard deviation
- TD:
-
Transversal diameter
- TEOAE:
-
Transient-evoked otoacoustic emissions
- TSH:
-
Thyroid stimulating hormone
- US:
-
Ultrasonography
References
Biswal S, Parida P, Dubbudu A, Sharawat IK, Panda PK. Chromosome 1p31.1 Deletion Syndrome: Limited Expression. Ann Indian Acad Neurol. 2021;24(1):78–80.
Piro E, Serra G, Giuffrè M, Schierz IAM, Corsello G. 2q13 microdeletion syndrome: report on a newborn with additional features expanding the phenotype. Clin Case Rep. 2021;9:e04289.
Serra G, Antona V, Giuffré M, Li Pomi F, Lo Scalzo L, Piro E, Schierz IAM, Corsello G. Novel missense mutation of the TP63 gene in a newborn with Hay-Wells/Ankyloblepharon-Ectodermal Defects-Cleft Lip/Palate (AEC) syndrome: clinical report and follow-up. Ital J Pediatr. 2021;47:196.
Piro E, Nardello R, Gennaro E, Fontana A, Taglialatela M, Mangano GD, Corsello G, Mangano S. A novel mutation in KCNQ3-related benign familial neonatal epilepsy: electroclinical features and neurodevelopmental outcome. Epileptic Disord. 2019;21(1):87–91.
Rivera-Pedroza CI, Barraza-García J, Paumard-Hernández B, Nevado J, Orbea-Gallardo C, Sánchez Del Pozo J, Heath KE. Chromosome 1p31.1p31.3 Deletion in a Patient with Craniosynostosis, Central Nervous System and Renal Malformation: Case Report and Review of the Literature. Mol Syndromol. 2017;8(1):30–5.
Badve CA, Mallikarjunappa MK, Iyer RS, Ishak GE, Khanna PC. Craniosynostosis: imaging review and primer on computed tomography. Pediatr Radiol. 2013;43:728–42.
World Health Organization. Child growth standards. 2021. https://www.who.int/tools/child-growth-standards/standards.
Bene M, Duca-Marinescu A, Ioan D, Maximilian C. De novo interstitial deletion del(1)(p21p32). J Med Genet. 1979;16(4):323–7.
Campbell CGN, Wang H, Hunter GW. Interstitial microdeletion of chromosome 1p in two siblings. Am J Med Genet. 2002;111(3):289–94.
Mircher C, Rethore MO, Lespinasse F, Fert-Ferrer S, Lundsteen C, Kirchoff M. Interstitial deletion of the short arm of chromosome 1: attempt to establish a clinical phenotype (46, XX, del (1)(p22p32)). Am J Med Genet A. 2003;118A(2):176–9.
Tassano E, Gamucci A, Celle ME, Ronchetto P, Cuoco C, Gimelli G. Clinical and Molecular Cytogenetic Characterization of a de novo Interstitial 1p31.1p31.3 Deletion in a Boy with Moderate Intellectual Disability and Severe Language Impairment. Cytogenet Genome Res. 2015;146(1):39–43.
Tassano E, Uccella S, Giacomini T, Fiorio P, Tavella E, Malacarne M, Gimelli G, Coviello D. Ronchetto P 1p31.1 microdeletion including only NEGR1 gene in two patients. Eur J Med Genet. 2020;63(6):103919.
Yieh L, Ramo C, Feist C, Shaffer BL, Kim A. Monozygotic dichorionic diamniotic twins with large interstitial deletion of chromosome 1p. Clin Case Rep. 2019;7(9):1735–40.
Liu W, Li W, Cai X, Yang Z, Li H, Su X, Song M, Zhou DS, Li X, Zhang C, Shao M, Zhang L, Yang Y, Zhang Y, Zhao J, Chang H, Yao YG, Fang Y, Lv L, Li M, Xiao X. Identification of a functional human-unique 351-bp Alu insertion polymorphism associated with major depressive disorder in the 1p31.1 GWAS risk loci. Neuropsychopharmacology. 2020;45(7):1196–206.
Genovese A, Cox DM, Butler MG. Partial Deletion of Chromosome 1p31.1 Including only the Neuronal Growth Regulator 1 Gene in Two Siblings. J Pediatr Genet. 2015;4(1):23–8.
Koehler U, Holinski-Feder E, Ertl-Wagner B, Kunz J, von Moers A, von Voss H, Schell-Apacik C. A novel 1p31.3p32.2 deletion involving the NFIA gene detected by array CGH in a patient with macrocephaly and hypoplasia of the corpus callosum. Eur J Pediatr. 2010;169(4):463–8.
Ji J, Salamon N, Quintero-Rivera F. Microdeletion of 1p32-p31 involving NFIA in a patient with hypoplastic corpus callosum, ventriculomegaly, seizures and urinary tract defects. Eur J Med Genet. 2014;57(6):267–8.
Chen CP, Su YN, Chen YY, Chern SR, Liu YP, Wu PC, Lee CC, Chen YT, Wang W. Chromosome 1p32-p31 deletion syndrome: prenatal diagnosis by array comparative genomic hybridization using uncultured amniocytes and association with NFIA haploinsufficiency, ventriculomegaly, corpus callosum hypogenesis, abnormal external genitalia, and intrauterine growth restriction. Taiwan J Obstet Gynecol. 2011;50(3):345–52.
Rao A, O’Donnell S, Bain N, Meldrum C, Shorter D, Goel H. An intragenic deletion of the NFIA gene in a patient with a hypoplastic corpus callosum, craniofacial abnormalities and urinary tract defects. Eur J Med Genet. 2014;57(2–3):65–70.
Revah-Politi A, Ganapathi M, Bier L, Cho MT, Goldstein DB, Hemati P, Iglesias A, Juusola J, Pappas J, Petrovski S, Wilson AL, Aggarwal VS, Anyane-Yeboa K. Loss-of-function variants in NFIA provide further support that NFIA is a critical gene in 1p32-p31 deletion syndrome: A four patient series. Am J Med Genet A. 2017;173(12):3158–64.
Labonne JD, Shen Y, Kong IK, Diamond MP, Layman LC, Kim HG. Comparative deletion mapping at 1p31.3-p32.2 implies NFIA responsible for intellectual disability coupled with macrocephaly and the presence of several other genes for syndromic intellectual disability. Mol Cytogenet. 2016;17:9–24.
Lu W, Quintero-Rivera F, Fan Y, Alkuraya FS, Donovan DJ, Xi Q, Turbe-Doan A, Li QG, Campbell CG, Shanske AL, Sherr EH, Ahmad A, Peters R, Rilliet B, Parvex P, Bassuk AG, Harris DJ, Ferguson H, Kelly C, Walsh CA, Gronostajski RM, Devriendt K, Higgins A, Ligon AH, Quade BJ, Morton CC, Gusella JF, Maas RL. NFIA haploinsufficiency is associated with a CNS malformation syndrome and urinary tract defects. PLoS Genet. 2007;3(5):e80.
Thakur M, Taha D, Misra VK. A Case of Congenital Hypopituitarism Associated With a 1p31 Microdeletion: A Possible Role for LEPR and JAK1. J Endocr Soc. 2017;1(4):278–82.
Serra G, Memo L, Antona V, Corsello G, Favero V, Lago P, Giuffrè M. Jacobsen syndrome and neonatal bleeding: report on two unrelated patients. Ital J Pediatr. 2021;47:147.
Corsello G, Antona V, Serra G, Zara F, Giambrone C, Lagalla L, Piccione M, Piro E. Clinical and Molecular Characterization of 112 Single-Center Patients With Neurofibromatosis Type 1. Ital J Pediatr. 2018;44(1):45.
Serra G, Antona V, Corsello G, Zara F, Piro E, Falsaperla R. NF1 microdeletion syndrome: case report of two new patients. Ital J Pediatr. 2019;45(1):138.
Serra G, Memo L, Coscia A, Giuffrè M, Iuculano A, Lanna M, Valentini D, Contardi A, Filippeschi S, Frusca T, Mosca F, Ramenghi LA, Romano C, Scopinaro A, Villani A, Zampino G, Corsello G. Recommendations for neonatologists and pediatricians working in first level birthing centers on the first communication of genetic disease and malformation syndrome diagnosis: consensus issued by 6 Italian scientific societies and 4 parents’ associations. Ital J Pediatr. 2021;47(1):94.
Acknowledgements
Not applicable.
Funding
No funding was granted for this research.
Author information
Authors and Affiliations
Contributions
GC conceptualized the report, revised the manuscript and gave final approval of the version to be submitted. GS drafted the manuscript and took care of the patient. VA contributed to the acquisition of genetical data. MG revised the manuscript. EP performed neurological and developmental assessment. SS performed instrumental investigations. MS contributed in drafting the manuscript and took care of the patient. All authors approved the final manuscript as submitted.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Written informed consent was obtained from parents at admission of their newborn. The study was approved by the Mother and Child Department of the University of Palermo (Palermo, Italy). All procedures performed in this report were in accordance with the ethical standards of the institutional and national research committee, and with the 1964 Helsinki declaration and its later amendments, or comparable ethical standards.
Consent for publication
Written informed consent for publication was obtained.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Serra, G., Antona, V., Giuffrè, M. et al. Interstitial deletions of chromosome 1p: novel 1p31.3p22.2 microdeletion in a newborn with craniosynostosis, coloboma and cleft palate, and review of the genomic and phenotypic profiles. Ital J Pediatr 48, 38 (2022). https://doi.org/10.1186/s13052-022-01232-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13052-022-01232-7