
Abstract
Background
Germline mutations in PALB2 have been identified in approximately 1% of familial breast cancer (BC) in several populations. Nevertheless its contribution in the South-American population is unknown. The goal of this study was to determine the prevalence of PALB2 mutations in the Chilean population.
Methods
100 Chilean BRCA1/2-negatives familial BC cases were included for the PALB2 mutation analysis. We use conformational sensitive gel electrophoresis and direct sequencing. Using a case-control design, we studied the identified variants in 436 BC cases and 809 controls to evaluate their possible association with BC risk.
Results
No pathogenic mutations were detected. We identified three variants, the variant c.1861C > A not previously described was found in one of the 436 cases and none of the 809 controls. The bioinformatic analyses indicate that this variant probably is not pathogenic. PALB2 c.1676A > G (rs152451A/G) and c.2993C > T (rs45551636C/T) variants were significantly associated with increased BC risk only in cases with a strong family history of BC (OR = 1.9 [CI 95% 1.3-2.8] p < 0.01 and OR = 3.3 [CI 95% 1.4-7.3] p < 0.01, respectively). The rs152451A/G-rs45551636C/T composite genotype produce increase of the BC risk in cases with a strong family history of BC (OR = 3.6 [CI 95% 1.7-8.0] p = 0.003). The rs152451-G/rs45551636-C and rs152451-G/rs45551636-T haplotypes were associated with an increased BC risk only in cases with a strong family history of BC (OR = 1.6 [CI 95% 1.0-2.5] p = 0.05 and OR = 3.7 [CI 95% 1.8-7.5] p < 0.001, respectively).
Conclusion
Our results suggest that PALB2 c.1676A > G and c.2993C > T play roles in BC risk in women with a strong family history of BC.
Similar content being viewed by others

Background
Genetic factors play an important role in breast cancer (BC) development, including the presence of BRCA1, BRCA2, ATM, and other genes [1], but only 5% of BC incidence can be explained by mutation in these high-penetrance genes [2]. Moreover, these genes are responsible for only about 16-20% of the risk for familial BC. Therefore, the genetic basis of 80% of familial cases remains unexplained [3]. The remaining risk is likely to involve mutations in moderate- and/or low-penetrance susceptibility genes.
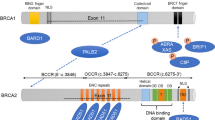
The PALB2 (partner and localizer of BRCA2) protein interacts with BRCA2, stabilizing the intranuclear accumulation of BRCA2 proteins at sites of DNA damage [4]. PALB2 is also recruited by BRCA1 in response to DNA damage and serves as a linker between BRCA1 and BRCA2, necessary for BRCA2-mediated homologous-recombination repair [5,6]. Thus, BRCA1, BRCA2, and PALB2 are key BC susceptibility genes that function together in the same DNA damage response pathway [5,7]. Biallelic loss of PALB2 causes increased predisposition to cancers, increased sensitivity to DNA-damaging agents, and Fanconi anemia [8].
Germline PALB2 mutations are rare, but have been associated with increased risk for breast and other cancers. In families with multiple breast cancer cases, germline PALB2 mutations have been reported in populations from Western Europe, the United Kingdom, Finland, French Canada, and Australia [9-12]. In familial BC, germline PALB2 mutations are associated with a 2.3- to 6.0-fold increased risk for BC [9,13,14]. Germline PALB2 mutations have also [13] been reported in a lower frequency in unselected female BC cases from Finland, French Canada, Australia, China, Malaysia, and Singapore [10-12,15,16]. No PALB2 mutations have been identified in Jewish families of either Ashkenazi or non-Ashkenazi origin [17,18]. The contribution of PALB2 mutations to BC in the South American population is unknown. The Chilean population is the result of admixture between Asian and Spanish populations; therefore, whether germline PALB2 mutations contribute to Chilean BC risk is unknown.
In this study, we screened the coding regions and exon-intron boundaries of the PALB2 gene in BRCA1/2-negative women with familial BC, to determine the contribution of PALB2 variation in the Chilean population. Also, using a case-control design, we studied the c.1676A > G (exon 4), c.1861C > A (exon 5), and c.2993C > T (exon 9) variants to evaluate their possible association with BC susceptibility.
Methods
Families
A total of 436 BC patients belonging to 436 high-risk BRCA1/2-negative Chilean families were selected from the files of the Servicio de Salud del Area Metropolitana de Santiago, Corporación Nacional del Cáncer (CONAC) and other private services in the Metropolitan Area of Santiago. All index cases were tested for BRCA1 and BRCA2 mutations as described [19]. Pedigrees were constructed on the basis of an index case considered to have the highest probability of being a deleterious mutation carrier. None of the families met the strict criteria for other known syndromes involving BC, such as Li-Fraumeni, ataxia-telangiectasia, or Cowden disease.
Table 1 shows the specific characteristics of the families selected according to the inclusion criteria. All families participating in the study self-reported Chilean ancestry dating from several generations, after extensive interviews with several members of each family from different generations. In the selected families, 12.4% (54/436) had cases of bilateral BC; 8.0% (35/436) had cases of both BC and ovarian cancer (OC); and 2.1% (9/436) had male BC. In the BC group, the mean age of diagnosis was 42.7 years, and 75.2% had age of onset < 50 years.
This study was approved by the Institutional Review Board of the School of Medicine of the University of Chile. Informed consent was obtained from all participants.
Control population
The sample of healthy Chilean controls (n = 809) was recruited from CONAC files. DNA samples were taken from unrelated individuals with no personal or familial history of cancer who gave their consent for anonymous testing. These individuals were interviewed and informed as to the aims of the study. DNA samples were obtained according to all ethical and legal requirements. The control sample was matched by age and socioeconomic strata with respect to the cases. Over 90% of cases and controls lived in the city of Santiago.
Mutation analysis
Genomic DNA was extracted from peripheral blood lymphocytes of 436 cases belonging to the high-risk selected families and 809 controls. Samples were obtained according to the method described by Chomczynski and Sacchi [20].
PALB2 complete sequence analysis
This analysis was performed in 100 cases belonging to families with a) three or more family members with breast and/or ovarian cancer and b) two family members with breast and/or ovarian cancer. The whole coding sequence and exon–intron boundaries of the PALB2 gene were amplified by polymerase chain reaction (PCR) using previously-described primers [21]. For exon 9, we designed primers using Web Primer3 version 0.4.0 [22]. We amplified 16 amplicons, whose sizes are shown in Additional file 1: Table S1. The fragments obtained were analyzed for sequence variants using conformational sensitive gel electrophoresis (CSGE) [23]. Amplified samples were denaturated at 95°C for 5 minutes and 65°C for 30 minutes to generate heteroduplex. The products were diluted 1:2 in sucrose buffer and loaded in a partially denaturing MDE® gel (Cambrex, UK) at constant power of 7 W during different time periods depending on the size of the fragment. Gels were silver-stained and dried on a vacuum gel dryer.
Any fragment showing a mobility shift was directly sequenced, to identify the variant. Sequencing was performed in an ABI 3730XL automated fluorescence-based cycle sequencer and a BigDye v.3.1 terminator system (Applied Biosystems, Foster City, CA). The sequences utilized for naming PALB2 sequence variants, obtained from the NCBI RefSeq database, were NM_024675.3 (transcript) and NP_078951.2 (protein).
PALB2 rs152451 (1676A > G), rs45551636 (2993C > T), and c. 1861C > A analysis
Genotyping for rs152451 (c.1679A > G), rs45551636 (c.2993C > T), and c.1861C > A was carried out using TaqMan SNP Genotyping Assays (Applied Biosystems, Foster City, CA) (assay ID C_2392113_10, C_86371270_10, and a customized assay, respectively). The reaction was performed in a 10 uL final volume containing 5 ng of genomic DNA, 1X TaqMan Genotyping Master Mix, and 1X TaqMan SNP Genotyping Assay. Polymerase chain reaction was carried out in a StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, CA). The thermal cycles were initiated for 10 minutes at 95°C, followed by 40 cycles each of 92°C for 15 seconds and 60°C for 1 minute. Each genotyping run contained DNA controls confirmed by sequencing. The alleles were assigned using the SDS 2.2 software (Applied Biosystems, Foster City, CA). As a quality control, we repeated the genotyping on ~10% of the samples, and all genotype scoring was performed and checked separately by two reviewers unaware of the case-control status.
Bioinformatics and statistical analyses
The Hardy-Weinberg equilibrium assumption was assessed in the control sample using a goodness-of-fit chi-square test (HWChisq function included on “HardyWeinberg”.package v.1.4.1). Fisher’s exact test was used to test the association of genotypes and/or alleles between cases and controls. p < 0.05 was used as the criterion of significance. The odds ratios (OR) and their 95% confidence interval (CI) were calculated to estimate the strength of the association between cases and controls (odds ratio fisher function included on “epitools” package v.0.5-6). Haplotype estimation was carried out using UNPHASED v.3.1.5 software, which uses a maximum likelihood approach [24]. In silico analyses of the effect of missense mutations on protein function were carried out using PolyPhen-2 (Polymorphism Phenotyping version 2) [25], SIFT [26] and PROVEAN [27]. To evaluate protein stability we used I-Mutant 3.0 tools (http://gpcr2.biocomp.unibo.it/cgi/predictors/I-Mutant3.0/I-Mutant3.0.cgi). We obtained the wildtype structure of the PALB2 protein from RASMOL (http://rasmol.org/) and used MODELLER v.9.12 (http://salilab.org/modeller/) to analyze the 3D-structure and function of the PALB2 protein.
Results
We analyzed the complete coding sequence and splice-boundary region of PALB2 in 100 probands from BC families negative for BRCA1 and BRCA2 point mutations with the aim of identifying PALB2 sequence variation in a Chilean population. We identified three sequence variants: c.1676A > G (p.Q559R, rs152451) (exon 4), c.1861C > A (p.P621M) (exon 5), and c.2993C > T (p.G998E, rs45551636) (exon 9). The variants detected were analyzed in 436 BC BRCA1/2-negative cases and 809 controls. For the analysis, the whole sample was subdivided into two groups: cases belonging to families with two or more family members with BC and/or OC (n = 273) (subgroup A) and non-familial early-onset BC (≤50 years) (n = 163) (subgroup B).
The variant c. 1861C > A, not previously described in the literature nor in the PALB2 variation database (http://www.lovd.nl/PALB2), was found in only one of the 436 cases and none of the 809 controls. Therefore, this mutation corresponds to a new mutation not previously described.
The c.1676A > G and c.2993C > T variants correspond to previously-described variants (rs152451 and rs45551636, respectively). Table 2 shows the genotype distributions and allele frequencies of rs152451 and rs45551636 variants in the whole data set and in subgroups A and B with respect to the controls. The observed genotype frequencies for the two variants were all in Hardy–Weinberg equilibrium in the controls (P = 0.46 for rs152451, P = 0.68 for rs45551636, respectively).
In the single locus analysis, the genotype and allele distribution for rs152451 did not indicate an association of this variant with increased BC risk either in the whole sample or in subgroup B. However, in the familial BC cases (subgroup A), the minor allele frequency (MAF) (allele G) was higher in cases than in controls (0.12 and 0.09, respectively, p = 0.03). Furthermore, in subgroup A, allele G carriers (AG + GG) were associated with a significantly increased BC risk (OR = 1.5 [CI 95% 1.1-2.1], p = 0.02) (Table 2). We also analyzed the relationship between rs152451 and BC risk within patients with BC familial history according the number of BC cases in the family (Table 3). No association between rs152451 and BC risk was found in cases belonging to families with two BC and/or OC cases. Nevertheless, BC risk was significantly increased in cases belonging to families with 3 or more members affected by BC and/or OC. In these families, the frequency of allele G was 0.16 in BC cases versus 0.09 in controls (OR = 1.9 [CI 95% 1.3-2.8] p < 0.01), and G allele carriers (AG and AG + GG) were associated with a significantly increased BC risk (OR = 2.0 [CI 95% 1.3-3.2] p < 0.01).
With respect to rs45551636, the genotype and allele distribution did not indicate an association of this variant with non-familial early-onset BC (≤50 years) (Table 2). Nevertheless, in the whole sample, the MAF (T allele) was higher in cases (0.03) than controls (0.01), and the difference was statically significant (OR = 2.0 [CI 95% 1.1-3.7], p = 0.03). This result indicates that the T allele is associated with a significantly increased BC risk. We also observed increased risk of BC for T allele carriers (CT + TT) in the whole sample and in subgroup A (OR = 1.9 [CI 95% 1.0-3.6] p = 0.04 and OR = 2.4 [CI 95% 1.2-4.7] p = 0.01, respectively) (Table 2). When we analyze the effect of allele T considering the number of BC cases in the families, no association between rs45551636 and BC risk was found in cases belonging to families with two BC cases. Nevertheless, BC risk increased 3.0-fold in the heterozygous group (OR = 3.0 [CI 95% 1.2-6.8] p < 0.01) and 3.3-fold in allele T carriers (CT + TT) belonging to families with 3 or more cases of BC (OR = 3.3 [CI 95% 1.4-7.3] p < 0.01) (Table 3).
Table 4 shows the composite genotype analysis for rs152451 and rs45551636. The rs152451 A/G - rs45551636 C/T composite genotype showed a higher frequency in cases compared to controls in the whole sample and in subgroup A (OR = 1.9 [CI 95% 1.0–3.6] p = 0.04 and OR = 2.6 [CI 95% 1.4–4.5] p = 0.01 respectively) (Table 4). Nevertheless, the frequency of A/G-C/T composite genotype did not differ between non-familial early-onset BC (≤50 years) versus controls (OR = 0.9 [CI 95% 0.3–2.7] p = 1.0). When we analyzed the relationship between the composite genotype and BC risk within patients with familial BC history according the number of BC cases in the family (Table 4), we observed that the A/G-C/T composite genotype, which includes the two risk alleles, was associated with a significantly increased BC risk both in cases belonging to families with two or more BC and/or OC cases and in cases belonging to families with three or more BC and/or OC cases, with a stronger effect of the composite genotype in the latter group of cases "(OR = 2.6 [CI 95% 1.4-5.0] p = 0.01) and OR = 3.6 [CI 95% 1.7-8.0] p = 0.003, respectively). No association was observed in the families belonging to subgroup B. In conclusion, the A/G-C/T composite genotype produces a higher increase of BC risk in cases with a strong family history of BC and/or OC. This finding raises the possibility that these variants are in strong linkage disequilibrium.
Although phase for these variants could not be determined directly from the screening data, haplotypes were constructed from genotype data using UNPHASED software, which uses a maximum-likelihood approach. Theoretically, the total number of haplotypes with two SNPs is four, but we observed only three: wildtype (rs152451-A - rs45551636-C), rs152451-G - rs45551636-C, and rs152451-G - rs45551636-T. The haplotype estimation suggested a strong linkage disequilibrium between the two markers (coefficient of linkage disequilibrium, D' = 1). When we analyzed the haplotypes and BC risk considering the number of BC cases in the families (Table 5), we observed that rs152451-G - rs45551636-C and rs152451-G - rs45551636-T haplotypes were associated with a significantly increased BC risk in the families with a strong family history of BC and/or OC (OR = 1.6 [CI 95% 1.0-2.5] p = 0.05 and OR = 3.7 [CI 95% 1.8-7.5] p < 0.001, respectively). No association was observed in the families belonging to subgroup B.
Prediction of functional effect of PALB2 variants
The variant c.1861C > A produces the change of Pro to Met at position 621. The multiple alignment analysis with the PALB2 protein sequence indicates that the amino acid in position 621 is conserved in various species. The case-control study showed that this variant was detected in only one of 436 cases and none of the 809 controls. This results suggest that the variant c.1861C > A could be pathogenic. In order to predict the possible effect of this amino acid change in the structure and function of the PALB2 protein, we used PolyPhen-2, SIFT and PROVEAN software, which predicted that this variant is not likely to be pathogenic. To evaluate protein stability, we used the i-Mutant program. The amino acid change decreases the protein stability (-0.55 Kcal/mol). Considering the case-control study and in silico analyses, the c.1861C > A missense mutation can be classified as a variant of unknown significance (VUS), or as a rare variant.
The variant c.1676A > G (p.Q559R) corresponds to a previously-described variant (rs152415). The three pathogenicity programs predicted that it is likely non-pathogenic. The i-Mutant program shows that the amino acid change slightly decreases the protein stability (-0.06 Kcal/mol). The variant c.2993C > T (p.G998E) is also a previously-described variant (rs45551636). The results of the bioinformatic predictor programs indicated that it is probably pathogenic. The results obtained with the i-Mutant program predicted that the mutation decreases the protein stability (-0.41 Kcal/mol). In order to analyze the possible effect of this variant on the 3D-structure and function of the PALB2 protein, we carried out homology modeling using MODELLER v.9.12 (Additional file 2). The model obtained was compared in RASMOL with the structure of the wildtype protein, with results predicting that the amino acid change might affect this region and then modify PALB2 function. Considering there are currently no published functional studies, this variant could be classified as a variant of unknown significance.
Discussion
Mutations in BRCA1 and BRCA2 are associated with susceptibility to breast and ovarian cancer. At present, however, those mutations account for only a portion of familial cases, and consequently there is an intensive search for additional targets. PALB2 mutations have been identified as BC susceptibility alleles by both case-control and family studies [9,10,13,17,21,28-35], and are closely associated with BRCA2. Since the initial identification of PALB2 as a BC susceptibility gene [9], several investigators have screened for the gene in BRCA1/2-negative families with BC and/or women diagnosed with early-onset BC from various ethnic backgrounds, such as Australian [12,36], Chinese [16], German [32], Italian [33,37,38], Dutch [39], North American [17,21,40-44], Polish [31], Russian [32], South African [45], and Spanish [46,47] populations. However, the prevalence of PALB2 variants in the South American population had not yet been established. In the present study, we analyzed the complete coding sequence and exon-intron boundaries of PALB2 in Chilean BRCA1/2-negative BC patients; nevertheless, no pathogenic mutations were observed in the PALB2 coding sequence. Pathogenic mutations in PALB2 are rare (varying from 0.1% to 2.7%) and vary in frequency depending on the population [48]. No PALB2 germline mutations have been observed in the geographically-confined population of Iceland, for instance [49]. The contemporary Chilean population stems from the admixture of Amerindian peoples with the Spanish settlers in the 16th and 17th centuries. Later migrations in the 19th century of other populations (e.g., Germans, Italians, Arabs, and Croatians) have had only a minor impact on the overall population (≤4% of the total population) and are restricted to the specific locations of the country where they originally settled [50]. The relationships among ethnicity, Amerindian admixture, genetic markers, and socioeconomic strata have been extensively studied in Chile [51-53]. Thus, it is probable that in the mixed Chilean population, PALB2 is not a significant contributor to BC in high-risk BC families.
We identified three sequence variants: c.1676A > G (exon 4), c.1861C > A (exon 5), and c.2993C > T (exon 9). With respect to c.1861C > A (exon 5), this variant is not previously described in the literature or in the PALB2 mutation and polymorphism database (http://www.lovd.nl/PALB2). It corresponds to a missense mutation that produces the change of proline to methionine in the 621 position (p.P621M). It was found in only one of the 436 cases and in none of 809 controls. The patient carrier of this variant belongs to a BRCA1/2-negative family with three BC cases. Three sisters were screened for the variant, two with BC (diagnosed at 45 and 49 years) and the other healthy. The c.1861C > A carrier was the sister with BC with the earliest age of diagnosis. The bioinformatic analyses using PolyPhen-2, SIFT and PROVEAN tools predicted that this variant is not pathogenic, and therefore we classified this missense mutation as a new mutation of unknown significance.
The variants c.1676A > G (p.Q559R) and c.2993C > T (p.G998E) correspond to previously-described variants (rs152451 and rs45551636, respectively). These have been described in Spanish [46], Australian [29], Asian (Malaysian and Singaporean) [15], African–American [54], German and Russian [32], Italian [33], Chinese [16], and Finnish [10] populations. It is widely accepted that according to bioinformatic analyses, the c.1676A > G variant corresponds to a benign polymorphism while c.2993C > T affects PALB2 protein function. In the present study, using a case-control design, we evaluated the impact of PALB2 rs152451 and rs45551636 variants in Chilean women with familial and non-familial early-onset BC who are negative for BRCA1/2 point mutations. In both cases and controls, the allelic frequencies of both variants were similar to those reported by Blanco et al. (2013) [46] in a Spanish population. The MAF of rs152451 and rs45551636 in cases (0.10 and 0.03, respectively) differed from those reported in German (0.06 and 0.03, respectively) [32], Russian (0.06 and 0.01, respectively) [32], and African-American (0.16 and 0.005, respectively) [54] populations. In controls, the MAF of rs152451 and rs45551636 were 0.09 and 0.01 respectively, differing from those reported in the NHBLI ESP6500 database for African-Americans (0.22 and 0.006 respectively) but similar to those reported for European-Americans (0.09 and 0.02 respectively). Therefore, MAF frequencies vary by ethnicity. In the present study, both variants were significantly associated with increased risk of familial BC but not non-familial early-onset BC. On the other hand, when we consider the number of BC cases in the family, we found a significantly increased BC risk only in the carriers of allele G (AG + GG) in families with 3 or more members affected with BC and/or OC (OR = 2.0 [CI 95% 1.3-3.2], p < 0.01). Therefore, we propose that these two variants are associated with increased BC risk in families with a strong family history of BC. These results are in agreement with those published by Antoniou et al. [55], who concluded that the pathogenic mutations in PALB2 confer an absolute risk of 58% for those women with two or more first-degree relatives with BC at 50 years of age, and that the risk is lower for women with no family history of BC. This result was confirmed by the composite genotype analysis, suggesting that these two variants could be in linkage disequilibrium. The haplotype analysis also suggested strong linkage disequilibrium between the two markers. Also, we observed that rs152451-G/rs45551636-C and rs152451-G/rs45551636-T were associated with significantly increased BC risk in families with a strong family history of BC. In the literature, there are no publications with the analysis performed in the present manuscript.
The prediction of functional effects revealed that rs152451 is probably non-pathogenic. Nevertheless, the region of this variant is found inside the PALB2 motif, which interacts with chromatin [56]. This region was not crystallized; therefore, no 3D-structure information is available to evaluate the possible impact of this mutation. However, the analyses of the amino acid change properties (p.Q559R) lead us to propose that this variant might have an impact on PALB2 function. Functional analyses are needed to elucidate the effect of c.1676A > C on PALB2 protein function, and thus to confirm the results obtained in the case-control study. Regarding rs45551636, the bioinformatic analysis using PolyPhen-2, SIFT and PROVEAN tools predict that this variant is probably pathogenic. c.2993C > T is located within the PALB2 WD40-domain, which is involved in the interaction of the PALB2 protein with BRCA2 and RAD51 [56]. To analyze the effect of the amino acid change on PALB2 protein function, the model obtained using MODELLER v.9.12 was compared with the wildtype PALB2 structure. We observed that the wildtype structure Gly, which is smaller and very flexible, takes part in the loop between WD 2 and 3 repetitions. On the other hand, in the mutated structure, glutamic acid, which is more voluminous and negatively-charged, is protruded towards the interior of the hydrophobic pocket formed by WD40 repetitions resulting in an unusual torsion angle localized at the loop that connects these regions. In addition, with the evidence obtained from the bioinformatic tools, the structure-modeling analysis strongly suggests that this variant could affect the protein function.
Conclusions
Our data shows that PALB2 pathogenic germline mutations are not present in a Chilean population, and that rs152451 and rs45551636 are associated with increased BC risk in BRCA1/2- negative families with a strong family history of BC but not in non-familial early-onset BC. These results are in agreement with Southey et al. (2010) [12].
References
Walsh T, King MC. Ten genes for inherited breast cancer. Cancer Cell. 2007;11:103–5.
Chen P, Liang J, Wang Z, Zhou X, Chen L, Li M, et al. Association of common PALB2 polymorphisms with breast cancer risk: a case-control study. Clin Cancer Res. 2008;14:5931–7.
Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458:719–24.
Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, et al. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell. 2006;22:719–29.
Sy SMH, Huen MSY, Chen JJ. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc Natl Acad Sci U S A. 2009;106:7155–60.
Zhang F, Ma JL, Wu JX, Ye L, Cai H, Xia B, et al. PALB2 Links BRCA1 and BRCA2 in the DNA-Damage Response. Curr Biol. 2009;19:524–9.
Zhang F, Fan Q, Ren K, Andreassen PR. PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. Mol Cancer Res. 2009;7:1110–8.
Reid S, Schindler D, Hanenberg H, Barker K, Hanks S, Kalb R, et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat Genet. 2007;39:162–4.
Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet. 2007;39:165–7.
Erkko H, Xia B, Nikkilae J, Schleutker J, Syrjaekoski K, Mannermaa A, et al. A recurrent mutation in PALB2 in Finnish cancer families. Nature. 2007;446:316–9.
Foulkes WD, Ghadirian P, Akbari MR, Hamel N, Giroux S, Sabbaghian N, et al. Identification of a novel truncating PALB2 mutation and analysis of its contribution to early-onset breast cancer in French-Canadian women. Breast Cancer Res. 2007;9(6):R83.
Southey MC, Teo ZL, Dowty JG, Odefrey FA, Park DJ, Tischkowitz M, et al. A PALB2 mutation associated with high risk of breast cancer. Breast Cancer Res. 2010;12(6):R109.
Erkko H, Dowty JG, Nikkila J, Syrjakoski K, Mannermaa A, Pylkas K, et al. Penetrance analysis of the PALB2 c.1592delT founder mutation. Clin Cancer Res. 2008;14:4667–71.
Hollestelle A, Wasielewski M, Martens JW, Schutte M. Discovering moderate-risk breast cancer susceptibility genes. Curr Opin Genet Dev. 2010;20:268–76.
Phuah SY, Lee SY, Kang P, Kang IN, Yoon SY, Thong MK, et al. Prevalence of PALB2 mutations in breast cancer patients in multi-ethnic Asian population in Malaysia and Singapore. PLoS One. 2013;8:e73638.
Cao AY, Huang J, Hu Z, Li WF, Ma ZL, Tang LL, et al. The prevalence of PALB2 germline mutations in BRCA1/BRCA2 negative Chinese women with early onset breast cancer or affected relatives. Breast Cancer Res Treat. 2009;114:457–62.
Casadei S, Norquist BM, Walsh T, Stray S, Mandell JB, Lee MK, et al. Contribution of inherited mutations in the BRCA2-interacting protein PALB2 to familial breast cancer. Cancer Res. 2011;71:2222–9.
Catucci I, Milgrom R, Kushnir A, Laitman Y, Paluch-Shimon S, Volorio S, et al. Germline mutations in BRIP1 and PALB2 in Jewish high cancer risk families. Familial Cancer. 2012;11:483–91.
Gonzalez-Hormazabal P, Gutierrez-Enriquez S, Gaete D, Reyes JM, Peralta O, Waugh E, et al. Spectrum of BRCA1/2 point mutations and genomic rearrangements in high-risk breast/ovarian cancer Chilean families. Breast Cancer Res Treat. 2011;126:705–16.
Chomczynski P, Sacchi N. The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: twenty-something years on. Nat Protoc. 2006;1:581–5.
Tischkowitz M, Xia B, Sabbaghian N, Reis-Filho JS, Hamel N, Li GL, et al. Analysis of PALB2/FANCN-associated breast cancer families. Proc Natl Acad Sci U S A. 2007;104:6788–93.
Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, et al. Primer3-new capabilities and interfaces. Nucleic Acids Res. 2012;40(15):e115.
Korkko J, Annunen S, Pihlajamaa T, Prockop DJ, Ala-Kokko L. Conformation sensitive gel electrophoresis for simple and accurate detection of mutations: Comparison with denaturing gradient gel electrophoresis and nucleotide sequencing. Proc Natl Acad Sci U S A. 1998;95:1681–5.
Dudbridge F. Likelihood-based association analysis for nuclear families and unrelated subjects with missing genotype data. Hum Hered. 2008;66:87–98.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–81.
Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012;7:e46688.
Teo ZL, Sawyer SD, James PA, Mitchell G, Trainer AH, Lindeman GJ, et al. The incidence of PALB2 c.3113G>A in women with a strong family history of breast and ovarian cancer attending familial cancer centres in Australia. Familial Cancer. 2013;12:587–95.
Teo ZL, Park DJ, Provenzano E, Chatfield CA, Odefrey FA, Nguyen-Dumont T, et al. Prevalence of PALB2 mutations in Australasian multiple-case breast cancer families. Breast Cancer Res. 2013;15:R17.
Tischkowitz M, Sabbaghian N, Hamel N, Pouchet C, Foulkes WD, Mes-Masson AM, et al. Contribution of the PALB2 c.2323C > T [p.Q775X] Founder mutation in well- defined breast and/or ovarian cancer families and unselected ovarian cancer cases of French Canadian descent. BMC Med Genet. 2013;14:5.
Dansonka-Mieszkowska A, Kluska A, Moes J, Dabrowska M, Nowakowska D, Niwinska A, et al. A novel germline PALB2 deletion in Polish breast and ovarian cancer patients. BMC Med Genet. 2010;11:20.
Bogdanova N, Sokolenko AP, Iyevleva AG, Abysheva SN, Blaut M, Bremer M, et al. PALB2 mutations in German and Russian patients with bilateral breast cancer. Breast Cancer Res Treat. 2011;126:545–50.
Balia C, Sensi E, Lombardi G, Roncella M, Bevilacqua G, Caligo MA. PALB2: a novel inactivating mutation in a Italian breast cancer family. Familial Cancer. 2010;9:531–6.
Hellebrand H, Sutter C, Honisch E, Gross E, Wappenschmidt B, Schem C, et al. Germline mutations in the PALB2 gene are population specific and occur with low frequencies in familial breast cancer. Hum Mutat. 2011;32:E2176–88.
Tischkowitz M, Capanu M, Sabbaghian N, Li LL, Liang XL, Vallee MP, et al. Rare germline mutations in PALB2 and breast cancer risk: a population-based study. Hum Mutat. 2012;33:674–80.
Wong MW, Nordfors C, Mossman D, Pecenpetelovska G, Avery-Kiejda KA, Talseth-Palmer B, et al. BRIP1, PALB2, and RAD51C mutation analysis reveals their relative importance as genetic susceptibility factors for breast cancer. Breast Cancer Res Treat. 2011;127:853–9.
Papi L, Putignano AL, Congregati C, Piaceri I, Zanna I, Sera F, et al. A PALB2 germline mutation associated with hereditary breast cancer in Italy. Familial Cancer. 2010;9:181–5.
Peterlongo P, Catucci I, Pasquini G, Verderio P, Peissel B, Barile M, et al. PALB2 germline mutations in familial breast cancer cases with personal and family history of pancreatic cancer. Breast Cancer Res Treat. 2011;126:825–8.
Adank MA, van Mil SE, Gille JJ, Waisfisz Q, Meijers-Heijboer H. PALB2 analysis in BRCA2-like families. Breast Cancer Res Treat. 2011;127:357–62.
Ding YC, Steele L, Kuan CJ, Greilac S, Neuhausen SL. Mutations in BRCA2 and PALB2 in male breast cancer cases from the United States. Breast Cancer Res Treat. 2011;126:771–8.
Ding YC, Steele L, Chu LH, Kelley K, Davis H, John EM, et al. Germline mutations in PALB2 in African-American breast cancer cases. Breast Cancer Res Treat. 2011;126:227–30.
Ghadirian P, Robidoux A, Zhang P, Royer R, Akbari M, Zhang S, et al. The contribution of founder mutations to early-onset breast cancer in French-Canadian women. Clin Genet. 2009;76:421–6.
Tischkowitz MD, Sabbaghian N, Hamel N, Borgida A, Rosner C, Taherian N, et al. Analysis of the gene coding for the BRCA2-interacting protein PALB2 in familial and sporadic pancreatic cancer. Gastroenterology. 2009;137:1183–6.
Hofstatter EW, Domchek SM, Miron A, Garber J, Wang M, Componeschi K, et al. PALB2 mutations in familial breast and pancreatic cancer. Familial Cancer. 2011;10:225–31.
Sluiter M, Mew S, van Rensburg EJ. PALB2 sequence variants in young South African breast cancer patients. Familial Cancer. 2009;8:347–53.
Blanco A, de la Hoya M, Osorio A, Diez O, Miramar MD, Infante M, et al. Analysis of PALB2 gene in BRCA1/BRCA2 negative Spanish hereditary breast/ovarian cancer families with pancreatic cancer cases. PLoS One. 2013;8:e67538.
Garcia MJ, Fernandez V, Osorio A, Barroso A, Llort G, Lazaro C, et al. Analysis of FANCB and FANCN/PALB2 fanconi anemia genes in BRCA1/2-negative Spanish breast cancer families. Breast Cancer Res Treat. 2009;113:545–51.
Southey MC, Teo ZL, Winship I. PALB2 and breast cancer: ready for clinical translation! Appl Clin Genet. 2013;6:43–52.
Gunnarsson H, Arason A, Gillanders EM, Agnarsson BA, Johannesdottir G, Johannsson OT, et al. Evidence against PALB2 involvement in Icelandic breast cancer susceptibility. J Negat Results Biomed. 2008;7:5.
Cruz-Coke R. Ethnic origin and evolution of the Chilean population. Rev Med Chil. 1976;104:365–8.
Valenzuela CY, Harb Z. Socioeconomic assortative mating in Santiago, Chile: a demonstration using stochastic matrices of mother-child relationships applied to ABO blood groups. Soc Biol. 1977;24:225–33.
Valenzuela CY, Acuna MP, Harb Z. [Sociogenetic gradient in the Chilean population]. Rev Med Chil. 1987;115:295–9.
Valenzuela CY, Carvajal T, Marinkovic K. [Differences in hematologic variables according to ABO blood group]. Rev Med Chil. 1988;116:981–4.
Zheng Y, Zhang J, Niu Q, Huo D, Olopade OI. Novel germline PALB2 truncating mutations in African American breast cancer patients. Cancer. 2012;118:1362–70.
Antoniou AC, Foulkes WD, Tischkowitz M. Breast-cancer risk in families with mutations in PALB2. N Engl J Med. 2014;371:1651–2.
Sy SMH, Huen MSY, Chen JJ. MRG15 is a novel PALB2-interacting factor involved in homologous recombination. J Biol Chem. 2009;284:21127–31.
Acknowledgements
The authors thank the many families who participated in the research studies described in this article. We acknowledge the Breast Cancer Group of CONAC: Maria Teresa Barrios, Angelica Soto, Rossana Recabarren, Leticia Garcia, Karen Olmos and Paola Carrasco; and Lorena Seccia for her technical assistance. Grant sponsor: Fondo Nacional de Desarrollo Científico y Tecnológico (FONDECYT). Grant number: 1110081. Grant sponsor: Corporación Nacional del Cáncer.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interest
The authors declare that they have no competing interests.
Authors’ contributions
LJ conceived of the study, and participated in its design and coordination; YL and NL carried out the molecular genetic studies; YL and JCT performed the bioinformatic analysis; LJ, RB, RFR and SM prepared the manuscript, PGH participated in the design of the study and performed the statistical analysis; JMR, TB, OP, FG, EW, GI selected familial breast cancer cases from different oncology services, TB was responsible of controls selection, JP and GG curated the subject's database. All authors read and approved the final manuscript.
Additional files
Additional file 1: Table S1.
Primer sequences to amplify the whole coding sequence and exon–intron boundaries of the PALB2 gene.
Additional file 2:
Comparison of the WD40 domain’s three-dimensional structure in the wildtype PALB2 protein and the p.G998E (c.2993C > T) variant.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Leyton, Y., Gonzalez-Hormazabal, P., Blanco, R. et al. Association of PALB2 sequence variants with the risk of familial and early-onset breast cancer in a South-American population. BMC Cancer 15, 30 (2015). https://doi.org/10.1186/s12885-015-1033-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-015-1033-3