
Abstract
Background
PALB2 protein was recently identified as a partner of BRCA1 and BRCA2 which determines their proper function in DNA repair.
Methods
Initially, the entire coding sequence of the PALB2 gene with exon/intron boundaries was evaluated by the PCR-SSCP and direct sequencing methods on 70 ovarian carcinomas. Sequence variants of interest were further studied on enlarged groups of ovarian carcinomas (total 339 non-consecutive ovarian carcinomas), blood samples from 334 consecutive sporadic and 648 consecutive familial breast cancer patients, and 1310 healthy controls from central Poland.
Results
Ten types of sequence variants were detected, and among them four novel polymorphisms: c.2996+58T>C in intron 9; c.505C>A (p.L169I), c.618T>G (p.L206L), both in exon 4; and c.2135C>T (A712V) in exon 5 of the PALB2 gene. Another two polymorphisms, c.212-58A>C and c.2014G>C (E672Q) were always detected together, both in cancer (7.5% of patients) and control samples (4.9% of controls, p = 0.2). A novel germline truncating mutation, c.509_510delGA (p.R170fs) was found in exon 4: in 2 of 339 (0.6%) unrelated ovarian cancer patients, in 4 of 648 (0.6%) unrelated familial breast cancer patients, and in 1 of 1310 controls (0.08%, p = 0.1, p = 0.044, respectively). One ovarian cancer patient with the PALB2 mutation had also a germline nonsense mutation of the BRCA2 gene.
Conclusions
The c.509_510delGA is a novel PALB2 mutation that increases the risk of familial breast cancer. Occurrence of the same PALB2 alteration in seven unrelated women suggests that c.509_510delGA (p.R170fs) is a recurrent mutation for Polish population.
Similar content being viewed by others


Background
PALB2 protein [OMIM #610355, a partner and localizer of BRCA2] was recently identified in a complex with BRCA2 (Breast Cancer 2) protein [1]. PALB2 supports BRCA2 stability and determines its localization in the nucleus after DNA damage [1]. Relocation of PALB2 and BRCA2 to damaged chromatin is regulated by BRCA1 (Breast Cancer 1) protein [2]. These three proteins form a complex in which PALB2 acts as a bridge between BRCA1 and BRCA2 [2]. In cells depleted of PALB2, DNA repair pathway dependent on the BRCA1/2 is disrupted [1, 2]. This suggests that inactivation of the PALB2 gene may cause similar biological and phenotypic changes as inactivation of the BRCA1 or BRCA2 genes; the latter is responsible for a fraction of breast and ovarian cancers, and a dysfunction of BRCA2 alone - for the Fanconi anemia (FA) syndrome.
FA syndrome is a genetic disorder caused by germline biallelic mutations in one of 13 genes (including FANCD1/BRCA2 and FANCN/PALB2) of the so-called FA/BRCA pathway; this pathway controls the repair of double strand-breaks and the response to DNA crosslinking agents [3, 4]. Studies on FA patients provide evidence that biallelic mutations of the PALB2 gene are responsible for one of the syndrome's subtypes, which is very similar to the one caused by inactivation of the BRCA2 gene [5, 6].
Mutations of the BRCA1 and BRCA2 genes are the best known alterations of the FA/BRCA pathway. Heterozygous germline mutations of these genes predispose to breast and ovarian cancers. The cumulative risk of ovarian cancer to age 70 years for women with a BRCA1 or BRCA2 mutation is estimated at about 40% and 10%, respectively [7]. The cooperation between BRCA2 and PALB2 during the DNA repair process and similarity in clinical features of the FA syndrome caused by biallelic mutations of BRCA2 and PALB2 genes suggest the role of PALB2 alterations in carcinogenesis. To date, PALB2 alterations have been associated with familial breast [8–13] and pancreatic [14, 15] cancers. To our knowledge, PALB2 gene has not been extensively studied in ovarian cancer patients.
We demonstrate a novel inherited monoallelic deletion of the PALB2 gene in ovarian and breast cancer patients which seems to be a recurrent mutation for the population of central Poland.
Methods
Subjects
The study was performed on a series of 339 non-consecutive patients with ovarian cancer and 982 patients with breast cancer; all patients were Caucasian women from central Poland.
Ovarian cancer patients
The analysis was performed on tumor fragments (n = 339); patients' blood samples were used to confirm germline origin of the detected variants only. The samples were collected in two Warsaw hospitals (Institute of Oncology and Brodnowski Hospital) between the years 1995 and 2007. These hospitals treat cancer patients from central Poland. Mean age of patients was 56 years (range 17-88). Distribution of cases in appropriate age intervals in the group studied was comparable to that in the whole population of ovarian cancer patients in Poland [16]. Tumors were classified histologically according to the criteria of the World Health Organisation [17]. There were 231 (68%) serous carcinomas, 33 (10%) endometrioid, 15 (4%) mucinous, 26 (8%) clear cell, 17 (5%) undifferentiated and 17 (5%) other type carcinomas. BRCA1 mutational status (exons 2, 5, 11 and 20) had been previously determined in the majority of ovarian carcinomas (n = 243), and 39 (16%) were positive [18]. A few carcinomas were also studied for BRCA2 mutations in exons 2, 3, 11 (c.3035-6629) and 25 (Moes et al., data not published).
Breast cancer patients
DNA from the peripheral blood leukocytes was analyzed in 334 sporadic and 648 familial breast cancer cases, the latter with at least one first-, second- or third-degree relative affected by breast and/or ovarian cancer. In 40 families with third-degree relative affected, additional criteria were employed: carcinomas developing in a proband and/or a relative before the age of 50, metachronous or synchronous cancers diagnosed in a proband or a relative, or non-breast and non-ovarian cancers diagnosed in first- or second-degree relatives. The probands were patients of the following departments of the Institute of Oncology: Breast Tumors and Reconstructive Surgery, and Genetic Counselling. BRCA1 mutations (exons 2, 5, 11 and 20) had been previously detected for the purpose of counselling in 75 of 982 breast cancer patients (8.3%). The mean age of patients was 50 (range 21-81) and 48 years (range 24-85) for the sporadic and familial breast cancers, respectively.
Controls
As a control, we used 1310 blood samples from unrelated Caucasian women from central Poland. These samples were anonymously collected in Warsaw blood donation centers between November 2005 and February 2009 (n = 1142). A part of control blood samples (n = 168) were collected among employees and cancer-free patients from Brodnowski Hospital in Warsaw. Mean age of females from the control group was 32 years (range 18 to 75).
Patients gave their written informed consent to be included in the study. Anonymous blood donors gave informed consent. The study was approved by the bioethics committee of the Institute of Oncology (ref. no. 39/2007).
Molecular genetic methods
All 13 PALB2 exons with intron/exon boundaries were initially screened for variants in 70 non-consecutive ovarian carcinomas by the PCR-SSCP and sequencing methods. Sequence variants of interest were further studied in larger groups of ovarian carcinomas and in control blood samples (see: Results). The germline origin of changes detected in ovarian carcinomas was confirmed in blood samples from those patients. The analyses were performed in the Department of Molecular Pathology. Blood from breast cancer patients was screened only for the c.509_510delGA PALB2 deletion in the Department of Endocrinology, with the use of the dHPLC method.
DNA extraction
Fresh ovarian cancer specimens were snap-frozen and stored at -68°C. Cryostat sections were cut and evaluated by a pathologist (JK) as to the sufficient content of tumor tissue. Blood samples were collected in vials which contained EDTA, frozen and stored at -68°C. DNA from both ovarian carcinomas and blood samples was extracted with the use of proteinase K and the QIAamp Mini kit (Qiagen), and stored in the AE buffer (Qiagen). DNA from blood samples of breast cancer patients was isolated with the Genomic Midi AX kit (A & A Biotechnology, Gdansk, Poland).
PCR - Polymerase Chain Reaction
DNA fragments were amplified by the PCR method. Primers (presented in Table 1) were designed using the free Primer3 software [19] and the PALB2 genomic sequence, obtained from the NCBI genome browser [GenBank: NG_007406.1].
PCR mixture was prepared according to a standard method (Applied Biosystems PCR kit). PCR reaction was carried out for 36 cycles in a programmable thermal cycler (Biometra) with denaturation at 94°C, annealing at 56-58°C (depending on the exon) and extension at 72°C, for 30 sec each.
Single strand conformational polymorphism analysis (SSCP)
All thirteen coding exons of the PALB2 gene were screened by the SSCP method. In our experience, this method detects 90% of all alterations, and 100% of deletions and insertions [20]. PCR products were denatured with 0.1 M NaOH containing 2 mM EDTA, at 55°C for 15 min. Immediately after 95% formamide, 0.05% xylene cyanol and 0.05% bromphenol blue were added, the samples were loaded to polyacrylamide gels (1:39 N, N'-methylenebisacrylamide to acrylamide in 0.5 × TBE with 10% glycerol). Electrophoresis was performed at 100 V, for 16-24 hours at room temperature. DNA bands were visualised by the silver-staining method compilated from several procedures.
Denaturing High-Performance Liquid Chromatography
DNA from breast cancer patients was screened for PALB2 c.509_510delGA only (with Ex 4b-F and Ex 4b-R primers), using the dHPLC method carried out on automated dHPLC instrumentation (Transgenomic Inc). Crude PCR products were eluted with linear acetonitryle gradient. The gradient and the temperature required for successful resolution of heteroduplex molecules was determined with the use of the dHPLC melting algorithm (Transgenomic Inc). Mutation positive and negative controls for the screened mutation were always run with each set of samples. The heterozygous profiles were identified by visual inspection of the chromatograms, on the basis of appearance of additional, earlier eluting peaks.
Sequencing
We sequenced all variants detected by the SSCP and dHPLC. PCR products were purified by exonuclease I and alkaline phosphatase treatment, and then sequenced with the use of fluorescent automated method (BigDye Terminator Cycle Sequencing Kit v3.1, Applied Biosystems) with ABI PRISM 377 DNA sequencer.
PALB2 Haplotyping
DNA extracted from blood samples of PALB2 mutation carriers was genotyped with the use of three markers applied by Foulkes et al. [8]: D16S841 [UniSTS:2638], D16S403 [UniSTS:150021], and D16S417 [UniSTS:67206]. Primer sequences were obtained from the NCBI [21] UniSTS database. Marker and PALB2 gene positions were reckoned from NCBI [21] Homo sapiens chromosome 16 genomic contig (reference sequence: NT_010393.16). The loci were amplified by PCR (as described earlier, at the annealing temperature of 58°C) with the use of fluorescently labelled (6-FAM dye) forward primer. PCR products were diluted in sterile water, in a volume depending on reaction efficiency. The dilution at the volume of 0.8 μl was mixed with 8 μl of formamide (SIGMA) and 0.4 μl of the standard (Gene Scan 500 ROX, Applied Biosystems). The mixture was denaturated for 5 min at 95°C and then cooled on ice. Electrophoresis was performed in the ABI PRISM 377 DNA sequencer. Data were analyzed using the Peak Scanner Software v1.0 (Applied Biosystems).
Immunohistochemical stainings
Markers of basal/luminal types were evaluated immunohistochemically with the use of the following antibodies: anti-CK5/6 (DAKO, clone D5/16B4), anti-CK14 (Novocastra, clone NCL-L-LL002), anti-CK17 (DAKO, clone IR620), anti-EGFR (DAKO, clone K1994), anti-CD117 (DAKO, clone A4502). All immunostainings were performed against negative controls. Non-neoplastic mammary gland structures served as intrinsic positive controls.
Statistical analysis
Differences between the compared groups were analyzed by the two-sided Fisher's exact test [22]. The level of statistical significance was set at <0.05.
Results
The initial screening of 70 ovarian carcinomas revealed nine substitutions and one deletion of the PALB2 gene. These alterations, and the final number of the samples studied, are shown in Table 2.
Substitutions (studied in ovarian cancer patients only)
Nine substitutions were detected, four of which are novel (Table 2). Two sequence variants, i.e. c.212-58A>C in intron 3 and c.2014G>C in exon 5 were always detected together, both in cancer and control samples (p = 0.2). The majority of carcinomas and controls carrying these alterations were heterozygous for exon 5 and intron 3. Only one tumor and one control DNA were homozygous, and there were alleles C in both polymorphic sites.
c.509_510delGA deletion
We discovered a novel 2 base-pair deletion c.509_510delGA in exon 4 of the PALB2 gene (Figure 1, Table 2). It resulted in p.R170fs and created a premature stop codon at position 183.

The c.509_510delGA detected in the PALB2 gene in ovarian carcinomas. SSCP gel (in the middle) - shifted bands are indicated by arrows; on sequencing diagrams the deletion site is indicated by arrows.
Ovarian cancer patients
The alteration c.509_510delGA was detected in 2 of 339 ovarian cancers (0.6%) and in 1 of 1310 controls (0.08%; p = 0.1). The deletion was present in both tumor and blood DNA of the two patients. The analysis of sequencing diagrams (Figure 1) of tumor sample number 375 (about 30% of stromal cell contamination) and blood DNA of the same patient suggests the loss of heterozygosity at this locus. LOH was also observed at two of the three studied microsatellite loci (D16S481 and D16S403, data not shown).
One of the two patients with the deletion (tumor number 293) was diagnosed with a poorly differentiated endometrioid carcinoma at the age of 61 (The Federation Internationale de Gynecologie et d'Obstetrique staging system - FIGO IIIC, histological grade 3). She reported a history of cancers in her father's family. The father had a stomach cancer diagnosed at the age of 67, his sister had a lung cancer (age unknown), his brother had an unknown cancer at an unknown age. This patient also carried a nonsense germline mutation i.e. c.C4513T (Q1429X) in the BRCA2 gene.
The other patient (tumor number 375) was diagnosed with a poorly differentiated serous carcinoma at the age of 54 (FIGO IIIC, histological grade 3). She reported a history of cancers in her mother and her mother's sisters (thyroid and pancreatic carcinomas mentioned, age unknown). No mutations in exons 2, 3, 11 (c.3035-6629) and 25 of the BRCA2 gene were found.
Both of these patients died and we did not have a chance to confirm these data.
Breast cancer patients
Screening of 982 consecutive sporadic or familial breast cancer patients for c.509_510delGA resulted in detection of the additional four mutation carriers. The deletion was found in 4/648 (0.6%) familial breast cancer patients versus 1/1310 controls (p = 0.044).
Histopathological features of breast carcinomas from four of the deletion carriers are presented in Table 3. All but one had triple (ER, PR, HER2)-negative phenotype. To characterize breast carcinomas with the PALB2 mutation more specifically, we evaluated the expression of CK5/6, CK14, CK17, EGFR and CD117 (Table 3). Two carcinomas were of the basal type and two of the luminal type.
Pedigree diagrams were available for three patients (Figure 2). The pedigree was not available for the patient number 893, but she stated a breast cancer in her grandmother at the age of 87.

Pedigrees of breast cancer families with PALB2 mutations. Probands with confirmed mutation are indicated by an arrow. Br -breast cancer, Sto -stomach cancer, Liv -liver cancer, Lu - lung cancer, Pan - pancreatic cancer.
Haplotype analysis
Genotyping of the seven c.509_510delGA deletion carriers was performed with three microsatellite markers: D16S417 which is distal to PALB2, and D16S481 and D16S403 that are proximal to this gene. Genotypes are presented in Table 4. There was no common haplotype for all PALB2 mutations carriers with regard to the three markers analyzed.
Discussion
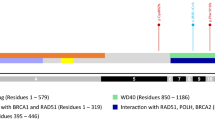
We discovered a novel PALB2 germline deletion, c.509_510delGA, located in exon 4. It may be expected that due to this deletion PALB2 protein is shortened to 169 amino acids from 1186 amino acids of the wild type and it is devoid of C-terminal domain (with WD40 repeats). WD40 motifs are thought to be responsible for protein-protein interactions and they seem to be necessary for BRCA2/PALB2 complex formation [11]. It was shown that PALB2 mutants (229delT, 2521delA and c.1592delT) that lacked C-terminal domain had highly reduced BRCA2 binding capacity and were defective in the repair of double-strand breaks and mitomycin C-induced damages [10, 11]. To date, all PALB2 gene alterations detected in families with breast cancer or FA disease were frameshift or nonsense changes, leading to the expression of a truncated protein. These data strongly support the assumption that the deletion detected in this study results in the production of an inactive protein. Silent and missense sequence variants of the PALB2 gene have been detected in previous studies in both cancer samples and in control tissues, but none of those variants have been strongly associated with cancer susceptibility [9, 10, 12].
Our study proves that PALB2 alterations contribute to the familial but not to the sporadic breast cancer in Poland. This is in agreement with previous reports, in which alterations of the PALB2 gene were predominantly associated with familial breast cancer [9–13]. Some authors reported PALB2 mutations also in pancreatic cancer [14, 15], male breast cancer [9] and in a single prostate cancer family [10]. Pancreatic cancer was mentioned at least twice in the family histories of our PALB2 mutation carriers.
The low frequency of PALB2 mutations (0.6%) observed in our study is similar to that noticed for breast cancer by other authors [8, 9, 11–13]. Only Erkko et al. [10] found a higher rate of mutations in the familial breast cancer: 2.7%. In their study, however, the rate of the mutations in the control population was also relatively high (0.2%). No other study except Erkko's et al. [10] and our study found PALB2 truncating mutations in control populations.
We are the first to present a PALB2 truncating mutation in a patient with medullary breast carcinoma. To date, such alterations have been detected predominantly in ductal breast carcinomas. In agreement with data presented by Heikkinen et al. [23], our study suggests that breast cancers with PALB2 mutations are predominantly triple-negative ones; however, their phenotype does not completely overlap with the basal type.
In our study, PALB2 alterations did not associate with ovarian cancer risk. In addition, the PALB2 deletion was accompanied by a germline BRCA2 nonsense mutation in one ovarian cancer patient (Moes et al., unpublished data). A similar observation was published by Heikkinen et al. [23] who found two breast cancer patients with a PALB2 deletion among 104 BRCA2 mutation carriers. Interestingly, the presence of these two alterations in carcinomas appears to be more frequent than just one occurring by chance, considering the low frequency of PALB2 and BRCA2 mutations in control populations.
To date, PALB2 gene alterations have not been extensively studied in ovarian cancer patients. Erkko et al. [24] found three (0.5%) ovarian cancer patients with PALB2 gene mutations in 593 unselected ovarian cancer cases screened for the PALB2 c.1592delT alteration, identified earlier in breast cancers [10]. Another dysfunction of PALB2 detected in ovarian cancer was its expression silencing by promoter hypermethylation (in 4 of 53 sporadic cases) [25]. These studies, together with our data suggest that PALB2 protein may be defective or insufficient in some rare ovarian carcinomas; however, a possible contribution of this insufficiency to ovarian cancer development needs elucidation.
It is unclear how the PALB2 deficiency contributes to cancer development. It has been suggested that PALB2 participates in breast carcinogenesis through haploinsufficiency and/or the dominant-negative effect. In the majority of breast cancers with PALB2 gene mutations, the loss of heterozygosity (LOH) was not observed [10, 11]. The only evidence of deletion of the wild-type allele was presented by Garcia et al. [12]. In our study, there was a suspicion of LOH at PALB2 locus in one ovarian carcinoma with PALB2 deletion. The regular allele of PALB2 gene may also be eliminated by promoter hypermethylation (as above). Such dysfunction was revealed not only in ovarian but also in sporadic breast cancers [25]. Since wild-type PALB2 proteins oligomerize at damaged DNA [26], truncated PALB2 mutants may possibly complex with wild type protein, thus, disturbing its proper function. In addition, other molecular changes may cooperate with PALB2 deficiency in cancer development.
Some of PALB2 gene alterations in breast cancer were suggested to be founder mutations for other ethnic groups [8, 10]. The presence of the same deletion in seven unrelated women in our study might suggest that this was a founder mutation for the examined population from central Poland. Although the genotype analysis of the mutation carriers showed differences in the haplotypes, one cannot exclude an ancient founder mutation. More detailed analysis is necessary to determine the origin of this alteration.
Our study had some limitations. The entire coding sequence of the PALB2 gene was screened in 70 non-consecutive ovarian cancers only; the sensitivity of the SSCP ranges from 70% to 95%, according to different publications [27, 28], and it is 90% in our laboratory [20]. Thus, some PALB2 alterations (particularly of the missense type) could have been missed. Nevertheless, a practical value of this study is that the c.509_510delGA should be searched for in Polish families with breast cancer.
Conclusions
The c.509_510delGA is a novel PALB2 mutation that increases the risk of familial breast cancer. Occurrence of the same PALB2 alteration in seven unrelated women suggests that c.509_510delGA (p.R170fs) is a recurrent mutation for population from Poland. It may be useful to include this PALB2 mutation to a list of alterations that should be searched for in Polish families with breast cancer.
References
Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, Liu X, Jasin M, Couch FJ, Livingston DM: Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell. 2006, 22: 719-729. 10.1016/j.molcel.2006.05.022.
Zhang F, Ma J, Wu J, Ye L, Cai H, Xia B, Yu X: PALB2 Links BRCA1 and BRCA2 in the DNA-Damage Response. Curr Biol. 2009, 19: 524-529. 10.1016/j.cub.2009.02.018.
Jacquemont C, Taniguchi T: The Fanconi anemia pathway and ubiquitin. BMC Biochem. 2007, 8 (Suppl 1): S10-10.1186/1471-2091-8-S1-S10.
Taniguchi T, D'Andrea AD: Molecular pathogenesis of Fanconi anemia: recent progress. Blood. 2006, 107: 4223-4233. 10.1182/blood-2005-10-4240.
Xia B, Dorsman JC, Ameziane N, de Vries Y, Rooimans MA, Sheng Q, Pals G, Errami A, Gluckman E, Llera J, et al: Fanconi anemia is associated with a defect in the BRCA2 partner PALB2. Nat Genet. 2007, 39: 159-161. 10.1038/ng1942.
Reid S, Schindler D, Hanenberg H, Barker K, Hanks S, Kalb R, Neveling K, Kelly P, Seal S, Freund M, et al: Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat Genet. 2007, 39: 162-164. 10.1038/ng1947.
Antoniou A, Pharoah PD, Narod S, Risch HA, Eyfjord JE, Hopper JL, Loman N, Olsson H, Johannsson O, Borg A, et al: Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. 2003, 72: 1117-1130. 10.1086/375033.
Foulkes WD, Ghadirian P, Akbari MR, Hamel N, Giroux S, Sabbaghian N, Darnel A, Royer R, Poll A, Fafard E, et al: Identification of a novel truncating PALB2 mutation and analysis of its contribution to early-onset breast cancer in French-Canadian women. Breast Cancer Res. 2007, 9: R83-10.1186/bcr1828.
Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, Reid S, Spanova K, Barfoot R, Chagtai T, et al: PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet. 2007, 39: 165-167. 10.1038/ng1959.
Erkko H, Xia B, Nikkila J, Schleutker J, Syrjakoski K, Mannermaa A, Kallioniemi A, Pylkas K, Karppinen SM, Rapakko K, et al: A recurrent mutation in PALB2 in Finnish cancer families. Nature. 2007, 446: 316-319. 10.1038/nature05609.
Tischkowitz M, Xia B, Sabbaghian N, Reis-Filho JS, Hamel N, Li G, van Beers EH, Li L, Khalil T, Quenneville LA, et al: Analysis of PALB2/FANCN-associated breast cancer families. Proc Natl Acad Sci USA. 2007, 104: 6788-6793. 10.1073/pnas.0701724104.
Garcia MJ, Fernandez V, Osorio A, Barroso A, Llort G, Lazaro C, Blanco I, Caldes T, de la Hoya M, Ramon YCT, et al: Analysis of FANCB and FANCN/PALB2 fanconi anemia genes in BRCA1/2-negative Spanish breast cancer families. Breast Cancer Res Treat. 2009, 113: 545-551. 10.1007/s10549-008-9945-0.
Cao AY, Huang J, Hu Z, Li WF, Ma ZL, Tang LL, Zhang B, Su FX, Zhou J, Di GH, et al: The prevalence of PALB2 germline mutations in BRCA1/BRCA2 negative Chinese women with early onset breast cancer or affected relatives. Breast Cancer Res Treat. 2009, 114: 457-462. 10.1007/s10549-008-0036-z.
Jones S, Hruban RH, Kamiyama M, Borges M, Zhang X, Parsons DW, Lin JC, Palmisano E, Brune K, Jaffee EM, et al: Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009, 324: 217-10.1126/science.1171202.
Tischkowitz MD, Sabbaghian N, Hamel N, Borgida A, Rosner C, Taherian N, Srivastava A, Holter S, Rothenmund H, Ghadirian P, et al: Analysis of the gene coding for the BRCA2-interacting protein PALB2 in familial and sporadic pancreatic cancer. Gastroenterology. 2009, 137: 1183-6. 10.1053/j.gastro.2009.06.055.
Didkowska J, Wojciechowska U, Tarkowski W, Zatonski W: Cancer in Poland. 2007, Warsaw: The Maria Sklodowska-Curie Memorial Cancer Center and Institute Of Oncology, 2005
Tavassoli FA, Devilee P: Cancer IAfRo: Pathology and Genetics of Tumours of the Breast and Female Genital Organs. 2003, Lyon: IARC Press-WHO
Kolasa IK, Rembiszewska A, Janiec-Jankowska A, Dansonka-Mieszkowska A, Lewandowska AM, Konopka B, Kupryjanczyk J: PTEN mutation, expression and LOH at its locus in ovarian carcinomas. Relation to TP53, K-RAS and BRCA1 mutations. Gynecol Oncol. 2006, 103: 692-697. 10.1016/j.ygyno.2006.05.007.
Primer3. [http://frodo.wi.mit.edu/primer3/]
Dansonka-Mieszkowska A, Ludwig AH, Kraszewska E, Kupryjanczyk J: Geographical variations in TP53 mutational spectrum in ovarian carcinomas. Ann Hum Genet. 2006, 70 (Pt 5): 594-604. 10.1111/j.1469-1809.2006.00257.x.
NCBI. [http://www.ncbi.nlm.nih.gov/]
Fisher Exact. [http://www.quantitativeskills.com/sisa/statistics/fisher.htm]
Heikkinen T, Karkkainen H, Aaltonen K, Milne RL, Heikkila P, Aittomaki K, Blomqvist C, Nevanlinna H: The breast cancer susceptibility mutation PALB2 1592delT is associated with an aggressive tumor phenotype. Clin Cancer Res. 2009, 15: 3214-3222. 10.1158/1078-0432.CCR-08-3128.
Erkko , et al: Occurrence of germline PALB2 mutations in ovarian cancer: 23-25. 2007, October : American Society of Human Genetics meeting
Potapova A, Hoffman AM, Godwin AK, Al-Saleem T, Cairns P: Promoter hypermethylation of the PALB2 susceptibility gene in inherited and sporadic breast and ovarian cancer. Cancer Res. 2008, 68: 998-1002. 10.1158/0008-5472.CAN-07-2418.
Sy SM, Huen MS, Zhu Y, Chen J: PALB2 Regulates Recombinational Repair through Chromatin Association and Oligomerization. J Biol Chem. 2009, 284: 18302-18310. 10.1074/jbc.M109.016717.
Jordanova A, Kalaydjieva L, Savov A, Claustres M, Schwarz M, Estivill X, Angelicheva D, Haworth A, Casals T, Kremensky I: SSCP analysis: a blind sensitivity trial. Hum Mutat. 1997, 10 (1): 65-70. 10.1002/(SICI)1098-1004(1997)10:1<65::AID-HUMU9>3.0.CO;2-L.
Nataraj AJ, Olivos-Glander I, Kusukawa N, Highsmith WE: Single-strand conformation polymorphism and heteroduplex analysis for gel-based mutation detection. Electrophoresis. 1999, 20 (6): 1177-85. 10.1002/(SICI)1522-2683(19990101)20:6<1177::AID-ELPS1177>3.0.CO;2-2.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2350/11/20/prepub
Acknowledgements
We express our gratitude to Dr Agnieszka Ludwig-Galezowska and Dr Agnieszka Timorek for providing a part of the control group, and to the staff of the Departments of Gynecologic Oncology and Pathology for help with tissue collection. Special thanks are directed to Dr Dariusz Piotrowski, a director, and to employees of the Regional Blood and Hemotherapy Center in Warsaw, as well as to employees of blood centers located in Central Clinical Hospital of the Ministry of Internal Affairs and Administration and Children's Memorial Health Institute Hospital for fast collection of a huge number of controls. We also thank to Alina Rembiszewska MSc, Danuta Szczypiorska-Wrede MSc and Elzbieta Kulczycka MSc for performing immunohistochemical stainings, to Aneta Balabas MSc for assistance in BRCA2 analysis, to Renata Zub MSc for providing sequencing electrophoresis data, and to Magdalena Chechlinska PhD for the linguistic correction.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
ADM designed and coordinated the study, carried out molecular analyses of ovarian cancers and controls, coordinated the collection of control blood samples, drafted the manuscript. AK carried out molecular analyses of breast cancer. JM participated in the design of the study, carried out a part of molecular analyses of ovarian cancers and controls. MD participated in molecular analyses of breast cancer. DN collected and characterized the breast cancer material, performed pedigree diagrams. AN collected and characterized the breast cancer material. PD participated in collection of ovarian cancer samples from the Institute of Oncology. KC participated in collection of ovarian cancer samples and controls from the Brodnowski Hospital. JK characterized the ovarian cancer material, analyzed immunohistochemical stainings, participated in the design and coordination of the study, critically reviewed and drafted the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Dansonka-Mieszkowska, A., Kluska, A., Moes, J. et al. A novel germline PALB2 deletion in Polish breast and ovarian cancer patients. BMC Med Genet 11, 20 (2010). https://doi.org/10.1186/1471-2350-11-20
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2350-11-20