
Abstract
Background
Oxidative stress is involved in neuronal cell death and mitochondrial dysfunction in neurodegenerative diseases. Liriope platyphylla (LP) has been suggested to have anti-inflammation, anti-bacterial, and anti-cancer effects. However, whether LP exerts neuroprotective effects on neuronal cells is unknown.
Methods
The present study was performed to investigate the neuroprotective effects of LP extract (LPE) against hydrogen peroxide (H2O2)-induced injury in human neuroblastoma cells SH-SY5Y. To test neuroprotective effects of LPE, we performed cell viability assay, flow cytometry analysis and western blot analysis. In addition, mitochondrial membrane potential (MMP) and oxidative stress were performed to evaluate the anti-apoptotic and anti-oxidant effects.
Results
LPE pretreatment conferred significant protection against the H2O2-induced decrease of SH-SY5Y cell viability. H2O2-induced increases of intracellular oxidative stress and mitochondrial dysfunction were attenuated by LPE pretreatment. Therefore, LPE pretreatment prevented SH-SY5Y cell injury. Treatment with H2O2 significantly induced poly(ADP ribose) polymerase (PARP) and caspase-3 cleavage, which was blocked by LPE. We found that p38 activation was involved in the neuroprotective effects of LPE.
Conclusions
Current findings suggest that LPE exerts neuroprotective effects against H2O2-induced apoptotic cell death by modulating p38 activation in SH-SY5Y cells. Therefore, LPE has potential anti-apoptotic effects that may be neuroprotective in neurodegenerative diseases and aging-related dementia.
Similar content being viewed by others


Background
Oxidative stress is involved in neuronal cell death, which is one of the major causes of neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis [1–4]. Some reactive oxygen species (ROS) are spontaneously generated, such as superoxide, peroxide, and hydroxyl radical, while ROS can also be generated due to exogenous factors, such as radiation or drug exposure [5–7]. ROS are generally found in the course of apoptotic cell death caused by intracellular microenvironmental changes. In this context, inhibiting ROS generation can be a useful way to protect normal neuronal cells from damage or death that lead to neurodegenerative diseases and aging-related cognitive decline [8–11].
Liriope platyphylla (LP) is a traditional herbal medicine, the roots of which have been widely used to brain-associated diseases such as forgetfulness and palsy in Donguibogam. Previous studies suggested that LP and its active compounds may exert beneficial effects in cases of viral infection, inflammation, asthma, diabetes, and obesity by modulating the mitogen-activated protein kinase (MAPK)/nuclear translocation of nuclear factor-κB (NF-κB) signaling pathway, as well as inflammatory proteins [12–16]. LP and red LP extract were reported to decrease amyloid-beta (Aβ1–42) peptide levels in the brain and increase nerve growth factor (NGF) levels in the serum of NSE/hAPPswe transgenic mice and Tg2576 mice respectively [17, 18]. However, the anti-apoptotic and neuroprotective effects of LP against hydrogen peroxide (H2O2)-induced neuronal cell loss have not been studied.
Therefore, the present study was performed to investigate whether LP extract (LPE) has neuroprotective effects against H2O2-induced neuronal cell loss in SH-SY5Y neuroblastoma cells. We examined LPE-induced anti-apoptotic and anti-inflammatory effects, as well as the related signaling pathways.
Methods
Preparation of Liriope platyphylla Extracts (LPE)
Dried roots of LP were purchased from local vendor Hyundai Herbal Market (Yeongcheon, Korea) and deposited in the herbal bank of KM-Application Center, Korea Institute of Oriental Medicine (KIOM; Daejeon, Korea) after verifying by Professor Ki Hwan Bae of the College of Pharmacy, Chungnam National University (Daejeon, Korea). Ethanolic extract of LP was extracted in 70 % ethanol (50 g/390 ml) at 40 °C in shaking incubator for 24 h. After extraction, the solution was filtered through filter paper (Whatman filter paper #1), and then the filtrate was lyophilized (yield; 65.9303 %). The freeze-dried LPE powder (100 mg) was then dissolved in 1 ml 50 % DMSO (v/v) and filtered through a 0.22 μm syringe filter.
Cell culture
SH-SY5Y cells (kindly provided by Prof. Jaewon Lee, Pusan National University, Korea) are human neuroblastoma-derived cell line and had neuron-like characteristic. These cells can differentiate into the neurons by induction of retinoic acid (RA). SH-SY5Y cells were cultured in a humidified 5 % CO2 incubator at 37 °C with RPMI 1640 media (Lonza, Walkersville, MD, USA) supplemented with heat inactivated 10 % fetal bovine serum (HyClone Laboratories, Utah, USA), 2 mM glutamine, and 1 % penicillin/streptomycin antibiotic mixture (Corning Incorporated, NY, USA).
Cell viability analysis
Cell viability was evaluated by Cell Counting Kit-8 (CCK) assay (Dojindo Laboratories, Kumamoto, Japan) and MTT assay. Cells (1 × 104 cells/ml) were seeded in 96-well plates. After 24 h, the cells were pretreated with different concentrations of LPE (0.5, 5, 50 μg/ml) for 6 h, cotreated with 100 μM H2O2 for 24 h, incubated in CCK solution for 90 min at 37 °C incubator. Color development was measured at 450 nm using ELISA microplate reader. For MTT assay, 100 μl of 0.25 mg/ml MTT solution in PBS was added to each well. After incubation at 37 °C for 2 h, MTT solution was removed, and cells were lysed by solubilization solution (1:1 DMSO:ethanol). Color development was measured at 560 nm using ELISA microplate reader. To identify the molecule critical for the neuroprotective effects of LPE, cells were treated with inhibitor 30 min prior to treatment of LPE and H2O2.
Preparation of cellular protein extraction and western blot analysis
Whole cell lysates were prepared using RIPA buffer (Millipore Corporation, Billerica, MA, USA) by adding protease inhibitor cocktail and phosphatase inhibitors (Roche Diagnostics, Basel, Switzerland). After washing cells twice with PBS, cells were harvested and collected by centrifugation at 12,000 rpm for 15 min. The pellets were resuspended in RIPA buffer. Protein concentrations were determined using bicinchoninic acid (BCA) assay kit with bovine serum albumin standard. Samples (30 μg protein per lane) were then separated in SDS-polyacrylamide gels and transferred electrophoretically to Immobilon-PSQ transfer membranes (Millipore Corporation, Billerica, MA, USA). Membranes were then placed immediately into a blocking solution (5 % nonfat milk) at room temperature for 30 min, and incubated with the following diluted primary antibodies; PARP, Caspase-3, p-p38, p38, β-actin (Cell Signaling technology, MA, USA) in TBS-T buffer (Tris–HCl based buffer containing 0.2 % Tween 20, pH 7.5) overnight at 4 °C. After washing (4 × 10 min), membranes were incubated with secondary antibody (monoclonal anti-mouse antibody or polyclonal anti-rabbit antibody (Santa Cruz Biotechnology, TX, USA) in TBS-T buffer at room temperature for 90 min. Horseradish-conjugated secondary antibody labeling was detected by enhanced chemiluminescence and blots were exposed to radiographic film. Pre-stained blue markers were used to determine molecular weights. Band intensities were measured using FluorChem™ SP software (Alpha. Innotech, San Leandro, CA, USA). Band intensities were normalized to β-actin or total p38.
ROS production
ROS production was measured by fluorogenic dye 2’, 7’-dichlorodihydrofluorescein diacetate (H2-DCFDA), which is oxidized by intracellular ROS. Cells (5 × 103 cells/well) were seeded in black 96-well plates and pretreated with vehicle or LPE for 6 h, treated with 50 μM H2-DCFDA for 30 min, and then cells were incubated with 100 μM H2O2 for 30 min. The cells were washed with PBS and fluorescent compound was detected by fluorescence microplate reader (SpectraMax i3, Molecular devices, CA, USA) with excitation and emission of 495 nm and 529 nm, respectively.
Mitochondrial membrane potentials assay
Mitochondrial membrane potential depolarization, an early process in the apoptotic cell death, was measured using a cationic carbocyanine dye JC-1. JC-1 dye exists in the cytosol as a monomeric form (green) and also accumulated as a J-aggregates form (red) in the mitochondria. However, in apoptotic cells, JC-1 exists in monomeric form and stains the cytosol green. The monomers emit a green fluorescence (excitation and emission of 490 nm and 530 nm) and J-aggregates a red fluorescence (excitation and emission of 490 nm and 590 nm). Cells were seeded in confocal dish (coverglass-bottom dish). After pretreatment with vehicle or LPE for 6 h, cells was then cotreated with 100 μM H2O2 for 1 h. Cells were further incubated with JC-1 (chloride salt, Biotium, Hayward, CA, USA) staining solution (5 μg/ml) at 37 °C incubator for 15 min and rinsed with culture media. Mitochondrial membrane potential was estimated by measuring the fluorescence of free JC-1 monomers (green) to JC-1 aggregates in mitochondria (red) by FV10i FLUOVIEW Confocal Microscope (Olympus, Tokyo, Japan). Mitochondrial depolarization is indicated by increase in the proportion of cells emitting green fluorescence. To measure the red and green fluorescence intensity ratio, 1024 × 1024 pixels images were collected (n = 8). Red and green fluorescence intensity, respectively, in the individual cells were quantified using FV10i software (Olympus).
Flow cytometry analysis
FITC Annexin V Apoptosis Detection Kit I (BD Biosciences, CA, USA) was used to detect the cell death. In brief, after treatment with vehicle or H2O2, H2O2 + LPE for 24 h, cells were trypsinized and resuspended in binding buffer (0.1 M HEPES/NaOH pH 7.4, 1.4 M NaCl and 25 mM CaCl2). 5 μl of Annexin V-FITC and 5 μl of propidium iodide (PI) were added and incubated for 15 min at room temperature in the dark. Cells were analyzed using flow cytometry (FACSCalibur, Becton Dickinson, CA, USA).
Statistical analysis
The statistical analysis of the differences between the vehicle, H2O2 and LPE-treated groups was determined by one-way analysis of the variance (ANOVA) with Dunnett’s test. The analyses were performed using GraphPad PRISM software® (GraphPad PRISM software Inc., Version 5.02, CA, USA). Results are expressed as means ± standard errors (SE), and p values of < 0.05 were considered as significant.
Results
Effects of LPE on H2O2-induced cell loss in SH-SY5Y cells
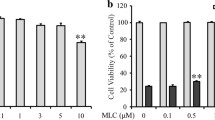
To determine the effects of LPE treatment on cell viability, SH-SY5Y cells were seeded in 96-well plates (5 × 104 cells/ml), cultured for 24 h, and then treated with LPE at concentrations ranging from 0.05 to 500 μg/ml for 24 h. LPE treatment had no significant effect on SH-SY5Y cell viability at concentrations up to 50 μg/ml. High concentrations of LPE (100 μg/ml – 500 μg/ml) decreased cell viability (Fig. 1a). Therefore, we chose an LPE concentration range of 0.5 μg/ml – 50 μg/ml that did not induce cytotoxicity in SH-SY5Y cells. To assess its neuroprotective effects against H2O2, cells were pretreated with LPE for 6 h and then exposed to 100 μM H2O2 for 24 h. CCK analysis showed that H2O2 decreased SH-SY5Y cell viability, while 50 μg/ml LPE pretreatment conferred significant protection against H2O2-induced cell loss (Fig. 1b).

Liriope platyphylla extract (LPE) exerted neuroprotective effects against H2O2-induced cell loss in SH-SY5Y neuroblastoma cells. a SH-SY5Y cells were seeded in 96-well plates (5 × 104 cells/ml) and cultured for 24 h. Cells were treated with the indicated concentrations of LPE for 24 h. LPE treatment at concentrations up to 50 μg/ml had no significant effect on SH-SY5Y cell viability. However, high LPE concentrations (100 – 500 μg/ml) decreased cell viability. The values shown are means ± standard errors (SE; n = 8). *p < 0.05, **p < 0.01 compared to vehicle b Neuroprotective effect of LPE against H2O2-induced cytotoxicity in SH-SY5Y cells. Cells were pretreated with LPE for 6 h, and then co-treated with LPE and 100 μM H2O2 for 24 h. The values shown are means ± SE (n = 8). **p < 0.01, compared to vehicle without H2O2, ##p < 0.01, compared to vehicle with H2O2
LPE suppressed H2O2-induced intracellular oxidative stress and disruption of mitochondrial membrane potential (MMP)
Exogenously added H2O2 induced intracellular ROS generation and affected the accompanying cellular metabolic responses [19]. We evaluated the effects of LPE treatment on H2O2-induced endogenous ROS in SH-SY5Y cells. To measure the levels of intracellular ROS in H2O2-treated SH-SY5Y cells, we used the fluorescent dye H2-DCFDA, which is oxidized to fluorescent DCF by ROS. H2O2 treatment caused a marked increase of intracellular ROS generation; however, 0.5 μg/ml – 50 μg/ml LPE pretreatment significantly reduced ROS production (Fig. 2a). Therefore, LPE had an anti-oxidative effect on H2O2-induced oxidative stress.

Effects of LPE on H2O2-induced oxidative stress and mitochondrial dysfunction in SH-SY5Y cells. a Total intracellular reactive oxygen species (ROS) levels were measured using the dichlorofluorescein diacetate (DCFDA) method. SH-SY5Y cells were exposed to LPE pretreatment for 6 h, and then labeled with 50 μM DCFDA for 30 min. Cells were then treated with 100 μM H2O2 and analyzed immediately using a fluorescent plate reader. Values are reported as means ± SE (n = 8). **p < 0.01, compared to vehicle without H2O2, ##p < 0.01, compared to vehicle with H2O2b Mitochondrial membrane potential (MMP) was assessed by confocal microscopy using JC-1 staining. Representative images showing red fluorescence (aggregated form) and green fluorescence (monomeric form). Cells were observed under 132× magnification. Scale bar = 30 μm. c The graph shows the red/green fluorescence intensity ratio quantitative analysis. Values are reported as means ± SE (n = 8). **p < 0.01, compared to vehicle without H2O2, ##p < 0.01, compared to vehicle with H2O2
ROS-mediated mitochondrial permeability transition pore (mPTP) opening causes mitochondrial dysfunction due to mitochondrial matrix swelling and outer membrane rupture. Therefore, we evaluated the effects of LPE on H2O2-induced disruption of MMP by JC-1 staining. Aggregated JC-1 exhibits red fluorescence in healthy mitochondria, while the monomeric form is characterized by green fluorescence when mitochondria are depolarized during apoptotic cell death. Intact mitochondria are indicated by an increase in the red/green fluorescence intensity ratio. In control cells (vehicle group), both red and green fluorescence were observed in healthy mitochondria, which were rod-shaped, in the cytoplasm (Fig. 2b and c). However, H2O2-treated cells showed decreased red fluorescence and increased green fluorescence, indicating loss of MMP. In addition, mitochondria in H2O2-treated cells were widely distributed in the cytoplasm (Fig. 2b and c). LPE pretreatment prevented the loss of MMP under H2O2-induced neurotoxic conditions. In particular, 50 μg/ml LPE pretreatment resulted in a JC-1 distribution similar to the vehicle group. These results suggest that the neuroprotective effects of LPE against H2O2-induced cell injury occurred via inhibition of MMP loss and oxidative stress.
LPE prevented H2O2-induced apoptotic cell death in SH-SY5Y cells
H2O2-induced ROS and mitochondrial dysfunction were reported to induce apoptotic cell death in various organs, resulting in physiological and pathological changes [20, 21]. As apoptotic cell death is closely associated with the breakdown of MMP, we examined whether LPE protected SH-SY5Y cells against H2O2-induced cell death. After treatment with H2O2 and/or LPE for 24 h, cells were stained to Annexin-V and PI for flow cytometry. Cells are represented in dot plot as healthy (Annexin-V−/PI−, lower left quadrant), early apoptosis (Annexin-V+/PI−, lower right quadrant), late apoptosis (Annexin-V+/PI+, upper right quadrant) and necrosis (Annexin-V−/PI+, upper left quadrant. The percentage of late apoptotic and necrotic cells increased in H2O2 treatment, indicating that both apoptosis and necrosis are major events involved in H2O2-induced cytotoxicity in SH-SY5Y cells. LPE pretreatment remarkably reduced H2O2-induced cell death in late apoptosis and necrotic cell populations (Fig. 3a and b). Exposure to H2O2 alone increased apoptotic markers, such as poly(ADP-ribose) polymerase (PARP) cleavage and caspase-3 cleavage, in SH-SY5Y cells (Fig. 4a and b). LPE pretreatment effectively blocked PARP and caspase-3 cleavage. These results showed that LPE pretreatment attenuated the cleavage of PARP and caspase-3 due to H2O2 treatment (Fig. 4a and b). Therefore, LPE has anti-apoptotic properties against H2O2-induced apoptosis in SH-SY5Y cells.

Effects of LPE on H2O2-induced cell death in SH-SY5Y cells. a Cells treated with vehicle, H2O2 or H2O2 + LPE (5 or 50 μg/ml) for 24 h were subjected to flow cytometry analysis after PI and Annexin-V staining. H2O2 treatment alone increased late apoptotic cells (R4, upper right quadrant) and necrotic cells (R5, upper left quadrant), but pretreatment of LPE protected against H2O2-induced cell death. b Quantitative data showed that percentage of healthy, early apoptotic, late apoptotic, and necrotic cells according to treatment. Values are reported as means ± SE (n = 3). **p < 0.01, compared to vehicle without H2O2, ##p < 0.01, compared to vehicle with H2O2

LPE attenuates H2O2-induced cleavage of PARP and caspase-3 in SH-SY5Y cells. Cells were pretreated with LPE for 6 h, and then co-treated with LPE and 100 μM H2O2 for 24 h. PARP a and caspase-3 b were examined using western blotting. H2O2 treatment alone resulted in PARP and caspase-3 cleavage, indicating that H2O2 induces apoptotic cell death in SH-SY5Y cells. LPE pretreatment protected against H2O2-induced cleavage of PARP and caspase-3. β-Actin was used as a protein loading control. A representative blot is shown from three independent experiments that yielded similar results. Quantification of band densities for cleaved PARP and caspase-3 were measured. Data are expressed as means ± SE (n = 3). **p < 0.01, compared to vehicle without H2O2, ##p < 0.01, compared to vehicle with H2O2
p38 MAPK modulation was involved in the neuroprotective mechanism of LPE against H2O2-induced cell injury
Previous studies suggested that ROS may activate the MAPK signaling pathway [22–25]. We investigated whether the MAPK signaling pathway, including extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38 MAP kinase, was involved in the neuroprotective effects of LPE against H2O2-induced cell injury. Specifically, SH-SY5Y cells were pretreated with LPE for 6 h, and then exposed to 100 μM H2O2 for 24 h. Activation of the MAPK signaling pathway was analyzed by western blotting. H2O2 treatment resulted in significant p38 phosphorylation; however, H2O2 treatment alone did not induce ERK or JNK activation (data not shown). Interestingly, we found that LPE only inhibited p38 phosphorylation in H2O2-treated SH-SY5Y cells (Fig. 5a). LPE did not affect the phosphorylation of ERK or JNK in H2O2-treated SH-SY5Y cells (data not shown). In addition, to determine whether the p38 signaling pathway was involved in the neuroprotective effects of LPE against H2O2-induced oxidative and toxic conditions, we used the p38 inhibitor, SB203580. Cells were seeded in 96-well plates (5 × 104 cells/ml), cultured for 24 h, pretreated with SB203580 (5 μM) for 30 min, treated with LPE (50 μg/ml) for 6 h, and exposed to 100 μM H2O2 for 24 h. Cell viability was determined by MTT assay. H2O2 markedly decreased cell viability; however, LPE dramatically reversed the cell loss in the absence of SB203580 (Fig. 5b). In the H2O2 treatment group, cell viability was significantly improved in the presence of SB203580, indicating that the p38 signaling pathway mediated the H2O2-induced cell loss. No additional or synergistic protective effects were observed in cells co-treated with LPE and SB203580. These results indicated that LPE had neuroprotective effects against H2O2-induced cytotoxicity via attenuation of p38 phosphorylation.

LPE exerts neuroprotective effects on H2O2-treated SH-SY5Y cells by modulating the p38 MAPK signaling pathway. a Cells were pretreated with LPE for 6 h, and then co-treated with LPE and 100 μM H2O2 for 24 h. Western blotting results demonstrated that H2O2 treatment activated phosphorylated (p)-p38; however, LPE pretreatment attenuated the p-p38 level. β-Actin was used as a protein loading control. A representative blot is shown from three independent experiments that yielded similar results. Quantification of band densities for phosphorylated p38/total p38 was measured. Data are expressed as means ± SE (n = 3). **p < 0.01, compared to vehicle without H2O2, ##p < 0.01, compared to vehicle with H2O2b Cells were pretreated with SB203580 (p38 MAPK inhibitor, 5 μM) for 30 min, 50 μg/ml LPE for 6 h, 100 μM H2O2 for 24 h, and then subjected to the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Values are shown as means ± SE (n = 8). **p < 0.01, compared to vehicle without H2O2, ##p < 0.01, compared to vehicle with H2O2, ††p < 0.01, compared to H2O2 with inhibitor.
Discussion
LP is a traditional herbal medicine in Asian countries and has been used as a therapeutic drug for treatment of cough, inflammation, airway inflammation, obesity, and diabetes [12–16]. Furthermore, previous studies suggested that LP activated the neurogenic signaling pathway, and ameliorated the symptoms of neurodegenerative disease. LP prevented Aβ1–42 peptide deposition and increased NGF production in Tg2576 mice [17, 18]. Previous reports evaluated the relationship between the active compounds in LP and neuronal differentiation/brain-derived neurotrophic factor (BDNF)-mediated memory. Manufactured red LP increased NGF secretion and ERK phosphorylation in the B35 neuronal cell line and in PC12 cells [26]. Previous studies demonstrated that LP contains several active compounds, including spicatoside A, ophiopogonin A – D, and methylophiopogonanone A, B. Spicatoside A exerts its neurogenic effects by inducing neurite outgrowth and activating ERK and the phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway in PC12 cells [27]. Memory consolidation via upregulation of hippocampal BDNF levels was enhanced in spicatoside A-treated mice [28]. Ophiopogonin D has antioxidant and anti-inflammatory effects, and inhibits autophagic cell death in cardiomyocytes and human umbilical vein endothelial cells (HUVECs) [29–31]. Previous studies revealed that methylophiopogonanone B inhibited melanocyte dendrite elongation and induced actin cytoskeletal reorganization via Rho activation in normal human epidermal melanocytes and HeLa cells [32, 33]. However, there have been no previous reports regarding the neuroprotective effects of LP against H2O2-induced cell injury. The results of the present study indicated that LP ethanol extract had neuroprotective and anti-apoptotic effects against H2O2-induced cell injury via attenuation of mitochondrial dysfunction and intracellular oxidative stress, as well as p38 phosphorylation in human neuroblastoma SH-SY5Y cells.
Human neuroblastoma SH-SY5Y cells are widely used as an in vitro model in neuroscience research, including studies of neurobiology, neuronal differentiation, and neuroprotective events. Interestingly, SH-SY5Y cells differentiate into functional and mature neurons following RA treatment. Exogenous H2O2 treatment has been used to induce oxidative stress, because oxidative stress is related to neurodegenerative diseases and aging [34–36]. Exogenous H2O2 inhibited cell growth in various cell lines [37–40]. To determine the final concentration of H2O2, SH-SY5Y cells were treated with H2O2 at concentrations ranging from 10 μM to 500 μM for 24 h (data not shown). The cell viability was not affected at 10 or 50 μM H2O2, however, up to this concentration, it was decreased a concentration-dependent manner, with significant cytotoxicity being observed at concentrations > 250 μM. We thought that up to 250 μM was too high to evaluate the neuroprotective effects of LPE because only 20 − 50 % cells were survived in SH-SY5Y cells treated with up to 250 μM. Therefore, we used 100 μM H2O2 (20 − 30 % inhibition) to examine the neuroprotective effects of LPE in SH-SY5Y cells. Our data determined that LPE pretreatment prevented H2O2-induced cell loss and morphological changes, where 100 μM H2O2 decreased cell viability. Therefore, LPE had a neuroprotective effect on H2O2-treated SH-SY5Y cells.
Oxidative stress may affect cell proliferation, differentiation, and survival by activating signaling pathways. However, prolonged or high levels of oxidative stress cause neurotoxicity and neuronal cell death in neurodegenerative diseases and aging [41–43]. It is well known that H2O2-induced oxidative stress disrupts MMP and results in mitochondrial dysfunction [41–44]. In the present study, our data confirmed that H2O2 markedly increased both the intracellular ROS levels and mitochondrial dysfunction, and LPE pretreatment restored the ROS levels and MMP in H2O2-treated SH-SY5Y cells. Moreover, we examined whether H2O2-induced oxidative stress resulted in cell death. We confirmed that H2O2 treatment induced cell death by cleavage of caspase-3/PARP and Annexin-V+/PI+ staining. However, we found that LPE pretreatment reduced the cell death in SH-SY5Y cells. These results suggested that LPE acted as an anti-apoptotic agent by modulating ROS and mitochondrial function under H2O2-induced neurotoxic conditions.
Previous papers reported that H2O2 had cytotoxic effects in various in vitro model by a mechanism involving pro-apoptotic factors (Bax, caspases, PARP) activation and intracellular signaling pathway [45–51]. Especially, MAPK signaling pathway has been suggested as an important mechanism in oxidative stress-mediated neurodegenerative diseases [52, 53]. To address the possible mechanism of LPE-mediated neuroprotective effects, we determined whether LPE inhibited the activation of MAPK signaling pathway (ERK, JNK, p38) in H2O2-treated SH-SY5Y cells. As showed our result, H2O2 only activated p38 (Fig. 5a) not ERK and JNK (data not shown). In addition, p38 inhibitor blocked the cell loss in H2O2-treated SH-SY5Y cells; therefore, H2O2 required p38 activation for the induction of cytotoxicity. Our results demonstrated that LPE protected the cell growth against H2O2-induced cytotoxicity through inhibition of p38 phosphorylation. In addition, LPE only treatment did not affect the level of phosphorylated p38 in SH-SY5Y cells (data not shown). Therefore, we conjectured that LPE has neuroprotective effects by inhibition of p38 phosphorylation in a background of stressful state by H2O2-induced oxidative stress and apoptotic/necrotic cell death. Our results indicated that H2O2-induced p38 phosphorylation was sustained for over 120 min. We found that LPE suppressed p38 phosphorylation in H2O2-treated SH-SY5Y cells. These results demonstrated that p38 activation was required for H2O2-induced cell loss. In conclusion, LPE had anti-oxidant and anti-apoptotic effects via p38 MAPK downregulation.
Conclusions
Collectively, the findings of this study indicated that LPE ameliorated H2O2-induced cell injury in SH-SY5Y cells. The protective effects of LPE were due to its ability to modulate ROS levels, apoptosis-related markers, and mitochondrial dysfunction via p38 MAPK regulation. The present study suggests that LPE may be a neuroprotective agent that can be used to prevent neurodegenerative diseases and brain aging.
Abbreviations
- LP:
-
Liriope platyphylla
- LPE:
-
Liriope platyphylla extract
- H2O2:
-
Hydrogen peroxide
- ROS:
-
Reactive oxygen species
- MMP:
-
Mitochondrial membrane potential
- PARP:
-
Poly(ADP ribose) polymerase
References
Hsieh HL, Yang CM. Role of redox signaling in neuroinflammation and neurodegenerative diseases. Biomed Res Int. 2013;2013(484613):484613–8. 1–18.
Padurariu M, Ciobica A, Lefter R, Serban IL, Stefanescu C, Chirita R. The oxidative stress hypothesis in Alzheimer's disease. Psychiatr Danub. 2013;25(4):401–9.
Pollari E, Goldsteins G, Bart G, Koistinaho J, Giniatullin R. The role of oxidative stress in degeneration of the neuromuscular junction in amyotrophic lateral sclerosis. Front Cell Neurosci. 2014;8:131. 1–8.
Urrutia PJ, Mena NP, Nunez MT. The interplay between iron accumulation, mitochondrial dysfunction, and inflammation during the execution step of neurodegenerative disorders. Front Pharmacol. 2014;5:38. 1–12.
Shokoohinia Y, Hosseinzadeh L, Moieni-Arya M, Mostafaie A, Mohammadi-Motlagh HR. Osthole attenuates doxorubicin-induced apoptosis in PC12 cells through inhibition of mitochondrial dysfunction and ROS production. Biomed Res Int. 2014;2014:156848. 1–7.
Wu LH, Li P, Zhao QL, Piao JL, Jiao YF, Kadowaki M, et al. Arbutin, an intracellular hydroxyl radical scavenger, protects radiation-induced apoptosis in human lymphoma U937 cells. Apoptosis. 2014;19(11):1654–63.
Xie Y, Zhang Y, Zhang LT, Zeng SX, Guo ZB, Zheng BD. Protective effects of alkaloid compounds from Nelumbinis Plumula on tert-butyl hydroperoxide-induced oxidative stress. Molecules. 2013;18(9):10285–300.
Budzynska B, Boguszewska-Czubara A, Kruk-Slomka M, Skalicka-Wozniak K, Michalak A, Musik I, et al. Effects of imperatorin on scopolamine-induced cognitive impairment and oxidative stress in mice. Psychopharmacology. 2015;232(5):931–42.
Hou CW, Chang SY, Jeng KC. Protective effect of a sesamin derivative, 3-bis (3-methoxybenzyl) butane-1, 4-diol on Abeta-stressed PC12 cells. Arch Pharm Res. 2014;38(4):543–8.
Lin X, Huang Z, Chen X, Rong Y, Zhang S, Jiao Y, et al. Protective effect of Millettia pulchra polysaccharide on cognitive impairment induced by D-galactose in mice. Carbohydr Polym. 2014;101:533–43.
Murakami S, Miyazaki I, Sogawa N, Miyoshi K, Asanuma M. Neuroprotective effects of metallothionein against rotenone-induced myenteric neurodegeneration in parkinsonian mice. Neurotox Res. 2014;26(3):285–98.
Han Y, Jung HW, Lee DH, Kwon SY, Son KH, Park YK. Anti-inflammatory effects of prosapogenin III from the dried roots of Liriope platyphylla in LPS-stimulated RAW264.7 cells. J Asian Nat Prod Res. 2013;15(9):1038–49.
Huang TJ, Tsai YC, Chiang SY, Wang GJ, Kuo YC, Chang YC, et al. Anti-viral effect of a compound isolated from Liriope platyphylla against hepatitis B virus in vitro. Virus Res. 2014;192:16–24.
Lee HR, Kim JE, Goo JS, Choi SI, Hwang IS, Lee YJ, et al. Red Liriope platyphylla contains a large amount of polyphenolic compounds which stimulate insulin secretion and suppress fatty liver formation through the regulation of fatty acid oxidation in OLETF rats. International Journal of Molecular Medicine. 2012;30(4):905–13.
Lee YC, Lee JC, Seo YB, Kook YB. Liriopis tuber inhibit OVA-induced airway inflammation and bronchial hyperresponsiveness in murine model of asthma. J Ethnopharmacol. 2005;101(1–3):144–52.
Lee YK, Kim JE, Nam SH, Goo JS, Choi SI, Choi YH, et al. Differential regulation of the biosynthesis of glucose transporters by the PI3-K and MAPK pathways of insulin signaling by treatment with novel compounds from Liriope platyphylla. International Journal of Molecular Medicine. 2011;27(3):319–27.
Choi SI, Go J, Kim JE, Lee YJ, Kwak MH, Jung YJ, et al. Precautionary effects of Red Liriope platyphylla on NGF secretion and Abeta42 deposition under the preclinical stage of Alzheimer's disease in Tg2576 mice. Lab Anim Res. 2013;29(4):212–20.
Choi SI, Goo JS, Kim JE, Hwang IS, Lee HR, Lee YJ, et al. Effects of Red Liriope platyphylla on NGF secretion ability, NGF receptor signaling pathway and gamma-secretase components in NSE/hAPPsw transgenic mice expressing Alzheimer's Disease. Lab Anim Res. 2012;28(3):155–63.
Gough DR, Cotter TG. Hydrogen peroxide: a Jekyll and Hyde signalling molecule. Cell Death Dis. 2011;2, e213.
Navarro A, Boveris A. The mitochondrial energy transduction system and the aging process. Am J Physiol Cell Physiol. 2007;292(2):C670–86.
Onyango IG, Lu J, Rodova M, Lezi E, Crafter AB, Swerdlow RH. Regulation of neuron mitochondrial biogenesis and relevance to brain health. Biochim Biophys Acta. 2010;1802(1):228–34.
Dabrowski A, Boguslowicz C, Dabrowska M, Tribillo I, Gabryelewicz A. Reactive oxygen species activate mitogen-activated protein kinases in pancreatic acinar cells. Pancreas. 2000;21(4):376–84.
McCubrey JA, Lahair MM, Franklin RA. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid Redox Signal. 2006;8(9–10):1775–89.
Ruffels J, Griffin M, Dickenson JM. Activation of ERK1/2, JNK and PKB by hydrogen peroxide in human SH-SY5Y neuroblastoma cells: role of ERK1/2 in H2O2-induced cell death. Eur J Pharmacol. 2004;483(2–3):163–73.
Son Y, Cheong YK, Kim NH, Chung HT, Kang DG, Pae HO. Mitogen-activated protein kinases and reactive oxygen species: how can ROS activate MAPK pathways? J Signal Transduct. 2011;2011:792639. 1–6.
Choi SI, Goo JS, Kim JE, Nam SH, Hwang IS, Lee HR, et al. Differential effects of the steaming time and frequency for manufactured red Liriope platyphylla on nerve growth factor secretion ability, nerve growth factor receptor signaling pathway and regulation of calcium concentration. Mol Med Rep. 2012;6(5):1160–70.
Hur J, Lee P, Moon E, Kang I, Kim SH, Oh MS, et al. Neurite outgrowth induced by spicatoside A, a steroidal saponin, via the tyrosine kinase a receptor pathway. Eur J Pharmacol. 2009;620(1–3):9–15.
Kwon G, Lee HE, Lee DH, Woo H, Park SJ, Gao Q, et al. Spicatoside A enhances memory consolidation through the brain-derived neurotrophic factor in mice. Neurosci Lett. 2014;572:58–62.
Park SH, Lee HJ, Ryu J, Son KH, Kwon SY, Lee SK, et al. Effects of ophiopogonin D and spicatoside A derived from Liriope Tuber on secretion and production of mucin from airway epithelial cells. Phytomedicine. 2014;21(2):172–6.
Qian J, Jiang F, Wang B, Yu Y, Zhang X, Yin Z, et al. Ophiopogonin D prevents H2O2-induced injury in primary human umbilical vein endothelial cells. J Ethnopharmacol. 2010;128(2):438–45.
Zhang YY, Meng C, Zhang XM, Yuan CH, Wen MD, Chen Z, et al. Ophiopogonin D attenuates doxorubicin-induced autophagic cell death by relieving mitochondrial damage in vitro and in vivo. J Pharmacol Exp Ther. 2015;352(1):166–74.
Ito Y, Kanamaru A, Tada A. Effects of methylophiopogonanone B on melanosome transfer and dendrite retraction. J Dermatol Sci. 2006;42(1):68–70.
Ito Y, Kanamaru A, Tada A. A novel agent, methylophiopogonanone b, promotes Rho activation and tubulin depolymerization. Mol Cell Biochem. 2007;297(1–2):121–9.
Rosenzweig S, Carmichael ST. Age-dependent exacerbation of white matter stroke outcomes: a role for oxidative damage and inflammatory mediators. Stroke. 2013;44(9):2579–86.
Saez-Atienzar S, Bonet-Ponce L, Blesa JR, Romero FJ, Murphy MP, Jordan J, et al. The LRRK2 inhibitor GSK2578215A induces protective autophagy in SH-SY5Y cells: involvement of Drp-1-mediated mitochondrial fission and mitochondrial-derived ROS signaling. Cell Death Dis. 2014;5(e1368):1–10.
Wang X, Wang W, Li L, Perry G, Lee HG, Zhu X. Oxidative stress and mitochondrial dysfunction in Alzheimer's disease. Biochim Biophys Acta. 2014;1842(8):1240–7.
Hu XL, Gao LY, Niu YX, Tian X, Wang J, Meng WH, et al. Neuroprotection by Kukoamine A against oxidative stress may involve N-methyl-d-aspartate receptors. Biochim Biophys Acta. 2014;1850(2):287–98.
Park WH. The effects of exogenous H2O2 on cell death, reactive oxygen species and glutathione levels in calf pulmonary artery and human umbilical vein endothelial cells. International Journal of Molecular Medicine. 2013;31(2):471–6.
Park WH. Anti-apoptotic effect of caspase inhibitors on H(2)O(2)-treated HeLa cells through early suppression of its oxidative stress. Oncol Rep. 2014;31(5):2413–21.
Zhou X, Su CF, Zhang Z, Wang CY, Luo JQ, Zhou XW, et al. Neuroprotective effects of methyl 3,4-dihydroxybenzoate against H(2)O(2)-induced apoptosis in RGC-5 cells. J Pharmacol Sci. 2014;125(1):51–8.
Dai DF, Chiao YA, Marcinek DJ, Szeto HH, Rabinovitch PS. Mitochondrial oxidative stress in aging and healthspan. Longev Healthspan. 2014;3(6):1–22.
Dias V, Junn E, Mouradian MM. The role of oxidative stress in Parkinson's disease. J Parkinsons Dis. 2013;3(4):461–91.
Pinho CM, Teixeira PF, Glaser E. Mitochondrial import and degradation of amyloid-beta peptide. Biochim Biophys Acta. 2014;1837(7):1069–74.
Ma S, Liu X, Xun Q, Zhang X. Neuroprotective effect of Ginkgolide K against H2O2-induced PC12 cell cytotoxicity by ameliorating mitochondrial dysfunction and oxidative stress. Biol Pharm Bull. 2014;37(2):217–25.
Chen B, Yue R, Yang Y, Zeng H, Chang W, Gao N, et al. Protective effects of (E)-2-(1-hydroxyl-4-oxocyclohexyl) ethyl caffeine against hydrogen peroxide-induced injury in PC12 cells. Neurochem Res. 2015;40(3):531–41.
Cheng Y, Zhang L, Sun W, Tang J, Lv Z, Xu Z, et al. Protective effects of a wheat germ peptide (RVF) against H2O2-induced oxidative stress in human neuroblastoma cells. Biotechnol Lett. 2014;36(8):1615–22.
Gallelli L, Falcone D, Scaramuzzino M, Pelaia G, D'Agostino B, Mesuraca M, et al. Effects of simvastatin on cell viability and proinflammatory pathways in lung adenocarcinoma cells exposed to hydrogen peroxide. BMC Pharmacol Toxicol. 2014;15(67):1–12.
Garcimartin A, Merino JJ, Gonzalez MP, Sanchez-Reus MI, Sanchez-Muniz FJ, Bastida S, et al. Organic silicon protects human neuroblastoma SH-SY5Y cells against hydrogen peroxide effects. BMC Complementary and Alternative Medicine. 2014;14:384. 1–9.
Ismail N, Ismail M, Imam MU, Azmi NH, Fathy SF, Foo JB. Mechanistic basis for protection of differentiated SH-SY5Y cells by oryzanol-rich fraction against hydrogen peroxide-induced neurotoxicity. BMC Complementary and Alternative Medicine. 2014;14((1):467. 1–11.
Sakuma S, Abe M, Kohda T, Fujimoto Y. Hydrogen peroxide generated by xanthine/xanthine oxidase system represses the proliferation of colorectal cancer cell line Caco-2. J Clin Biochem Nutr. 2015;56(1):15–9.
Tian X, Guo LP, Hu XL, Huang J, Fan YH, Ren TS, et al. Protective effects of Arctium lappa L. roots against hydrogen peroxide-induced cell injury and potential mechanisms in SH-SY5Y cells. Cell Mol Neurobiol. 2015;35(3):335–44.
Kim EK, Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 2010;1802(4):396–405.
Li J, W O, Li W, Jiang ZG, Ghanbari HA. Oxidative stress and neurodegenerative disorders. Int J Mol Sci. 2013;14(12):24438–75.
Acknowledgments
This research was supported by a grant (No. K15280) from the Korea Institute of Oriental Medicine funded by the Ministry of Science, ICT, and Future Planning, Republic of Korea.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
HRP, HL, HP, JWJ, WKC and JYM participated in the design of the study and contributed to the interpretation of data; HRP and HL performed all experiments; HRP, HP and JYM analyzed the data and wrote the manuscript. All authors read and approved the final manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Park, H.R., Lee, H., Park, H. et al. Neuroprotective effects of Liriope platyphylla extract against hydrogen peroxide-induced cytotoxicity in human neuroblastoma SH-SY5Y cells. BMC Complement Altern Med 15, 171 (2015). https://doi.org/10.1186/s12906-015-0679-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12906-015-0679-3