
Abstract
Background
The rich yellow-orange to vividly deep red bark of willow (Salix spp.) branches have high ornamental and economic value. However, the mechanism underlying the regulation of willow branch color remains unknown. Therefore, we performed metabolomics and transcriptomics analyses of purple, green, and red willow barks to elucidating the mechanisms regulating color development.
Results
Seven anthocyanins were isolated; pelargonidin, petunidin 3-O-rutinoside, and cyanin chloride were the most abundant in red bark, whereas pelargonin chloride was most abundant in purple bark. The green bark contained the highest level of malvidin; however, the malvidin level was not significantly higher than in the red bark. The purple bark contained the largest amount of canthaxanthin, a carotenoid pigment. The integrated pathways of flavonoid biosynthesis, carotenoid biosynthesis, and porphyrin and chlorophyll metabolism were constructed for the willow barks. Among the three barks, the expression of the structural genes ANS, ANR, and BZ1, which are involved in anthocyanin synthesis, was the highest in red bark, likely causing anthocyanin accumulation. The expression of CrtZ, which participates in the carotenoid pathway, was the highest in purple bark, likely leading to canthaxanthin accumulation. The high expression of DVR, POR, and CRD1 may be associated with green pigment synthesis in the chlorophyll biosynthesis pathway.
Conclusions
Purple bark color is co-regulated by anthocyanins and carotenoids, whereas red bark is characterized by anthocyanin accumulation and chlorophyll degradation. The green pigment is regulated by maintaining chlorophyll synthesis. BZ1 and CrtZ are candidate genes regulating anthocyanin and canthaxanthin accumulation in red and purple barks respectively. Collectively, our results may facilitate the genetic breeding and cultivation of colorful willows with improved color and luster.
Similar content being viewed by others


Background
Willows, belonging to the genus Salix of the Salicaceae family, are important landscape trees in China. The characteristics of willow, including their variable forms, branching morphology, and seasonally changing colors, have made them increasingly popular in gardening, horticulture, and landscape architecture [1]. Willow, particularly the weeping willow (Salix babylonica), has been cultivated for thousands of years. In the past 20 years, the golden weeping willow with bright golden branches in winter has increased in popularity. The colors of willow bark are diverse, including green, purple, yellow, white, brown, and red [2]. In recent years, the sale of colorful branches has thrived well in the landscape industry because of their novelty, leading to considerable economic profits. Therefore, the production of colorful willow branches is the primary focus of willow breeding. Although this variation is rich in hybrid generations, the underlying genetic mechanism is not well-understood.
The secondary metabolites determining color development in plants include three classes of compounds as follows: flavonoids, betalains, and carotenoids [3, 4]. Flavonoids, which include anthocyanins, produce pale yellow, red, violet, and purple to blue colors in flowers, seeds, and fruit skin [5]. The six most common anthocyanins that accumulate in flowers and fruits are cyanidin, petunidin, delphinidin, peonidin, pelargonin, and malvidin [6]. Accordingly, these compounds can change the color of plants from pink to blue-violet [7, 8]. Peonidin and cyanidin result in purple to red color in the skin, whereas pelargonin is responsible for brick red-colored skin. The presence of delphinidin, malvidin, and petunidin results in blue and purple colors. Betalains, which originate from tyrosine, are found in plants of the order Caryophyllales and in beetroot, where they produce yellow-to-red coloring [9]. Xanthophylls are the main carotenoids endowing flower petals and fruits with a pale to deep yellow color [10,11,12]. Combinations of flavonoids and carotenoids in the same organ increases color variety.
The flavonoid biosynthesis pathway has been well-characterized in many plants, including Arabidopsis [13], yam [14], and grapevine [15]. Flavonoids are not only synthesized as defense metabolites in response to abiotic stress in trees, such as poplars and ginkgo [16, 17], but also affect fruit quality and nutrition, such as in tomato [18] and kiwi fruit [19]. Anthocyanins, as a subgroup of flavonoids, render color to plants by modifying the anthocyanidins with sugars and acyl acids. The synthesis of precursors is catalyzed by the enzymes chalcone isomerase (CHI), flavonoid 3′, 5′-hydroxylase (F3′5′H), chalcone synthase (CHS), and flavonoid 3′-hydroxylase (F3′H). Subsequently, dihydrokaempferol, dihydroquercetin, and dihydromyricetin are converted into colorless anthocyanins. The catalytic function of dihydroflavonol 4-reductase (DFR) and anthocyanidin synthase (ANS) results in the production of anthocyanidin [20, 21]. Anthocyanins are the pigments generated by this complex pathway through glycosylation reactions catalyzed by UDP-glucosyltransferase and glutathione S-transferase [22].
Chlorophyll is a fat-soluble green pigment involved in photosynthesis in plants. Chlorophyll and heme have the same primary biosynthesis steps, beginning at 5-aminolaevulinic acid and then branching from protoporphyrin IX catalyzed by the enzyme porphobilinogen deaminase (HEMC, HEMD, HEME, HEMF, and HEMG). Subsequently, protoporphyrin IX combines with Mg2+ to form Mg-protoporphyrin IX through Mg-chelatases CHLH, CHLI1, CHLI2, or CHLD. The catalytic action of the enzyme encoded by divinyl chlorophyllide a 8-vinyl-reductase (DVR) generates protochlorophyllide (Pchlide), which is converted to chlorophyllide a. Finally, chlorophyllide a is catalyzed by CHLG-producing chlorophyll a or converted to chlorophyll b by chlorophyllide a oxygenase (CAO) [23]. Carotenoids are among the most widely distributed colorants in flowers and fruits and are responsible for red, orange, and yellow colors [24]. Geranyl–geranyl pyrophosphate is synthesized by the enzyme geranyl–geranyl pyrophosphate synthase, after which phytoene is produced by phytoene synthase (CrtB). The key enzymes are carotenoid cleavage dioxygenases4, ζ-carotene desaturase (ZDS), β-carotene hydrolase, nine cis-epoxycarotenoid dioxygenases (NCED), phytoene dehydrogenase (PDS), and zeaxanthin epoxidase (ZEP). The products of each of these enzymes contribute to coloration [25].
The color variation in willow bark is rich, ranging from green and yellow to red. Red bark is rare in southern China; thus, studying the components and regulation of genes that influence the color of willow bark is essential for breeding. In this study, green, purple, and red willow bark were evaluated using metabolome and transcriptome profiling. Metabolites and genes associated with pigment formation in the flavonoid and carotenoid biosynthesis pathways were verified. Our results provide new resources for studying color development in willow bark and a basis for precise breeding to obtain desired willow bark colors.
Results
Metabolic analysis of colored willow barks
The metabolic components of green (G), purple (P), and red (R) willow branch barks were detected and analyzed (Fig. 1). We identified 1639 and 1026 metabolites in positive and negative modes, respectively (Table S1). In the positive mode, 23, 11, and 14 differential metabolites were identified in the three comparisons, whereas in the negative mode, there were seven, 11, and three differential metabolites (Fig. 2a). In the G vs. P, G vs. R, and P vs. R comparisons, 135, 109, and 129 metabolites were detected, of which 55 (40.7%), 68 (62.4%), and 70 (54.3%) were upregulated, respectively (Table S2). To verify the function of the differentially accumulated metabolites (DAMs), Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment was conducted to annotate the DAMs. Metabolites of flavonoid biosynthesis (ko00941) and flavone and flavonol biosynthesis (ko00944) were enriched in the three groups of G vs. P (Fig. 2B), G vs. R (Fig. 2C), and P vs. R (Fig. 2D). In flavone and flavonol biosynthesis pathway, there were eight, seven, and six enriched DAMs in the G vs. P, G vs. R, and P vs. R groups, respectively; in flavonoid biosynthesis, there were 13, 13, and seven metabolites in the G vs. P, G vs. R, and P vs. R groups, respectively. Porphyrin and chlorophyll metabolism pathway (ko00860) was enriched in groups G vs. P (Fig. 2B) and P vs. R (Fig. 2D). There were two, one, and two metabolites in the G vs. P, G vs. R, and P vs. R groups, respectively.

Images of the green, purple, and red willow barks

Analysis of differentially accumulated metabolites (DAMs) in willow bark. A Venn diagram of DAMs in green vs. purple, green vs. red, and purple vs. red bark comparisons in positive (left) and negative ion modes (right). KEGG pathway orthology of enriched DAMs in B green vs. purple bark, C green vs. red bark, and D purple vs. red bark
Identification of DAMs involved in flavonoid biosynthesis, carotenoid biosynthesis, and porphyrin and chlorophyll metabolism pathways
Differences in the accumulation of metabolites involved in flavonoid, anthocyanins, and flavone and flavonol biosynthesis are illustrated as a heatmap (Fig. 3). A total of 43 flavonoids were identified and quantified: 14 were present in large amounts in red bark, 15 in purple bark, and nine in green bark. The contents of quercetin, kaempferide, phloretin, neohesperidin, xanthohumol, luteolin, phloridzin, syringetin, rutin, myricetin-3-O-galactoside, and myricetin were the highest in purple willow bark among the three barks. Naringenin, vitexin, epigallocatechin, eriodictyol, pinocembrin, laricitrin, and prunin levels were the highest in green bark. Trifolin levels increased from green to purple and then red bark. Seven anthocyanins were detected, among which pelargonidin, petunidin 3-O-rutinoside, and cyanin chloride were the most abundant in red bark. Pelargonin chloride was the most abundant in purple bark. The level of malvidin was the highest in the green bark. These results suggest that anthocyanins regulate the production of pigment molecules in willow barks. Three compounds, canthaxanthin, abscisic acid glucose ester, and abscisic acid, were detected in carotenoid biosynthesis pathway (Fig. 3B). Six metabolites of porphyrin and the chlorophyll metabolic pathway were identified (Fig. 3C).

Heatmap of DAMs involved in flavonoid and carotenoid biosynthesis pathways. A Heatmap of DAMs involved in flavonoid biosynthesis pathway. The red DAMs are anthocyanin. B Heatmap of DAMs in carotenoid biosynthesis pathway. C Heatmap of DAMs in porphyrin and chlorophyll metabolism pathway. R, red bark; P, purple bark; G, green bark
Transcriptome profile characterization
To investigate the putative genes regulating willow bark color, transcriptome sequencing was conducted. A total of 448,839,796 raw reads were obtained with a Q30 > 93.15% (Table S3). After annotation, 7935, 6896, and 6790 differentially expressed genes (DEGs) were annotated in the G vs. P, G vs. R, and P vs. R comparison groups, including 4519, 3360, and 2705 upregulated genes and 3416, 3509, and 4085 downregulated genes, respectively (Table S4). In KEGG pathway functional analysis, we focused on pigment biosynthesis-associated pathways. Flavonoid biosynthesis (ko00941) was significantly enriched in the three group comparisons, with 27, 28, and 21 genes enriched in the G vs. P, G vs. R, and P vs. R groups, respectively (Fig. 4). Carotenoid biosynthesis was significantly enriched in the G vs. P and P vs. R groups but not in the G vs. R group (Fig. 4B–D). Isoflavonoid biosynthesis was enriched in P vs. R (Fig. 4D).

Characterization of transcriptome profiling. A Venn diagram of differentially expressed genes (DEGs) among the three colored bark comparison groups. B–D Enrichment bubble diagram of DEG–KEGG metabolic pathway analysis for (B) green vs. purple, (C) green vs. red, and (D) purple vs. red bark
Real-time quantitative polymerase chain reaction validation of DEGs in flavonoid and carotenoid biosynthesis pathways
To verify the quality of the transcriptome sequencing results and genes involved in flavonoid and carotenoid biosynthesis, 12 genes (DFR, FLS, ANS, CHS, CHI, LAR, CYP75B1, CHI3, CHS1, CrtB, ZEP, and NCED) were evaluated using real-time quantitative polymerase chain reaction (RT-qPCR). The gene expression trends determined using RNA-sequencing (RNA-seq) were the same as those determined using RT-qPCR for the G, P, and R bark samples (Fig. 5). These results demonstrate that the sequencing transcriptome data are reliable.

The relative expression level between RT-qPCR and RNA-seq data of genes involved in flavonoid and carotenoid biosynthesis. The relative expression value was calculated using the 2−ΔΔCt method with β-actin gene of Salix as internal control. Error bars represent the mean ± SD of three biological replicates (n = 3). FRKM, fragments per kilobase of exon per million mapped fragments; G, green bark; P, purple bark; R, red bark
Integration analysis of metabolites and transcripts in the flavonoid biosynthesis pathway
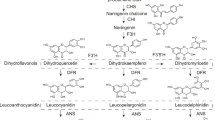
Integration of the results illustrating the changes in metabolite and transcript levels can improve the understanding of the regulatory pathway(s) mediating color development in willow bark. The flavonoid biosynthesis and flavone and flavonol biosynthesis pathways were significantly enriched in the three groups of bark color. As shown in Fig. 6, the gene expression levels of CHI, ANS, and BZ1 were the highest in the red bark. The expression of FLS was higher in the purple bark than in the other two barks. The expression levels of the downstream regulatory genes ANR and ANS were consistent with the contents of pelargonidin and epicatechin. In addition, the expression level of LAR was high in the green and purple barks, and the epigallocatechin content was highest in the green bark. These results support that different regulatory pathways produce different-colored pigments. Higher levels of pelargonidin, trifolin, phlorizin, and hesperetin accumulated in the red bark than in the purple or green bark (Fig. 6). Higher levels of quercetin, rutin, syringetin, myricetin, neohesperidin, phloretin, and xanthohumol were detected in the purple bark than in the green or red bark. The expression of CYP93B2 (IMYO5_C0793000200), CYP75A (IMYO5_009G0054300), FLS (IMYO5_019G0014000), and F3H (IMYO5_001G0309700) was consistent with the metabolite contents of luteolin, rutin, quercetin, myricetin, syringetin, neohesperidin, and phloretin in the purple bark. Thus, these genes may be involved in the accumulation of flavonoids in the purple bark. The highest expression of CHI ((IMYO5_016G0095300), CHS (IMYO5_003G0132300), ANS (IMYO5_016G0095300), BZ1 (IMYO5_013G0103500), and DFR (IMYO5_005G0181300) in red bark compared to that in the green and purple bark were consistent with the contents of phlorizin, hesperetin, naringin, cyanin chloride, cyanidin-3-O-glucoside, dihydromyricetin, pelargonidin, and petunidin-3-O-rutinoside.

Integrated analysis of flavonoid biosynthesis [26] based on metabolite and transcript analyses. The heatmap of the genes refers to the Z-score normalization of FPKM value of the samples, and the color scale from minimum (green) to maximum (red) corresponds with low to high gene expression, respectively. The three samples refers to the average value of the three groups (n = 3 each) for green bark (G), purple bark (P), and red bark (R); each sample was analyzed in biological triplicates. CHS, chalcone synthase; ANR, anthocyanidin reductase; ANS, anthocyanidin synthase; DFR, dihydroflavonol 4-reductase; CHI, chalcone flavanone isomerase; BZ1, bronze-1; LAR, leucoanthocyanidin reductase; FLS, flavonol synthase; CYP75A, flavonoid 3', 5'-hydroxylase; CYP75B1, flavonoid 3'-monooxygenase; CYP93B2, cytochrome P450 93B2; CYP73A, trans-cinnamate 4-monooxygenase; and F3H, flavanone 3-hydroxylase
Reconstruction of carotenoid biosynthesis and porphyrin and chlorophyll metabolic pathways for willow barks
In the carotenoid biosynthesis pathway, 15 DEGs were annotated, including one CrtB, one PDS, four ZDS, one DWARF27, one CrtZ, three ZEP, three NCED, and one AAO3, in the colored willow bark samples (Fig. 7A). Four genes showed the highest expression in purple bark: CrtZ, two ZEP, two NCED, and one AAO3. The levels of canthaxanthin and abscisic acid glucose ester metabolites were the highest in the purple bark compared to that in the other barks, supporting the role of various genes in the synthesis of carotenoids and their derivatives in different bark colors. In the porphyrin and chlorophyll metabolism pathway, 16 DEGs were enriched, of which one HCAR, one PPOX, and one chlH showed the highest expression in the purple bark (Fig. 7B).

Integrated analysis of carotenoid biosynthesis and porphyrin and chlorophyll metabolism pathways in willow barks. A Reconstruction of the carotenoid biosynthesis pathway [27]. B Reconstruction of the porphyrin and chlorophyll metabolism pathway [28]. The gene heatmap refers to the z-score normalization of the FPKM value of the samples, and the color scale from minimum (green) to maximum (red) corresponds with low to high gene expression, respectively. The three samples in order are G (green), P (purple), and R (red) bark. Each sample was analyzed in biological triplicates. Genes are defined as follows: CrtZ, beta-carotene 3-hydroxylase; CrtB, phytoene synthase; AAO3, abscisic aldehyde oxidase; ZDS, ζ-carotene desaturase; PDS, phytoene dehydrogenase; ZEP, zeaxanthin epoxidase; NCED, 9-cis-epoxycarotenoid dioxygenase; GGPP, geranylgeranyl pyrophosphate synthase; CRD1, magnesium-protoporphyrin IX monomethyl ester [oxidative] cyclase; hemH, ferrochelatase; HO, heme oxygenase; hemC, porphobilinogen deaminase; POR, NADPH-cytochrome P450 reductase; hemB, delta-aminolevulinic acid dehydratase; DVR, divinyl chlorophyllide a 8-vinyl-reductase; HCAR, 7-hydroxymethyl chlorophyll a reductase; CHLI, magnesium chelatase subunit chlI; CAO, chlorophyllide a oxygenase; SGR, stay green; PPOX, protoporphyrinogen oxidase; chlH, magnesium chelatase; NOL, chlorophyll(ide) b reductase; CLH2, chlorophyllase 2
Correlation between DEGs and DAMs associated with pigment regulation
A correlation network was constructed to investigate the correlation between pigment-associated metabolites and DEGs involved in flavonoid and carotenoid biosynthesis and porphyrin and chlorophyll metabolic pathways (Fig. 8). Malvidin, pelargonin, and cyanidin-3-O-glucoside were associated with numerous genes, indicating that genes influence these components. Cyanidin-3-O-glucoside and canthaxanthin were associated with genes, such as ANR, DFR, CHS, and PDS. Malvidin shares the same regulatory genes as canthaxanthin. Pelargonin was the hub metabolite associated with malvidin, canthaxanthin, cyanidin-3-O-glucoside, and petunidin-3-O-rutinoside (Fig. 8).

Correlation between differentially expressed genes and metabolites. Yellow line: positive correlation between DEGs and DAMs; blue line: negative correlation between DEGs and DAMs
Discussion
Integrated metabolome and transcriptome analyses have been increasingly applied in studies of color formation in plants [29]. To our knowledge, this is the first study to perform these analyses to elucidate the regulatory mechanism of willow bark color. Mining of DAMs and DEGs involved in pigmentation in willow bark can provide a fundamental basis for the genetic breeding of colorful willows.
Plant colors are the products of flavonoid, chlorophyll, and carotenoid synthesis [29]. Among the various colors, purple, blue, and red depend on the contents of flavonoids/anthocyanins, including cyanidin and peonidin in common [30]. Chlorophylls present in photosynthetic reaction systems are responsible for green coloration [31], and yellow to orange colors are attributed to carotenoids [32]. However, the regulation of pigment biosynthesis is complex and influenced by the species and environmental conditions and their interactions. In willow bark, four types of anthocyanin derivatives, including cyanidin, pelargonidin, petunidin, and malvidin, were identified. Pelargonidin is an anthocyanin that commonly produces red pigment in plants [33]; in our study, the accumulation of pelargonidin was correlated with red color in willow bark. Cyanidin-3-O-glucoside is the red-brown component of the ligulate flower [34] and the purple pigment encoded by dominant Pp alleles in black rice [35]. No significant difference in cyanidin-3-O-glucoside was observed between purple and red willow barks. The stability and types of pigments are influenced by temperature, hydrogen ion concentration (pH), illuminated environment, and anthocyanin structure [36]. Petunidin 3-O-rutinoside can change from dark red to purple and from blue to green and light yellow in a pH-dependent manner, providing a new approach for regulating the red color based on the soil pH. Pelargonidin mostly appeared red, whereas pelargonidin chloride was purple. Cyanin chloride mostly appeared as red, cyanidin O-rutinoside equally exhibited purple and green, and cyanidin-3-O-glucoside contributed to red and purple coloration. Thus, structural differences in the same anthocyanin lead to different colors in willow bark.
Carotenoids are also natural pigments that color vegetables, fruits, and flowers yellow, orange, and red and supplement the flavonoid/anthocyanin content when their levels decline. Canthaxanthin is a ketocarotenoid that accumulates in Chlorella zofingiensis [37] but has not yet been detected in plants [38]. The reddish-orange coloration in trout flesh, flamingo feathers, egg yolks, and koi carp skin is attributed to canthaxanthin [39, 40]. Canthaxanthin was the only carotenoid detected in willow bark in this study. Its content in purple bark was 10- and sixfold higher than that in green and red barks. The purple color was likely determined by the type of carotenoid and anthocyanin combinations, and canthaxanthin plays a major role in the purple variety. Color mutations can be caused by damage to the chlorophyll biosynthesis pathway. Stercobilin, an intermediate in the chlorophyll biosynthesis pathway and a brown pigment, was detected in the red bark. In contrast, the levels of L-glutamic acid, an intermediate product, were higher in the red bark than in the green and purple barks.
Stable purple and red barks in willow are rare and have ornamental and economic value. The structural genes involved in color change related to flavonoid biosynthesis are CYP73A, ANS, ANR, CHI, CHS, DFR, and F3H [41]. In total, 13 structural genes were identified in the three colors of willow bark. CHS catalyzes the first step of chalcone synthesis, and CHI catalyzes the stereospecific cyclization of chalcone and 6-deoxychalcone to (2S)-liquiritigenin and (2S)-naringenin [42]. The expression of CHS and CHI may affect the accumulation of anthocyanins as a branch point [43]. One CHI (IMY05_010G0169800) and one CHS (IMY05_003G0132300) were expressed at high levels in the red bark, whereas CHI (IMY05_019G0069500) and two other CHS (IMY05_014G0118800, IMY05_016G0041700) were expressed at comparable levels. These genes may play different roles in flavonoid biosynthesis, including the synthesis of compounds such as phloretin (P bark), phlorizin (R bark), rutin (P bark), luteolin (P bark), apigenin (G bark), hesperetin (R bark) and neohesperidin (P bark). A change in DFR enzyme activity may lead to the production of red transgenic gentian flowers by reducing DHK. The low DHK metabolite content and high pelargonidin content indicate a role for DHK in red pigmentation. Enzyme encoded by ANS genes can directly convert the substrate of leucoanthocyanidins into colorful anthocyanidins. The activity of ANS and DFR may result in a high accumulation of pigmentation. The expression of ANS and ANR were significantly higher in the red barks than in the green and purple barks, which may result in anthocyanin accumulation. BZ1 encodes the final enzyme that converts UDP-glucose to water-soluble 3-hydroxyl group anthocyanins to achieve a more stable state [44]. BZ1 and ANS are responsible for anthocyanin accumulation in red-skinned pears [45]. The expression level of ANS in the red bark was 11-fold higher than that in the green bark. Regarding the two BZ1 genes, IMY05_013G0103500 in the red bark was 147- and 44-fold higher than that in the green and purple barks, respectively; IMY05_013G0118300, was 27- and 28-fold higher in the red and purple barks than in the green bark, respectively. This indicates that the expression levels of ANS and BZ1 determine red pigment accumulation in willow bark.
In the carotenoid synthesis pathway, most carotenoid pigments are derived from phytoene, which is synthesized in the first reaction of the pathway catalyzed by CrtB. Red lycopene is subsequently synthesized by PDS and ZDS. Lycopene cyclization produces β-carotene, α-carotene, and their derivatives in two branches [46]. In the primary synthesis steps, the expression of CrtB is the highest in red bark. Subsequently, one PDS and four ZDS were highly expressed in the green bark and partially expressed at high levels in the red bark, indicating that branching of the carotenoid synthesis pathway resulted in the production of different products. Only one gene, CrtZ (IMY05_017G0118300), was expressed at high levels in the purple bark compared to the other barks. This is consistent with the content of canthaxanthin. Overexpression of the β-carotene 3-hydroxylase gene (CrtZ) from Agrobacterium aurantiacum resulted in the accumulation of canthaxanthin [47]. The protein DWARF27 (D27) participates in strigolactone biosynthesis, which regulates rice branching [48]. Thus, D27 is a candidate gene that regulates the branching and phenotype of colorful willows.
The purple color of willow barks is likely conferred by the co-pigmentation interaction of carotenoid and anthocyanin, with carotenoid as a major factor. The expression of most key structural genes involved in the chlorophyll biosynthesis pathway, including hemH, hemB, POR, CRD1, DVR, and CLH2, was the highest in the green bark, indicating that the green color was determined by chlorophyll synthesis. POR is a key enzyme catalyzing the conversion of protochlorophyllide to chlorophyllide in chlorophyll synthesis [49]. CRD1 mainly functions in chloroplast movement. The higher expression of POR and CRD1 in the green bark may explain the maintenance of green pigmentation. Overexpression of the stay-green (SGR) protein in melon led to CHL degradation and leaf yellowing [50]. The highest expression of SGR (IMY05_005G0141100) was observed in the red bark, whereas another SGR (IMY05_016G0094700) was silenced in the green bark, causing green coloration. Degradation metabolites were detected at high levels in the red bark, indicating that chlorophyll was degraded by the SGR. CAO is vital for regulating the chlorophyll a/b ratio, and expression of 7-hydroxymethyl chlorophyll a reductase (HCAR) may limit chlorophyll b degradation [51]. Overexpression of HCAR accelerated chlorophyll degradation in cucumber [52]. Therefore, the highest expression of CAO and HCAR in the purple bark suggests that chlorophyll degradation occurs via the conversion of chlorophyll a to chlorophyll b. Based on these findings, we constructed a regulatory mode for willow bark coloration (Fig. 9).

A cartoon illustrating the proposed regulation network of color development in willow barks. ANR, anthocyanidin reductase; BZ1, bronze-1; CHS, chalcone synthase; DFR, dihydroflavonol 4-reductase; ANS, anthocyanidin synthase; CHI, chalcone flavanone isomerase; F3H, flavanone 3-hydroxylase; CrtZ, beta-carotene 3-hydroxylase; CAO, chlorophyllide a oxygenase; HCAR, 7-hydroxymethyl chlorophyll a reductase; hemB, delta-aminolevulinic acid dehydratase; hemC, porphobilinogen deaminase; hemH, ferrochelatase; CLH2, chlorophyllase 2; CRD1, magnesium-protoporphyrin IX monomethyl ester [oxidative] cyclase; POR, NADPH-cytochrome P450 reductase; DVR, divinyl chlorophyllide a 8-vinyl reductase
Conclusion
In this study, the pigmentation regulation of bark color in Salix was characterized through integrated metabolome and transcriptome analyses. Red bark contained more pelargonidin, petunidin 3-O-rutinoside, and cyanin chloride than purple and green barks. Purple bark contained more pelargonin chloride than red and green barks. Malvidin had the highest level in the green bark; however, there was no significant difference between malvidin levels in green and red bark. Canthaxanthin, a carotenoid metabolite, was detected in purple bark. The structural genes CHS, ANS, DFR, ANR, and BZ1 were annotated in the flavonoid biosynthesis pathway. The high expression of ANS and BZ1 in red bark likely led to anthocyanin accumulation. CrtZ exhibited the highest expression level in purple bark, indicating an association with canthaxanthin accumulation. The high expression of hemH, hemB, POR, CRD1, DVR, and CLH2 may explain the green pigmentation in the green bark. SGR was highly expressed in the red bark and is likely associated with the accumulation of stercobilin in the chlorophyll degradation pathway. CAO and HCAR expression were the highest in the purple bark, suggesting that chlorophyll degradation occurs through the conversion of chlorophyll a to chlorophyll b and may lead to the production of pheophorbide a. The connection network of anthocyanin and DEGs also suggest that the purple bark is co-regulated by anthocyanins and carotenoids; the red bark is characterized by anthocyanin accumulation and chlorophyll degradation; the green pigment is regulated by maintaining chlorophyll synthesis. Collectively, our results may facilitate the genetic breeding and cultivation of colorful willows with improved color and luster.
Methods
Plant materials
The three Salix species were collected and deposited in the willow nursery in Dafeng District, Yancheng City, Jiangsu province (33.20–33.35°N, 120.15–120.47°E). This area is in the transition zone between subtropical and warm, humid zones. It is approximately 2 km from the sea, with an altitude of approximately 3 m. The annual average temperature, frost-free period, annual precipitation, and sunshine time are 14.1℃, 213 days, 1042.2 mm, and 2238.9 h, respectively. The species were preserved by the authors who have permission to collect willow barks. The purple bark was from Salix × Jiangsuensis ‘J1053’, G bark from S. integra, and R bark from S. miyabeana. The voucher specimens of S. × Jiangsuensis ‘J1053’, S. integra, and S. miyabeana were deposited at the herbarium of Jiangsu academy of forestry under voucher numbers J1053, P61, and P76, respectively. They are all shrub willow species. S. × Jiangsuensis ‘J1053’ was hybridized by S. suchowensis × S. dasyclados. S. miyabeana with red bark is an excellent ornamenta variety selected from numerous clones introduced from the United States. It has strong adaptability, curved stems, and red branches, as well as desirable ornamental characteristics. S. integra is a robust parent clone with green bark. The red, green, and purple colors of the barks are evident and stable during the seedling stage. The email of the person coordinating sample collection is zjwin718@126.com.
Sample preparation
The willow bark was scraped from branches using scalpels in the nursery. The bark of each color was collected from six individuals of the same species. The samples were placed in microcentrifuge tubes, rapidly frozen in liquid nitrogen, and stored at − 80 °C until metabolite and RNA isolation. Six biological repeats were performed to identify the metabolites, and three biological repeats were used to determine the transcripts.
Metabolite extraction and ultra-high performance liquid chromatography-tandem mass spectrometry analysis
Willow bark powder (100 mg), which was ground in liquid nitrogen, was placed in a microcentrifuge tube, and 500 µL water containing 80% methanol was added. The mixture was resuspended, vortexed, and incubated for 5 min. The powder was centrifuged for 20 min at 1500 × g, 4 °C. The supernatant was transferred to a clean microcentrifuge tube containing liquid chromatography-mass spectrometry (LC–MS)-grade water containing 53% methanol and centrifuged for 20 min. The supernatant was absorbed and analyzed using an LC–MS system [53]. A vanquish ultra-high performance LC system (Thermo Fisher Scientific, Germering, Germany) and Orbitrap Q ExactiveTMHF-X mass spectrometer (Thermo Fisher Scientific, Germering, Germany) were used for ultra-high performance LC-tandem MS detection in Gene Denovo (Gene Denovo Co., Ltd., Guangzhou, China). A Hypesil Gold chromatography column (100 × 2.1 mm, 1.9 μm) was used at a column temperature gradient of 40 °C and flow rate of 0.2 mL/min. Eluent a was composed of 0.1% formic acid in positive mode and 5 mM ammonium acetate in negative mode. Eluent b was composed of methanol in both modes. The negative mode was eluent a, pH 9.0, and eluent b, methanol.
Identification and annotation of metabolites
Raw data were analyzed using Compound Discoverer 3.1 (CD3.1) for each metabolite quantitation. After peak extraction and peak area quantitation, the molecular formula was predicted based on the peaks of molecular and fragment ions. The molecules were searched against the mzCloud, mzVault, and Masslist databases for relative and accurate quantification of the final metabolites. Metabolite data were analyzed in positive and negative ion modes separately. The principle component analysis (PCA) for the quality check of metabolites and multivariate regression analysis were conducted in R language gmodels (v2.18.1) [54]. In the comparison groups, partial least squares discriminant analysis (PLS-DA) was applied using the R package ropls [55] (http://www.r-project.org/), and orthogonal projection to latent structures-discriminant analysis (OPLS-DA) was applied using R package models (http://www.r-project.org/). Differential metabolites were screened based on the variable importance in the projection (VIP) score of the (O)PLS model with a p-value of the t-test < 0.05 and VIP ≥ 1 between the two groups. The abundance of differential metabolites was normalized based on the z-score, and hierarchical clustering was performed using the R package pheatmap [56] between the two groups. The annotation and enrichment of differential metabolites were mapped to the KEGG pathway database.
Transcriptome sequencing and DEG identification
An Omega plant RNA kit (Omega Bio-Tek, Norcross, GA, USA) was used to extract total RNA, and cDNA libraries were constructed using an NEB#7530 kit (#E7530, New England Biolabs, Ipswich, MA, USA). The library quality was assessed using a DNA 1000 assay kit (#5067–1504, Agilent Technologies, Santa Clara, CA, USA). The library was sequenced on an Illumina HiSeq2500 platform (San Diego, CA, USA). Raw reads were filtered by fastp [57] (version 0.18.0) to obtain high-quality clean reads by removing reads containing more than 10% of unknown nucleotides (N) and more than 50% of low-quality (Q-value ≤ 20) bases. The clean reads were mapped to the Salix suchowensis (Salix suchowensis (ID 36,318) -Genome-NCBI (nih.gov)) genome using HISAT 2.2.4 [58] with the parameters set as “-rna-strandness RF” and others as a default. The mapped reads were assembled by StringTie v1.3.1 [59, 60]. An FPKM (fragment per kilobase of transcript per million mapped reads) value was calculated by StringTie software to quantify the expression abundance and variations of each transcript. DEGs analysis was performed using DESeq2 software [61] (and by edgeR within a group between two samples). Genes with a false discovery rate (FDR) adjusted P value of < 0.05 (FDR = 5%) and absolute fold change ≥ 2 were considered differentially expressed. Annotation of DEGs and KEGG functional enrichment followed the method described by Zhou et al. (2020) [62]. The abundance of DEGs were normalized based on the z-score and the heatmap was performed using Omicsmart, a dynamic real-time interactive online platform for data analysis (http://www.omicsmart.com).
Real-time quantitative PCR verification
The RNA used for RT-qPCR was reverse-transcribed using Hifair R III 1st Strand cDNA Synthesis SuperMix gDNA digester plus (Yeasen Biotechnology [Shanghai] Co., Ltd., Shanghai, China). RT-qPCR was performed using Genious 2 × SYBR Green Fast qPCR Mix (ABclonal, Wuhan, China) on a LightCycler® 480 II (Roche, Basel, Switzerland). The two-step cycling procedure was performed at 95 °C, 5 min; 40 cycles at 95 °C, 10 s; 60 °C, 30 s, and 40 cycles at 72 °C for one-point sensing. The melting curve cycle conditions were 95 °C (15 s), 60 °C (60 s), and 95 °C (15 s) to continuously obtain the signal. The expression values were analyzed using the 2−ΔΔCt method. The figure was drawn using Origin software. Primers were designed using Primer Premier 5 (Premier Biosoft, Palo Alto, CA, USA) and are listed in Table S5.
Metabolite and DEG correlation network construction
Pearson correlation coefficients were determined to integrate the transcriptomic and metabolomics correlation network. Gene and metabolite were ranked in descending order of their absolute correlation coefficients. Pairs of genes and metabolites involved in the flavonoid, chlorophyll, and carotenoid biosynthesis pathways (with the absolute pearson correlation > 0.5) were applied for metabolite-transcript network analysis using the igraph package in R software [63].
Availability of data and materials
The transcriptome sequence data were submitted to GenBank of NCBI (https://www.ncbi.nlm.nih.gov/) under the accession no. SRP375909. The associated BioProject, SRA, and Bio-Sample numbers are as follows: PRJNA839146, SAMN28511960, SAMN28511961, SAMN28511962, SAMN28511963, SAMN28511964, SAMN28511965, SAMN28511966, SAMN28511967, and SAMN28511968. The metabolome data were submitted to Metabolights (https://www.ebi.ac.uk/metabolights/) under the data number MTBLS4820.
References
Hanley SJ, Karp A. Genetic strategies for dissecting complex traits in biomass willows (Salix spp.). Tree Physiol. 2014;34(11):1167–80. https://doi.org/10.1093/treephys/tpt089.
Guo JH, Jiao ZY, He XD, ZhuGe Q, Zhou J. A comprehensive evaluation of ornamental characteristics and adaptability of willows based on analytic hierarchy processes. J Nanjing Forestry Univ (Natural Science Edition). 2021;45:169–76. https://doi.org/10.12302/j.issn.1000-2006.202106002.
Nishihara M, Nakatsuka T. Genetic engineering of flavonoid pigments to modify flower color in floricultural plants. Biotechnol Lett. 2011;33:433–41. https://doi.org/10.1007/s10529-010-0461-z.
Kanako I, Masumi T, Yoshikazu T. Functional analysis of Antirrhinum kelloggii flavonoid 3’-hydroxylase and flavonoid 3’, 5’-hydroxylase genes; critical role in flower color and evolution in the genus Antirrhinum. J Plant Res. 2012;125:451–6. https://doi.org/10.1007/s10265-011-0455-5.
Tanaka Y, Katsumoto Y, Brugliera F, Mason J. Genetic engineering in floriculture. Plant Cell Tiss Org. 2015;80:1–24. https://doi.org/10.1007/s11240-004-0739-8.
Tanaka Y, Sasaki N, Ohmiya A. Biosynthesis of plant pigments: anthocyanins, betalains and carotenoids. Plant J. 2008;54:733–49. https://doi.org/10.1111/j.1365-313X.2008.03447.x.
Kazuma K, Noda N, Suzuki M. Flavonoid composition related to petal color in different lines of Clitoria ternatea. Phytochemistry. 2003;64:1133–9. https://doi.org/10.1016/s0031-9422(03)00504-1.
Yu P, He X, Baer M. Plant flavones enrich rhizosphere Oxalobacteraceae to improve maize performance under nitrogen deprivation. Nat Plants. 2021;7:481–99. https://doi.org/10.1038/s41477-021-00897-y.
Kujala T, Loponen J, Pihlaja K. Betalains and phenolics in red beetroot (Beta vulgaris) peel extracts: extraction and characterisation. Z Naturforsch C J Biosci. 2001;56:343–8. https://doi.org/10.1515/znc-2001-5-604.
Nisar N, Li L, Lu S, Khin NC, Pogson BJ. Carotenoid metabolism in plants. Mol Plant. 2015;8(1):68–82. https://doi.org/10.1016/j.molp.2014.12.007.
Fraser PD, Bramley PM. The biosynthesis and nutritional uses of carotenoids. Prog Lipid Res. 2004;43:228–65. https://doi.org/10.1016/j.plipres.2003.10.002.
Khoo HE, Prasad KN, Kong KW, Jiang Y, Ismail A. Carotenoids and their isomers: color pigments in fruits and vegetables. Molecules. 2011;16:1710–38. https://doi.org/10.3390/molecules16021710.
Broun P. Transcriptional control of flavonoid biosynthesis: a complex network of conserved regulators involved in multiple aspects of differentiation in Arabidopsis. Curr Opin Plant Biol. 2005;8(3):272–9. https://doi.org/10.1016/j.pbi.2005.03.006.
Wu ZG, Jiang W, Mantri N, et al. Transciptome analysis reveals flavonoid biosynthesis regulation and simple sequence repeats in yam (Dioscorea alatam L.) tubers. BMC Genomics. 2015;16:346. https://doi.org/10.1186/s12864-015-1547-8.
Ferreira V, Matus JT, Pinto-Carnide O, Carrasco D, Arroyo-García R, Castro I. Genetic analysis of a white-to-red berry skin color reversion and its transcriptomic and metabolic consequences in grapevine (Vitis vinifera cv. ’Moscatel Galego’). BMC Genomics. 2019;20:952. https://doi.org/10.1186/s12864-019-6237.
Shi X, Zheng B, Ahmed U. Expression profiling of flavonoid biosynthesis genes and secondary metabolites accumulation in Populus under drought stress. Molecules. 2021;26:1–17. https://doi.org/10.3390/molecules26185546.
Xu F, Cheng H, Cai R, et al. Molecular cloning and function analysis of an anthocyanidin synthase gene from Ginkgo biloba, and its expression in abiotic stress responses. Mol Cells. 2008;26:536. https://doi.org/10.1080/19768354.2008.9647187.
Wang J, Li G, Li C, Zhang C, Cui L, Ai G, Wang X, Zheng F, Zhang D, Larkin RM, Ye Z, Zhang J. NF-Y plays essential roles in flavonoid biosynthesis by modulating histone modifications in tomato. New Phytol. 2021;229:3237–52. https://doi.org/10.1111/nph.17112.
Yu M, Man Y, Lei R. Metabolomics study of flavonoids and anthocyanin-related gene analysis in kiwifruit (Actinidia chinensis) and kiwiberry (Actinidia arguta). Plant Mol Biol Rep. 2020;38:353–69. https://doi.org/10.1007/s11105-020-01200-7.
Tohge T, de Souza LP, Fernie AR. Current understanding of the pathways of flavonoid biosynthesis in model and crop plants. J Exp Bot. 2017;68:4013–28. https://doi.org/10.1093/jxb/erx177.
Wang Z, Cui Y, Vainstein A, Chen S, Ma H. Regulation of fig (Ficus carica L.) fruit color: metabolomic and transcriptomic analyses of the flavonoid biosynthetic pathway. Front Plant Sci. 2017;8:1990. https://doi.org/10.3389/fpls.2017.01990.
Lu J, Zhang Q, Lang L, Jiang C, Wang X, Sun H. Integrated metabolome and transcriptome analysis of the anthocyanin biosynthetic pathway in relation to color mutation in miniature roses. BMC Plant Biol. 2021;21:257. https://doi.org/10.1186/s12870-021-03063-w.
Hollingshead S, Kopecna J, Armstrong DR, Bučinská L, Jackson PJ, Chen GE, Dickman MJ, Williamson MP, Sobotka R, Hunter CN. Synthesis of chlorophyll-binding proteins in a fully segregated Δycf54 strain of the Cyanobacterium Synechocystis PCC 6803. Front Plant Sci. 2016;7:292. https://doi.org/10.3389/fpls.2016.00292.
Howitt CA, Pogson BJ. Carotenoid accumulation and function in seeds and non-green tissues. Plant Cell Environ. 2006;29:435–45. https://doi.org/10.1111/j.1365-3040.2005.01492.x.
Yuan H, Zhang J, Nageswaran D, et al. Carotenoid metabolism and regulation in horticultural crops. Hortic Res. 2015;2:15036. https://doi.org/10.1038/hortres.2015.36.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. https://doi.org/10.1093/nar/28.1.27.
Kanehisa M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019;28:1947–51. https://doi.org/10.1002/pro.3715.
Kanehisa M, Furumichi M, Sato Y, Ishiguro-Watanabe M, Tanabe M. KEGG: integrating viruses and cellular organisms. Nucleic Acids Res. 2021;49:D545–51. https://doi.org/10.1093/nar/gkaa970.
Chen XJ. Comparison of metabolome and transcriptome of flavonoid biosynthesis pathway in a purple-leaf tea germplasm jinmingzao and a green-leaf tea germplasm huangdan reveals their relationship with genetic mechanisms of color formation. Int J Mol Sci. 2020;21(11):4167. https://doi.org/10.3390/ijms21114167.
Mizuno T, Sugahara K, Tsutsumi C. Identification of anthocyanin and other flavonoids from the green-blue petals of Puya alpestris (Bromeliaceae) and a clarification of their coloration mechanism. Phytochemistry. 2021;181: 112581. https://doi.org/10.1016/j.phytochem.2020.112581.
Renger T, May V, Kühn O. Ultrafast excitation energy transfer dynamics in photosynthetic pigment-protein complexes. Phys Rep. 2001;343(3):137–254. https://doi.org/10.1016/S0370-1573(00)00078-8.
Carvalho E, Fraser PD, Martens S. Carotenoids and tocopherols in yellow and red raspberries. Food Chem. 2013;139:744–52. https://doi.org/10.1016/j.foodchem.2012.12.047.
Ku SK, Yoon EK, Lee W. Antithrombotic and antiplatelet activities of pelargonidin in vivo and in vitro. Arch Pharm Res. 2016;39:398–408. https://doi.org/10.1007/s12272-016-0708-x.
Zhang Y, Hu Z, Chu G, Huang C, Tian S, Zhao Z, Chen G. Anthocyanin accumulation and molecular analysis of anthocyanin biosynthesis-associated genes in eggplant (Solanum melongena L.). J Agric Food Chem. 2014;62:2906–12. https://doi.org/10.1021/jf404574c.
Khan MI, Giridhar P. The berries of Santalum album L. as a new source of cyanidin-3-glucoside and chemical profiling during different stages of berry development. P Natl A Sci India B. 2014; 84: 689–694; https://doi.org/10.1007/s40011-014-0312-0.
Khoo HE, Azlan A, Tang ST, Lim SM. Anthocyanidins and anthocyanins: colored pigments as food, pharmaceutical ingredients, and the potential health benefits. Food Nutr Res. 2017;61(1):1361779. https://doi.org/10.1080/16546628.2017.1361779.
Pelah D, Sintov A, Cohen E. The effect of salt stress on the production of canthaxanthin and astaxanthin by Chlorella zofingiensis grown under limited light intensity. World J Microb Biot. 2004;20:483–6. https://doi.org/10.1023/B:WIBI.0000040398.93103.21.
Shimokoriya M, Hattori S. On the formation of carthamin in the flowers of Carthamus tinctorius. Arch Biochem Biophys. 1955;54:93–101. https://doi.org/10.1016/0003-9861(55)90011-4.
Rebelo BA, Farrona S, Ventura MR, Abranches R. Canthaxanthin, a red-hot carotenoid: applications, synthesis, and biosynthetic evolution. Plants. 2020;9:1039. https://doi.org/10.3390/plants9081039.
Esatbeyoglu T, Rimbach G. Canthaxanthin: from molecule to function. Mol Nutr Food Res. 2017;61:1–17. https://doi.org/10.1002/mnfr.201600469.
Fan W, Li B, Tian H, Li X, Ren H, Zhou Q. Metabolome and transcriptome analysis predicts metabolism of violet-red color change in Lilium bulbs. J Sci Food Agric. 2022;102:2903–15. https://doi.org/10.1002/jsfa.11631.
Yvonne CC, Mohamed ZSHT. Isolation and characterization of chalcone isomerase (CHI) gene from Boesenbergia rotunda. S Afr J Bot. 2020;130:475–82. https://doi.org/10.1016/j.sajb.2020.01.010.
Kang JH, McRoberts J, Shi FJ, Moreno E, Jones AD, Howe GA. The flavonoid biosynthetic enzyme chalcone isomerase modulates terpenoid production in glandular trichomes of tomato. Plant Physiol. 2014;164:1161–74. https://doi.org/10.1104/pp.113.233395.
Wu X, Gong Q, Ni X, Zhou Y, Gao Z. UFGT: the key enzyme associated with the petals variegation in Japanese apricot. Front Plant Sci. 2017;8:108. https://doi.org/10.3389/fpls.2017.00108.
Yang YN, Yao GF, Zheng D, Zhang SL, Wang C, Zhang MY, Wu J. Expression differences of anthocyanin biosynthesis genes reveal regulation patterns for red pear coloration. Plant Cell Rep. 2015;34:189–98. https://doi.org/10.1007/s00299-014-1698-0.
Zhang Y, Shi M, Mao X. Time-resolved carotenoid profiling and transcriptomic analysis reveal mechanism of carotenogenesis for astaxanthin synthesis in the oleaginous green alga Chromochloris zofingiensis. Biotechnol Biofuels. 2019;12:287. https://doi.org/10.1186/s13068-019-1626-1.
Araya-Garay JM, Ageitos JM, Vallejo JA, Veiga-Crespo P, Sánchez-Pérez A, Villa TG. Construction of a novel Pichia pastoris strain for production of xanthophylls. AMB Express. 2012;2:24. https://doi.org/10.1186/2191-0855-2-24.
Van ZA, Liu W, Xiao TT, et al. The strigolactone biosynthesis gene DWARF27 is co-opted in rhizobium symbiosis. BMC Plant Biol. 2015;15:260. https://doi.org/10.1186/s12870-015-0651-x.
Xu G, Guo H, Zhang D, et al. REVEILLE1 promotes NADPH: protochlorophyllide oxidoreductase a expression and seedling greening in Arabidopsis. Photosynth Res. 2015;126:331–40. https://doi.org/10.1007/s11120-015-0146-5.
Bade RG, Bao ML, Jin WY, Niu YD, Hasi A. Genome-wide identification and analysis of the SGR gene family in Cucumis melo L. Genet Mol Res. 2016;17:15. https://doi.org/10.4238/gmr15048485.
Tanaka A, Ito H, Tanaka R, et al. Chlorophyll a oxygenase (CAO) is involved in chlorophyll b formation from chlorophyll a. P Natl Acad Sci Usa. 1998;95(21):12719–23. https://doi.org/10.1073/pnas.95.21.12719.
Liu W, Chen G, Chen J, Jahan MS, Guo S, Wang Y, Sun J. Overexpression of 7-hydroxymethyl chlorophyll a reductase from cucumber in tobacco accelerates dark-induced chlorophyll degradation. Plants (Basel). 2021;10:1820. https://doi.org/10.3390/plants10091820.
Want EJ, Masson P, Michopoulos F, et al. Global metabolic profiling of animal and human tissues via UPLC-MS. Nat Protoc. 2012;8:17–32. https://doi.org/10.1038/nprot.2012.135.
Warnes GR, Bolker B, Lumley T, Johnson RC. Gmodels: Various R programming tools for model fitting. 2007; available online: https://CRAN.R-project.org/package=gmodels.
Thevenot EA. ropls: PCA, PLS (-DA) and OPLS (-DA) for multivariate analysis and feature selection of omics data. 2016.
Li Y, Chen QY, Xie XD, Cai Y, Li JF, Feng YL, Zhang YJ. Integrated metabolomics and transcriptomics analyses reveal the molecular mechanisms underlying the accumulation of anthocyanins and other flavonoids in cowpea pod (Vigna unguiculata L.). J Agr Food Chem. 2020;68(34):9260–75. https://doi.org/10.1021/acs.jafc.0c0185.
Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–90. https://doi.org/10.1093/bioinformatics/bty560.
Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357. https://doi.org/10.1038/nmeth.3317.
Pertea M, Pertea GM, Antonescu CM, et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33(3):290–5. https://doi.org/10.1038/nbt.3122.
Pertea M, Kim D, Pertea GM, et al. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc. 2016;11(9):1650. https://doi.org/10.1038/nprot.2016.095.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. https://doi.org/10.1186/s13059-014-0550-8.
Zhou J, Huang J, Tian X. Transcriptome analysis reveals dynamic changes in the salt stress response in Salix. J For Res. 2020;31:1851–62. https://doi.org/10.1007/s11676-019-00941-w.
Csardi G. The igraph software package for complex network research. Interjournal Complex Systems. 2006; 1695.
Acknowledgements
Not applicable.
Funding
This work was funded by the Jiangsu Agricultural Science and Technology Independent Innovation Fund (grant number CX (20) 3042); the Independent Innovation Projects of the Jiangsu Academy of Forestry (grant number ZZKY202101); and the Key Research and Development Program of the National Forestry and Grassland Administration (grant number GLM [2021] 83).
Author information
Authors and Affiliations
Contributions
Conceptualization, JZ; methodology, JHG, XDH; software, JZ; validation, JZ, JHG, PW; writing—original draft preparation, JZ; writing—review and editing, JZ; visualization, JZ, BSW, ZGQ; funding acquisition, JZ, BSW, QSC. All authors have read and agreed to the published version of the manuscript.
Authors’ information
National Willow Engineering Technology Research Center, Jiangsu Academy of Forestry, Nanjing 211153, Jiangsu, China; Nanjing Forestry University, Nanjing 210037, Jiangsu, China
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable. The study was fully compliant with local and national regulations on plant usage.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
TableS1. Information on all metabolites. Table S2. Up- and downregulatedmetabolites. Table S3. RNA-sequencing profiles. Table S4. Number of up- anddownregulated differentially expressed genes. Table S5. The primers designedfor RT-qPCR.
Additional file 2: TableS6.
The metabolites identified in positive ion mode.
Additional file 3: Table S7.
The metabolites identified in negative ionmode.
Additional file 4: Table S8.
The differential expressed genes identified inwillow barks.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhou, J., Guo, J., Chen, Q. et al. Different color regulation mechanism in willow barks determined using integrated metabolomics and transcriptomics analyses. BMC Plant Biol 22, 530 (2022). https://doi.org/10.1186/s12870-022-03909-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-022-03909-x