
Abstract
Background
Roses are famous ornamental plants worldwide. Floral coloration is one of the most prominent traits in roses and is mainly regulated through the anthocyanin biosynthetic pathway. In this study, we investigated the key genes and metabolites of the anthocyanin biosynthetic pathway involved in color mutation in miniature roses. A comparative metabolome and transcriptome analysis was carried out on the Neptune King rose and its color mutant, Queen rose, at the blooming stage. Neptune King rose has light pink colored petals while Queen rose has deep pink colored petals.
Result
A total of 190 flavonoid-related metabolites and 38,551 unique genes were identified. The contents of 45 flavonoid-related metabolites, and the expression of 15 genes participating in the flavonoid pathway, varied significantly between the two cultivars. Seven anthocyanins (cyanidin 3-O-glucosyl-malonylglucoside, cyanidin O-syringic acid, cyanidin 3-O-rutinoside, cyanidin 3-O-galactoside, cyanidin 3-O-glucoside, peonidin 3-O-glucoside chloride, and pelargonidin 3-O-glucoside) were found to be the major metabolites, with higher abundance in the Queen rose. Thirteen anthocyanin biosynthetic related genes showed an upregulation trend in the mutant flower, which may favor the higher levels of anthocyanins in the mutant. Besides, eight TRANSPARENT TESTA 12 genes were found upregulated in Queen rose, probably contributing to a high vacuolar sequestration of anthocyanins. Thirty transcription factors, including two MYB and one bHLH, were differentially expressed between the two cultivars.
Conclusions
This study provides important insights into major genes and metabolites of the anthocyanin biosynthetic pathway modulating flower coloration in miniature rose. The results will be conducive for manipulating the anthocyanin pathways in order to engineer novel miniature rose cultivars with specific colors.
Similar content being viewed by others


Background
Throughout human history, roses have held a high iconic cultural significance. Roses are used as garden ornamental plants and are employed in various industries such as food and cosmetic. There is a wide variation in flower colors of rose cultivars. In ornamental plants, the floral color is the main attribute and is closely related to the distribution of pigment types [1]. Natural pigments such as flavonoids, carotenoids, betalains, and alkaloids are well known in flower color formation [2,3,4].
Particularly, flavonoids have attracted more attention as important secondary metabolites in plants [5]. Flavonoids are known to play significant roles in various aspects of plant physiological processes, including protection from ultraviolet radiation, pigmentation, and plant defense [6]. Flavonoids are the products of the phenylpropanoid biosynthesis pathway which has been well characterized in Arabidopsis and petunia [7, 8]. Several genes such as phenylalanine lyase, cinnamic acid hydroxylase, 4-coumarate-CoA ligase, chalcone synthase, chalcone isomerase, flavonoid 3-hydroxylase, flavonol synthase, dihydroflavonol 4-reductase, and anthocyanidin synthase, participate at different steps of the pathways. Anthocyanins are the end-products of the flavonoid pathway, but they are unstable in the cytoplasm and require further glycosylation. Genes of the UDP-glucosyltransferase and glutathione S-transferase families enter into action [9, 10]. Conserved structural genes have been identified in the flavonoid biosynthetic pathway, but their regulatory mechanisms vary across plant species [5, 7, 8, 11,12,13,14].
As the key aesthetic characteristic of roses, flower color has been early investigated by breeders. Up to date, only few flavonoids have been reported in roses. Rose petals are known to contain anthocyanins, such as pelargonidin, cyanidin, and peonidin [15,16,17]. The presence of 3,5-diglycosyl anthocyanidins in association with 3-glycosylated flavonols confer the pink and red colors of roses [18, 19]. Three genes (RhGT1, RhGT2, and RhGT3) encoding flavonoid 3-glycosyltransferases were found to be involved in the formation of anthocyanin glycosides [20]. Our understanding of flower coloration mechanisms in roses is far to be complete since several structural genes and as well as transcription factors (TF) regulating the anthocyanin pathways are yet to be reported. Moreover, the large variation existing in modern roses could indicate varying molecular mechanisms of color formation among different roses [21, 22]. High-quality genomes of various Rosa species are now available and will facilitate fundamental research on these plants [23,24,25]. Although some genes involved in flower coloration in roses have been cloned, the lack of mutants for comparative studies makes it impossible to clearly understand the regulation of anthocyanin pathways [26,27,28,29,30,31,32,33,34,35,36,37,38].
Understanding the genetic and molecular mechanisms that modify coloration will not only answer a fascinating query involving fundamental rose biology but will also allow the manipulation of rose quality. By integrating metabolome and transcriptome analyses, researchers have identified key metabolites and genes (structural or TF) regulating color formation in plants. In this study, we used a miniature rose cultivar (Neptune King) and its natural color mutant (Queen) and investigated the molecular changes affecting anthocyanin composition in their petals.
Results
Metabolome profiling of petal samples
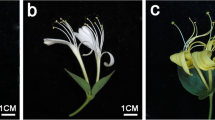
Fresh petals were collected from the miniature rose cultivar Neptune King (H) having light pink color and its natural color mutant Queen (S) displaying deep pink color (Fig. 1a, b). To compare the flavonoid content, petal samples were analyzed using UPLC-MS/MS. The flavonoid profiles of H and S flowers showed marked differences (Figures S1 and S2, Supplementary Materials). Using a local metabolite database, a total of 190 flavonoid-related metabolites were identified, including 18 anthocyanins, 61 flavonols, 8 dihydroflavones, 3 dihydroflavonols, 12 flavonoid carbonosides, 7 isoflavones, 67 other flavonoids, 11 proanthocyanidins, 1 chalcone, and 2 tannins (Table S1, Supplementary Materials). We used the metabolite quantification data to construct a hierarchical clustering heatmap. The results showed a close relationship between biological replicates, a sign of high quality metabolome quantification (Fig. 2a, b). Moreover, it could be observed a clear separation between H and S petal samples, indicating distinct flavonoid profiles in H and S samples.

Phenotypes of: a the wild type of miniature rose cultivar Neptune King (H); b Its natural mutant Queen (S)

a Principal component analysis based on FPKM data; b Clustered heatmap based on the 190 metabolites between the flower samples. The color scale (from red to green) represents normalized metabolite contents using the row Z-score. DH, Neptune King metabolites; DS, Queen metabolites
Differentially accumulated metabolites (DAMs) between H and S petal samples were determined based on the variable importance in projection ≥ 1 and a fold change ≥ 2 or ≤ 0.5. There were 45 metabolites whose accumulation was significantly different between the compared samples, and these differentially accumulated flavonoids (DAFs) were mainly anthocyanins, other flavonoids, procyanidins, and flavonols (Table 1). In particular, seven anthocyanins, namely, cyanidin 3-O-glucosyl-malonylglucoside, cyanidin O-syringic acid, cyanidin 3-O-rutinoside, cyanidin 3-O-galactoside, cyanidin 3-O-glucoside, peonidin 3-O-glucoside chloride, and pelargonidin 3-O-glucoside displayed significantly higher content in the S cultivar than in the H cultivar. The quantity of cyanidin O-syringic acid was high in the S cultivar but undetectable in the H cultivar. Additionally, the content of cyanidin 3-O-glucosyl-malonylglucoside was down-accumulated in the H cultivar compared to the S cultivar. The results indicate that these anthocyanins may play a crucial role for deep pink color formation in S flowers. Thus, the high accumulation of peonidin, pelargonidin, and cyanidin derivatives in the S cultivar may have led to its color-related changes.
Transcriptome sequencing and analysis
Global gene expression was further profiled in the petal samples of the two cultivars. From the six cDNA libraries (three biological replicates per sample), we obtained a total of 47.52 Gb of clean data, with more than 91% of bases scoring Q30 (Table S2, Supplementary Materials). The libraries had 46,782,302–55,533,348 clean reads, which were successfully mapped to the Rosa chinensis genome (https://lipm-brows-ers.toulouse.inra.fr/pub/RchiOBHm-V2/), with matching rates in the range of 80.49%–82.12%. Consequently, 5,149 novel genes were identified, 1,303 unique genes were found to be expressed in the rose petals (Table S3, Supplementary Materials). A total of 38,551 genes were detected in the rose samples and their expression levels was estimated based on the Fragments per kilobase of exon model per million reads mapped values method (Fig. 3a, Table S4, Supplementary Materials). Transcriptome based principal component analysis (PCA) of the data showed a clear distinction between H and S samples, which is similar to the results obtained based on the metabolome analysis (Fig. 3b). This implies that the DAMs in the two phenotypes were regulated by differentially expressed genes (DEGs). A total of 4,298 DEGs, including 1,851 upregulated and 2,447 downregulated genes (Figure S2, Supplementary Materials) were identified between the samples.

a Overall distribution of sample gene expression based on FPKM data; b Principal component analysis based on FPKM data. ZH, Neptune King genes; ZS, Queen genes
Expression of genes involved in the flavonoid biosynthesis pathway
The expression patterns of genes involved in the flavonoid biosynthesis pathways were analyzed. A total of 150 genes were assigned to this pathway (Table S5, Supplementary Materials). Comparison of the H and S data showed 15 flavonoid related genes differentially expressed. All genes involved in the specific anthocyanin biosynthetic pathways, except for ANS, were upregulated in the S cultivar, which may be determinant for color-related changes (Table 2).
Expression patterns of transcription factors
MYB and bHLH are particular transcription factors (TF) known to control the expression levels of structural genes involved in the biosynthesis of anthocyanins [39]. Hence, we searched or the DEGs encoding TF between the two cultivars. Only 30 DEGs encoding TFs were identified. Among these TFs, we identified one bHLH (RchiOBHmChr1g0361301) and two MYB (RchiOBHmChr1g0373541 and RchiOBHmChr1g0361191) (Table 3). RchiOBHmChr1g0361301 (bHLH) and RchiOBHmChr1g0361191 (MYB) were strongly up-regulated in the S cultivar, suggesting a positive regulation of anthocyanin biosynthetic structural genes. In contrast, RchiOBHmChr1g0373541 (MYB) was not expressed at all in the petals of the S cultivar but was expressed in petals of the H cultivar. Comparison of the sequences of this transcript among the two cultivars did not show any nucleotide change, therefore it is probable that a mutation has occurred in the regulatory regions.
Expression patterns of MATE and ABCC gene family members
It has been reported that members of the multidrug and toxic compound extrusion (MATE) and the ATP-binding cassette transporter sub-family C (ABCC) families play preponderant roles for the sequestration of the anthocyanins from the cytoplasm to vacuoles [40, 41]. We therefore expanded our study on the expression patterns of ABCC and MATE genes in miniature rose petals. A total of 55 and 64 ABCC and MATE genes were found expressed in the H and S samples. No ABCC gene was differentially expressed between both samples. In contrast, 10 MATE DEGs were identified, including 8 down- and 2 up-regulated MATE genes in H sample (Table S6, Supplementary Materials). The high number of down-regulated MATE genes in H samples suggests a weak sequestration of anthocyanins in vacuoles of the petals.
qRT-PCR validation of the expression patterns of anthocyanin related genes
To further validate the RNA-seq results, nine DEGs related to anthocyanin biosynthesis were selected, and their expression levels were analyzed in H and S petals at the blooming stage using qRT-PCR. The primer sequences of the genes are shown in Table S7, Supplementary Materials. The results confirmed that anthocyanin biosynthetic and regulatory genes, including DFR (RchiOBHmChr5g0037071, RchiOBHmChr6g0281711), CHI (RchiOBHmChr1g0372181), BZ1 (RchiOBHmChr4g0393121, RchiOBHmChr6g0302721), UGT79B1 (RchiOBHmChr5g0009571), MYB (RchiOBHmChr6g0252211, RchiOBHmChr7g0241861), and bHLH (RchiOBHmChr1g0361301) were upregulated in petals of the S cultivar (Figure S3, Supplementary Materials). We noticed a complete concordance between qRT-PCR and RNA-seq results, demonstrating that the RNA-seq data and DEG analysis in this study were reliable.
Association analysis of the metabolome and transcriptome
To demonstrate the synthetic and regulatory characteristics of the DAMs and DEGs, subnetworks were constructed to show transcript–metabolite correlations. Only pairs with a correlation coefficient > 0.8 were considered in the analysis.
Four transcript–metabolite correlation networks were constructed. Among these, Ko00941 consisted of 15 nodes and 31 edges, with 26 pairs showing a positive correlation and 5 pairs showing a negative correlation (Fig. 4a). Ko00942 consisted of five nodes and six edges, with four pairs showing a positive correlation and two pairs showing a negative correlation (Fig. 4b, Supplementary Materials). Ko00943 consisted of nine nodes and eight edges, with four pairs showing a positive correlation and four pairs showing a negative correlation (Fig. 4c). Ko00944 consisted of five nodes and six edges, with three pairs showing a positive correlation and three pairs showing a negative correlation (Fig. 4d). Detailed information on the gene–metabolite pairs involved in flavonoid biosynthesis is listed in Table S8-1–S8-4, Supplementary Materials.

Transcript–metabolite correlation networks: a Ko00941; b Ko00942; c Ko00943; d Ko00944. Green: Metabolite; Red: gene. Solid lines: positive correlation; Dashed lines: negative correlation
These results indicate that several classic flower pigmentation-related genes were highly correlated with their corresponding metabolites, which are involved in flavonoid biosynthesis, thereby reconfirming the importance of flavonoid biosynthesis in flower color.
Modulation of biosynthesis and accumulation of anthocyanins in rose flowers
The flavonoid pathway begins with the chalcone synthase (CHS), leading to the synthesis of naringenin chalcone from 4-coumaroyl-CoA and malonyl-CoA. Next, naringenin chalcone is isomerized by chalcone isomerase (CHI) to naringenin. Naringenin is converted into dihydrokaempferol by flavanone 3-hydroxylase (F3H). Later, dihydrokaempferol can be further hydroxylated into dihydroquercetin or dihydromyricetin by flavonoid 3′-hydroxylase (F3′H) or flavonoid 3′,5′-hydroxylase (F3′5′H), respectively. All these dihydroflavonols are converted by dihydroflavonol 4-reductase (DFR) into colorless leucoanthocyanidins and subsequently to colored anthocyanidins by anthocyanidin synthase (ANS). At the end, anthocyanidins are decorated and glycosylated by different glycosyltransferase enzymes, including, flavonoid 3-O-glucosyltransferase (UFGT) [19].
Based on our results, we reconstructed the anthocyanin biosynthetic pathway in miniature roses. The identified anthocyanins and their relevant intermediates were rearranged to their corresponding positions (Fig. 5). The key genes involved in the anthocyanin biosynthesis were significantly upregulated in petals of the S cultivar, which was consistent with the high accumulation of anthocyanins in S. Taken together, we deduce that the coordinated upregulation of CHS, DFR, FLS, BZ1, GT1, UGT78D2, and UGT79B1 together with the high expression levels of MATE genes play a crucial role in stimulating anthocyanin synthesis and accumulation, including cyanidin O-syringic acid, cyanidin 3-O-rutinoside, cyanidin 3-O-galactoside, cyanidin 3-O-glucoside, peonidin 3-O-glucoside chloride, and pelargonidin 3-O-glucoside, conferring the deep pink color in the S cultivar.

Flavonoid biosynthesis pathway and the differential metabolites of petals in miniature rose cultivar Neptune King (H) and natural mutant Queen (S). The red dots represent the up-regulated metabolites
Discussion
The aesthetic feature of flowers is of central importance in the ornamental quality and commercial value of orchids. Flower coloration has been the focus of biological studies [42,43,44], which have shown that there are species-specific peculiarities in the regulation of pigmentation. To contribute to the existing knowledge, species-specific studies of flower color formation and elucidation of specific regulatory mechanisms are necessary. In the present study, we investigated the underlying mechanism of color formation in two distinct flower phenotypes, the miniature rose cultivar Neptune King and its mutant, Queen rose.
Effects of the of anthocyanins distribution on flower coloration in the two rose cultivars
A total of 190 flavonoid-related metabolites were detected and categorized into eight subgroups, including 18 anthocyanins and 11 procyanidins. This provides the most complete view of the metabolites involved in flower coloration in miniature roses. Previous studies [15,16,17, 45, 46] on the rose coloration process have been limited to a few flavonoid metabolites. The composition of anthocyanidins and flavonols determines the diverse phenotypes of flowers [47, 48]. The analysis of the DAMs showed that the mutant Queen was characterized by a significant high accumulation of various flavonoids. In the present study, we detected 45 differential flavonoid-related metabolites, including 7 anthocyanins. The findings indicate that cyanidin O-syringic acid, cyanidin 3-O-rutinoside, cyanidin 3-O-galactoside, cyanidin 3-O-glucoside, peonidin 3-O-glucoside chloride, and pelargonidin 3-O-glucoside accumulated to a greater extent in mutant flowers. Therefore, cyanidin derivatives can be considered the key anthocyanins responsible for the color change. It was reported that cyanidin and peonidin are responsible for orange to red colors [3, 49,50,51,52,53,54].
Anthocyanin contents have been regarded as the major factors endowing transgenic tobacco with a pink or light red flower color [55,56,57,58,59]. Similarly, cyanidin and peonidin have been shown to be responsible for the red color in various plants, such as tree peony, strawberry, apple, Chrysanthemum, Rhododendron and Prunus mume [2, 3, 60,61,62,63]. In addition, a combination of several flavonoid metabolites results in a co-pigmentation effect, which leads to deeper colors [64,65,66]. In the present work, the levels of one luteolin derivative, three kaempferol, three quercetin derivatives, one apigenin, were simultaneously increased in S samples, indicating a co-pigmentation effect responsible for the deep pink color observed in the mutant Queen rose.
Major anthocyanin related genes affecting flower coloration in the two rose cultivars
Recently, high-throughput RNA-seq has been extensively used in studies on roses [67,68,69,70]. The pathways controlling flower coloration are properly characterized in plants, including roses [71, 72]. However, because of the extensive genetic variations observed in roses, varying mechanisms for color formation are expected.
In this study, the transcriptomes of Neptune King and its color mutant were analyzed, and differentially expressed flavonoid biosynthetic genes affecting the color formation were identified. The results of RNA-seq showed a significant increase in the expression of genes involved in the flavonoid biosynthetic pathway in S flowers, which strongly supported the findings of our metabolome analysis. Our findings indicated the upregulated expression of genes such as CHS, DFR, BZ1, UGT79B1, GT1, UGT78D2, FLS, and IF7MAT in S compared to H, implying that the high expression of these genes was the immediate cause for the higher accumulation of anthocyanins in mutant flowers. In previous reports, the expression of early or late biosynthetic genes, usually correlates positively with the anthocyanin content [73,74,75,76,77,78,79,80].
CHS is the primary rate-limiting enzyme that catalyzes the transformation of p-coumaroyl-CoA and malonyl-CoA into tetrahydroxy-chalcone in flavonoid biosynthesis. Weak expression of CHS restrains anthocyanin biosynthesis and leads to color changes in plant tissues. Variation of CHS expression levels confers color polymorphism in crabapple and arctic mustard flowers [81, 82]. In the present work, we deduce that the high expression levels of CHS in S flowers would provide sufficient amounts of precursor compounds for the higher anthocyanin content.
DFR is considered a critical late enzyme in anthocyanin biosynthesis. DFR catalyzes the conversion of dihydroquercetin into leucoanthocyanins which can supply flavonoids for the anthocyanin pathway. The modulation of DFR expression levels led to varying anthocyanins levels and pigmentations in plants such as Dianthus caryophyllus, Arabidopsis, crabapples, and rubellis [83,84,85,86,87,88,89].
Anthocyanidins are extremely unstable and degenerate easily; therefore, glycosylation is crucial to stabilize and transport these compounds to vacuoles, where they can function as pigments. This glycosylation role is played by UFGT genes [7, 90]. In the present study, RhGT1 expression was upregulated in S flowers, which may account for the deep pink color. In accordance to our study, Griesser et al. [91] demonstrated that a downregulation of GT1 resulted in of a significant reduction of anthocyanin pigments in ripe strawberry fruits.
MYB and bHLH are the major transcription factors regulating the expression of structural genes involved in the anthocyanin biosynthesis [39]. In this study, we identified three key genes that could be further investigated in order to facilitate the modulation of color in miniature roses.
Downregulation of MATE genes could reduce anthocyanin sequestration in the vacuoles of H flowers
Anthocyanins, particularly the hydrophobic aglycone forms, are toxic molecules for the cell machinery due to their high chemical reactivity. Hence, after synthesis, a mechanism of anthocyanin removal is immediately triggered, leading to a sequestration in the central vacuole. High vacuolar accumulation of anthocyanins confers the observed color phenotypes in plant tissues. MATE and ABCC members have been reported as key genes involved in anthocyanins sequestrations in the vacuoles [40, 41]. Debeaujon et al. [40] were the first to discover the gene TRANSPARENT TESTA12 (TT12), a MATE member, necessary for flavonoid sequestration in vacuoles of the seed coat endothelium in Arabidopsis. Later on, TT12 has been identified, isolated and functionally validated in various plant species such as Brassicas, grapevine and blueberry [92,93,94]. In this study, we identified eight TT12 upregulated in petals of the S cultivar, indicating that these genes may contribute to the high accumulation of anthocyanins observed in S.
Conclusions
In this study, higher contents of diverse flavonoids and alterations in gene expression patterns were observed in the rose mutant as compared to its wild type. The mutation induced a significant upregulation of the key genes participating in the anthocyanin synthesis pathway, glycosylation and sequestration, resulting in significantly increased anthocyanin accumulation and darker flower coloration in the S cultivar. Functional characterization of the candidate structural genes as well as the three major transcription factors will continue.
Methods
Plant materials and sampling
The miniature rose cultivar Neptune King (H) and its natural mutant Queen (S) were cultivated at the Liaoning Academy of Agricultural Sciences in Shenyang City (41°48′11.75″N, 123°25′31.18″E), China. The formal identification of the plant materials was undertaken by Professor Hongmei Sun. The plants are kept at the Key Laboratory of Protected Horticulture of Education Ministry and Liaoning Province, College of Horticulture, Shenyang Agricultural University, Shenyang, China. The petal samples were collected for the metabolome study, RNA-Seq and qRT-PCR analysis. Three biological replicates were gathered per sample, with 10 blooms flowers randomly collected from 10 plants in the same batch. The petals were frozen in liquid nitrogen, and saved at − 80 °C until further use. The H and S samples used for the metabolome analysis were designated DH and DS, respectively, and those used for the RNA-seq analysis were designated ZH and ZS, respectively.
Sample preparation and extraction
Flavonoid metabolites were extracted following a previous protocol [65], with some modifications. Petal samples were vacuum freeze-dried and ground (1.5 min at 30 Hz) to powder using a grinder. The powder (0.1 g) was weighed and dissolved in a 70% aqueous solution of methanol. To improve extraction efficiency the dissolved sample was swirled three times then stored at 4 °C overnight. The supernatant was filtered with a micropore membrane (0.22 μm pore size) after centrifugation (10,000 × g, 10 min), and stored in a sample injection bottle for UPLC-MS/MS analysis.
Flavonoid identification, quantification, and data analysis
All samples were analyzed by a UPLC-MS/MS system (Shim-pack UFLC SHIMADZU CBM30A and Applied Biosystems 6500 QTRAP). A scheduled multiple reaction monitoring method was used to metabolite quantification.
Based on the self-built MWDB database (Metware Biotechnology Co., Ltd., Wuhan, China) and a public database of metabolite information, the fundamental and secondary MS records had been qualitatively analyzed using Analyst 1.6.3. The identified metabolites with significant difference in content were set with 0.5 ≥ fold change ≥ 0 or a fold change ≥ 2, p-value < 0.05, and VIP ≥ 1 were considered DAFs. Finally, the KEGG pathway database (http://www.kegg.jp/kegg/pathway.html) and MWDB were centered on metabolic reactions and concatenated possible metabolic pathways.
RNA extraction, quantification, and sequencing
RNA extraction from petal samples, quantification and sequencing were conducted according to the procedures detailed by Dossa et al. [95, 96]. Six libraries representing the collected H and S petal samples (three biological replicates respectively) were prepared following standard procedures of Illumina HiSeq 4000 platform.
Transcriptome data analysis
The raw data were cleaned by removing adaptors and low-quality reads using Fastqc with default parameters. Then, the clean reads were mapped to the R. chinensis genome (https://lipm-brows-ers.toulouse.inra.fr/pub/RchiOBHm-V2/) using HISAT tool. Fragments per kilobase of exon model per million reads mapped values were used for gene/transcript measurement. Differential expressed gene (DEG) analysis was conducted using the DESeq2 tool. Genes with |log2Fold Change|≥ 1 and a p-value < 0.05, were described as DEGs [96]. GO and KEGG enrichment analyses of the DEGs were conducted in the R package ‘clusterProfiler’.
qRT-PCR analysis
Nine anthocyanin-related genes were targeted for qRT-PCR. The qRT-PCR was performed based on the same RNA samples used for sequencing [95]. The gene Actin was used as the internal control for transcript expression normalization. The primer pairs were designed using the Primer5 tool. The cDNAs were synthesized using SuperScript™ III First-Strand Synthesis SuperMix for qRT-PCR (Invitrogen). The qRT-PCR was conducted on an ABI 7500 Fast real-time detection system (Applied Biosystems) The reaction mix contained: 5 μl of FastStart Universal SYBR Green Master (Roche Life Sciences), 2 μl of 2 μM primer mix, 2 μl of a diluted 1:10 cDNA and ddH2O was added to a final volume of 10 μl. The cycling conditions were set as follow: 95 °C for 10 min, and 40 cycles of 95 °C for 15 s, and 60 °C for 1 min. The experiment was conducted with three biological replicates and three technical replicates. Data was analyzed according to the 2−∆∆Ct method [97].
Availability of data and materials
The RNA-seq data has been submitted to NCBI SRA: PRJNA684357.
Abbreviations
- ANS:
-
Anthocyanidin synthase
- CHI:
-
Chalcone isomerase
- CHS:
-
Chalcone synthase
- DAF:
-
Differentially accumulated flavonoid
- DAM:
-
Differentially accumulated metabolite
- DEG:
-
Differentially expressed gene
- DFR:
-
Dihydroflavonol 4-reductase
- F3H:
-
Flavonoid 3-hydroxylase
- FLS:
-
Flavonol synthase
- GO:
-
Gene Ontology
- GT:
-
Anthocyanidin 5,3-O-glucosyltransferase
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- KOG:
-
Eukaryotic Orthologous Groups
- OPLS-DA:
-
Orthogonal partial least squares discriminant analysis
- PC1:
-
First principal component
- PCA:
-
Principal component analysis
- qRT-PCR:
-
Quantitative real-time polymerase chain reaction
- RNA-seq:
-
RNA sequencing
- UGT:
-
UDP-glucosyltransferase
- UPLC-MS/MS:
-
Ultra-performance liquid chromatography–tandem mass spectrometry
- VIP:
-
Variable importance in projection
References
Zhao D, Tao J. Recent advances on the development and regulation of flower color in ornamental plants. Front Plant Sci. 2015;6:261.
Wang LS, Shiraishi A, Hashimoto F, Aoki N, Sakata Y. Analysis of petal Anthocyanins to investigate flower coloration of Zhongyuan (Chinese) and Daikon Island (Japanese) tree peony cultivars. J Plant Res. 2001;114:33–43.
Ma KF, Zhang QX, Cheng TR, Yan XL, Pan HT, Wang J. Substantial epigenetic variation causing flower color chimerism in the ornamental tree Prunus mume revealed by single base resolution methylome detection and transcriptome sequencing. Int J Mol Sci. 2018;19:2315.
Zhu HH, Yang JX, Xiao CH, Mao TY, Zhang J, Zhang HY. Differences in flavonoid pathway metabolites and transcripts affect yellow petal colouration in the aquatic plant Nelumbo nucifera. BMC Plant Biol. 2019;19:277.
Ferrer JL, Austin MB, Stewart C, Noel JP. Structure and function of enzymes involved in the biosynthesis of phenylpropanoids. Plant Physiol Biochem. 2008;46:356–70.
Forkmann G. Flavonoids as flower pigments: The formation of the natural spectrum and its extension by genetic engineering. Plant Breeding. 1991;106(1):1–26.
Saito K, Yonekura-Sakakibara K, Nakabayashi R, Higashi Y, Yamazaki M, Tohge T, Fernie AR. The flavonoid biosynthetic pathway in Arabidopsis: structural and genetic diversity. Plant Physiol Biochem. 2013;72:21–34.
Holton TA, Brugliera F, Tanaka Y. Cloning and expression of flavonol synthase from Petunia hybrida. Plant J. 2010;4:1003–10.
Jaakola L. New insights into the regulation of anthocyanin biosynthesis in fruits. Trends Plant Sci. 2013;18:477–83.
Winkel-Shirley B. Flavonoid biosynthesis. A colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant Physio. 2001;126:485–93.
Grotewold E. The genetics and biochemistry of floral pigments. Annu Rev Plant Biol. 2006;57:761–80.
Davies, KM, Schwinn KE. Molecular biology and biotechnology of flower pigments. Plant Dev Biol Biotech Perspect. 2010:161–87.
Ji T, Mengchen C, Jie Z, Ke-ting L, Tingting S, Xi Z, Yuncong Y. Characteristics of dihydroflavonol 4-reductase gene promoters from different leaf colored Malus crabapple cultivars. Hor Res. 2017;4:17070.
Quattrocchio F, Wing JF, van der Woude K, Mol JN, Koes R. Analysis of bHLH and MYB domain proteins: species-specific regulatory differences are caused by divergent evolution of target anthocyanin genes. Plant J. 1998;13:475–88.
Arisumi K. Studies on the flower colour in Rosa, with special references to the biochemical and genetic analyses and to the application of those results to the practical breeding I. Gakugei Zasshi Kyushu Daigaku Nogakubu. 1963;20:131–49.
Arisumi K. Studies on the flower colour in Rosa, with special references to the biochemical and genetic analyses and to the application of those results to the practical breeding II. Gakugei Zasshi Kyushu Daigaku Nogakubu. 1964;21:169–84.
Eugste CH, Marki-Fische E. The chemistry of Rose pigments. Angrew Chem Int Ed Engl. 1991;30:654–72.
Biolley JP, Jay M. Anthocyanins in modern roses—chemical and colorimetric features in relation to the color range. J Exp B. 1993;44:1725–34.
Ogata J, Kanno Y, Itoh Y, Tsugawa H, Suzuki M. Plant biochemistry: anthocyanin biosynthesis in roses. Nature. 2005;435:757–8.
Fukuchi-Mizutani M, Akagi M, Ishiguro K, Katsumoto Y, Fukui Y, Togami J, Nakamura N, Tanaka Y. Biochemical and molecular characterization of anthocyanidin/flavonol 3-glucosylation pathways in Rosa × hybrida. Plant Biotech. 2011;28:239–44.
Koes R, Verweij W, Quattrocchio F. Flavonoids: a colorful model for the regulation and evolution of biochemical pathways. Trends Plant Sci. 2005;10:236–42.
Jing M, Fougère-Danezan M, Zhang LB, Li DZ, Yi TS. Untangling the hybrid origin of the Chinese tea roses: evidence from DNA sequences of single-copy nuclear and chloroplast genes. Plant Syst. 2011;297:157–70.
Nakamura N, Hirakawa H, Sato S, Otagaki S, Matsumoto S, Tabata S, Tanaka Y. Genome structure of Rosa multiflora, a wild ancestor of cultivated roses. DNA Res. 2017;25:113–21.
Raymond O, Gouzy J, Just J, Badouin H, Verdenaud M, Lemainque A, Vergne P, Moja S, Choisne N, Pont C. The Rosa genome provides new insights into the domestication of modern roses. Nat Genet. 2018;50:772–7.
Hibrand SO, Ruttink T, Hamama L, Kirov I, Lakhwani D, Zhou NN, Bourke PM, Daccord N, Leus L, Schulz D. A high-quality genome sequence of Rosa chinensis to elucidate ornamental traits. Nat Plants. 2018;4:473–84.
Lou Q, Liu Y, Qi Y, Jiao S, Tian F, Jiang L, Wang Y. Transcriptome sequencing and metabolite analysis reveals the role of delphinidin metabolism in flower colour in grape hyacinth. J Exp Bot. 2014;65:3157.
Matus JT. Transcriptomic and metabolomic networks in the grape berry illustrate that it takes more than flavonoids to fight against ultraviolet radiation. Front Plant Sci. 2016;7:1337.
Wang Z, Cui Y, Vainstein A, Chen S, Ma H. Regulation of fig (Ficus carica L.) fruit color: metabolomic and transcriptomic analyses of the flavonoid biosynthetic pathway. Front Plant Sci. 2017;8:1990.
Wang Z, Du H, Zhai R, Song L, Ma F, Xu L. Transcriptome analysis reveals candidate genes related to color fading of “Red Bartlett” (Pyrus communis L.). Front Plant Sci. 2017;8:455.
Li Y, Fang J, Qi X, Lin M, Zhong Y, Sun L, Cui W. Combined analysis of the fruit metabolome and transcriptome reveals candidate genes involved in flavonoid biosynthesis in Actinidia arguta. Int J Mol Sci. 2018;19.
Xiaoan L, Na L, Ling F, Shouzhou Z. Integrated metabolome and transcriptome analysis uncovers the role of Anthocyanin metabolism in Michelia maudiae. Int J Genom. 2019.
Zhou C, Mei X, O’Neill Rothenberg D, Yang Z, Zhang W, Wan S, Yang H, Zhang L. Metabolome and transcriptome analysis reveals putative genes involved in Anthocyanin accumulation and coloration in white and pink tea (Camellia sinensis) flower. Molecules. 2020;25:190.
Dong T, Han R, Yu J, Zhu M, Zhang Y, Gong Y, Li Z. Anthocyanins accumulation and molecular analysis of correlated genes by metabolome and transcriptome in green and purple asparaguses (Asparagus officinalis L.). Food Chem. 2019;271:18–28.
Wang M, Chen L, Liang Z, He X, Liu W, Jiang B, Yan J, Sun P, Cao Z, Peng Q, Lin Y. Metabolome and transcriptome analyses reveal chlorophyll and anthocyanin metabolism pathway associated with cucumber fruit skin color. BMC Plant Biol. 2020;20:386.
Jiao F, Zhao L, Wu X, Song Z, Li Y. Metabolome and transcriptome analyses of the molecular mechanisms of flower color mutation in tobacco. BMC Genom. 2020;21:611.
Liu Y, Lv J, Liu Z, Wang J, Yang B, Chen W, Ou L, Dai X, Zhang Z, Zou X. Integrative analysis of metabolome and transcriptome reveals the mechanism of color formation in pepper fruit (Capsicum annuum L.). Food Chem. 2020;306:125629.
Zhang Q, Wang L, Liu Z, Zhao Z, Zhao J, Wang Z, Zhou G, Lio P, Liu M. Transcriptome and metabolome profiling unveil the mechanisms of Ziziphus jujuba Mill. peel coloration. Food Chem. 2020;312:125903.
Cho K, Cho K-S, Sohn H-B, Ha IJ, Hong S-Y, Lee H, Kim Y-M, Nam MH. Network analysis of the metabolome and transcriptome reveals novel regulation of potato pigmentation. J Exp B. 2016;67:1519–33.
Broun P. Transcriptional control of flavonoid biosynthesis: A complex network of conserved regulators involved in multiple aspects of differentiation in Arabidopsis. Curr Opin Plant Biol. 2005;8:272–9.
Debeaujon I, Peeters AJM, Léon-Kloosterziel KM, Koornneef M. The TRANSPARENT TESTA12 gene of arabidopsis encodes a multidrug secondary transporter-like protein required for flavonoid sequestration in vacuoles of the seed coat endothelium. Plant Cell. 2001;13:853–71.
Francisco RM, Regalado A, Ageorges A, Burla BJ, Bassin B, Eisenach C, Zarrouk O, Vialet S, Marlin T, Chaves MM, Martinoia E, Nagy R. ABCC1, an ATP binding cassette protein from grape berry, transports anthocyanidin 3-O-Glucosides. Plant Cell. 2013;25:1840–54.
Mol J, Grotewold E, Koes R. How genes paint flowers and seeds. Trends Plant. 1998;3:212–7.
Ben-Meir H, Zuker A, Weiss D, Vainstein A. Molecular control of floral pigmentation: anthocyanins. In Breeding for ornamentals: classical and molecular approaches, Vainstein A., Ed. Springer. 2002, 253–72.
Sobel JM, Streisfeld MA. Flower color as a model system for studies of plant evo-devo. Front Plant Sci. 2013;4:321.
Ping H, Furong L, Bin L, Yongqi Z. Hybrid-transcriptome sequencing and associated metabolite analysis reveal putative genes involved in flower color difference in rose mutants. Plants. 2019;8:267.
Suzuki K, Tsuda S, Fukui Y, Fukuchi-Mizutani M, Yonekura-Sakakibara K, Tanaka Y, Kusumi T. Molecular characterization of rose flavonoid biosynthesis genes and their application in Petunia. Biotechnol Biotechnol Equip. 2000;14:56–62.
Andersen OM, Jordheim M. Anthocyanins. Chichester: Wiley, 2010; pp. 1–12
GouldK, Davies K, Winefield C. Anthocyanins: biosynthesis, functions, and application. Springer; 2009.
Khoo HE, Azlan A, Tang ST, Lim SM. Anthocyanidins and anthocyanins: colored pigments as food, pharmaceutical ingredients, and the potential health benefits. Food Nutr. 2017;61:1361779.
Tanaka Y, Brugliera F, Chandler S. Recent progress of flower colour modification by biotechnology. Int J Mol Sci. 2009;10:5350–69.
Xue L, Wang ZG, Zhang W, Li YX, Wang J, Lei JJ. Flower pigment inheritance and anthocyanin characterization of hybrids from pink-flowered and white-flowered strawberry. Sci Hortic. 2016;200:143–50.
Holton TA, Cornish EC. Genetics and biochemistry of Anthocyanin biosynthesis. Plant Cell. 1995;7:1071–83.
Jun O, Yoshiaki K, Yoshio I, Hidehito T, Masahiko S. Plant biochemistry: Anthocyanin biosynthesis in roses. Nature. 2005;435:757–8.
Yang X, Xia X, Zhang Z, Nong B, Zeng Y, Wu Y, Xiong F, Zhang Y, Liang H, Pan Y, Dai G, Deng G, Li D. Identification of anthocyanin biosynthesis genes in rice pericarp using PCAMP. Plant Biotechnol J. 2019;17:1700–2.
Kumar V, Yadav SK. Overexpression of CsANR increased flavan-3-ols and decreased anthocyanins in transgenic tobacco. Mol Biotechnol. 2013;54:426–35.
Huang ZA, Zhao T, Wang N, Zheng SS. Ectopic expression of Lc differentially regulated anthocyanin biosynthesis in the floral parts of tobacco (Nicotiana tobacum L.) plants. Bot Stud. 2016;57:24.
Dasgupta K, Thilmony R, Stover E, Oliveira ML, Thomson J. Novel R2R3-MYB transcription factors from Prunus americana regulate differential patterns of anthocyanin accumulation in tobacco and citrus. GM Crops Food. 2017;8:85–105.
Nishihara M, Nakatsuka T, Yamamura S. Flavonoid components and flower color change in transgenic tobacco plants by suppression of chalcone isomerase gene. FEBS Lett. 2005;579:6074.
Yang CC, Yu JJ, Zhao Q, Zhu DY, Ao GM. Influence of maize Lc regulatory gene on flower colour of transgenic tobacco and petunia. J Agric Biotechnol. 2007;15:85–9.
Liu L, Zhang LY, Wang SL, Niu XY. Analysis of anthocyanins and flavonols in petals of 10 Rhododendron species from the Sygera Mountains in Southeast Tibet. Plant Physiol Biochem. 2016;104:250–6.
Schwinn KE, Markham KR, Giveno NK. Floral flavonoids and the potential for pelargonidin biosynthesis in commercial chrysanthemum cultivars. Phytochemistry. 1993;35:145–50.
Honda C, Kotoda N, Wada M. Anthocyanin biosynthetic genes are coordinately expressed during red coloration in apple skin. Plant Physiol Biochem. 2002;40:955–62.
Du H, Lai L, Wang F, Sun W, Zhang L, Li X, Wang L, Jiang L, Zheng Y. Characterization of flower coloration in 30 Rhododendron species via anthocyanin and flavonol identification and quantitative traits. Plant Biol. 2018;20:121–9.
Asen S, Stewart RN, Norris KH. Co-pigmentation of anthocyanins in plant tissues and its effect on color. Phytochemistry. 1972;11:1139–44.
Boulton R. The copigmentation of anthocyanins and its role in the color of red wine: a critical review. Am J Enol Vitic. 2001;52:67–87.
Mizuno T, Yabuya T, Kitajima J, Iwashina T. Identification of novel Cglycosylflavones and their contribution to flower colour of the Dutch iris cultivars. Plant Physiol Biochem. 2013;72:116–24.
Liu X, Cao X, Shi S, Zhao N, Li D, Fang P, Chen X, Qi W, Zhang Z. Comparative RNA-Seq analysis reveals a critical role for brassinosteroids in rose (Rosa hybrida) petal defense against Botrytis cinerea infection. BMC Genet. 2018;19:62.
Qi W, Chen X, Fang P, Shi S, Li J, Liu X, Cao X, Zhao N, Hao H, Li Y. Genomic and transcriptomic sequencing of Rosa hybrida provides microsatellite markers for breeding, flower trait improvement and taxonomy studies. BMC Plant Biol. 2018;18:119.
Dubois A, Carrere S, Raymond O, Pouvreau B, Cottret L, Roccia A, Onesto JP, Sakr S, Atanassova R, Baudino S. Transcriptome database resource and gene expression atlas for the rose. BMC Genom. 2012;13:638.
Guo X, Yu C, Luo L, Wan H, Zhen N, Xu T, Tan J, Pan H, Zhang Q. Transcriptome of the floral transition in Rosa chinensis ‘Old Blush.’ BMC Genom. 2017;18:199.
Koes R, Verweij W, Quattrocchio F. Flavonoids: a colorful model for the regulation and evolution of biochemical pathways. Trends Plant. 2005;10:236–42.
Bendahmane M, Dubois A, Raymond O, Le Bris M. Genetics and genomics of flower initiation and development in roses. J Exp Bot. 2013;64:847–57.
Honda C, Kotoda N, Wada M, Kondo S, Kobayashi S, Soejima J, Zhang Z, Tsuda T, Moriguchi T. Anthocyanin biosynthetic genes are coordinately expressed during red coloration in apple skin. Plant Physiol Biochem. 2002;40:955–62.
Ban Y, Kondo S, Ubi BE, Honda C, Bessho H, Moriguchi T. UDP-sugar biosynthetic pathway: contribution to cyanidin 3-galactoside biosynthesis in apple skin. Planta. 2009;230:871–81.
Aza-González C, Herrera-Isidrón L, Núñez-Palenius HG, Martínez De La Vega O, Ochoa-Alejo N. Anthocyanin accumulation and expression analysis of biosynthesis-related genes during chili pepper fruit development. Biologia Plantarum. 2013;57:49–55.
Rahim MA, Busatto N, Trainotti L. Regulation of anthocyanin biosynthesis in peach fruits. Planta. 2014;240(5):913–29.
Zheng T, Tan W, Yang H, Zhang L, Li T, Liu B, Zhang D, Lin H. Regulation of anthocyanin accumulation via MYB75/HAT1/TPL-mediated transcriptional repression. PLoS Genet. 2019;15.
Zhuang H, Lou Q, Liu H, Han H, Wang Q, Tang T, Ma Y, Wang H. Differential regulation of anthocyanins in green and purple turnips revealed by combined de novo transcriptome and metabolome analysis. Int J Mol Sci. 2019;20:4387.
Ying H, Shi J, Zhang S, Pingcuo G, Wang S, Zhao F, Cui Y, Zeng X. Transcriptomic and metabolomic profiling provide novel insights into fruit development and flesh coloration in Prunus mira Koehne, a special wild peach species. BMC Plant Biol. 2019;19:463.
Qiao Z, Liu S, Zeng H, Li Y, Wang X, Chen Y, Wang X, Cai N. Exploring the molecular mechanism underlying the stable purple-red leaf phenotype in Lagerstroemiaindica cv Ebony embers. Int J Mol Sci. 2019;20:5636.
Tai D, Tian J, Zhang J, Song T, Yao YA. Malus crabapple chalcone synthase gene, Mc CHS, regulates red petal color and flavonoid biosynthesis. PLoS ONE. 2014;9:110570.
Dick CA, Buenrostro J, Butler T, Carlson ML, Kliebenstein DJ, Whittall JB. Arctic mustard flower color polymorphism controlled by petal-specific downregulation at the threshold of the anthocyanin biosynthetic pathway. PLoS ONE. 2011;6:18230.
Stich K, Eidenberger T, Wurst F, Forkmann G. Enzymatic conversion of dihydroflavonols to flavan-3, 4-diols using flower extracts of Dianthus caryophyllus L. (carnation). Planta. 1992;187(1):103–8.
Feyissa DN, Løvdal T, Olsen KM, Slimestad R, Lillo C. The endogenous GL3, but not EGL3, gene is necessary for anthocyanin accumulation as induced by nitrogen depletion in Arabidopsis rosette stage leaves. Planta. 2009;230(4):747–54.
Tian J, Han Z-Y, Zhang J, Hu YJ, Song T, Yao Y. The balance of expression of dihydroflavonol 4-reductase and flavonol synthase regulates flavonoid biosynthesis and red foliage coloration in crabapples. Sci Rep. 2015;5(1):12228.
Davies KM, Albert NW, Schwinn KE. From landing lights to mimicry. The molecular regulation of flower colouration and mechanisms for pigmentation patterning. Funct Plant Biol. 2012;39:619–38.
Chen K, Liu H, Lou Q, Liu Y. Ectopic expression of the grape hyacinth (Muscari armeniacum) R2R3-MYB transcription factor gene, MaAN2, induces Anthocyanin accumulation in tobacco. Front Plant Sci. 2017;8:965.
Ishihara H, Tohge T, Viehöver P, Fernie AR, Weisshaar B, Stracke R. Natural variation in flavonol accumulation in Arabidopsis is determined by the flavonol glucosyltransferase BGLU6. J Exp Bot. 2016;67:1505–17.
Jie G, Rui R, Yonglu W, Jianpeng J, Sagheer A. Comparative metabolomic analysis reveals distinct flavonoid biosynthesis regulation for leaf color development of Cymbidium sinense ‘Red Sun.’ Int J Mol Sci. 1869;2020:21.
Yonekura-Sakakibara K, Tohge T, Matsuda F, Nakabayashi R, Takayama H, Niida R, Watanabe-Takahashi A, Inoue E, Saito K. Comprehensive flavonol profiling and transcriptome co expression analysis leading to decoding gene-metabolite correlations in Arabidopsis. Plant Cell. 2008;20:2160–76.
Griesser M, Hoffmann T, Bellido ML, Rosati C, Fink B, Kurtzer R, Aharoni A, Muñoz-Blanco J, Schwab W. Redirection of flavonoid biosynthesis through the down-regulation of an anthocyanidin glucosyltransferase in ripening strawberry fruit. Plant Physiol. 2008;146:1528–39.
Gomez C, Terrier N, Torregrosa L, Vialet S, Fournier-Level A, Verriès C, Souquet JM, Mazauric JP, Klein M, Cheynier V, Ageorges A. Grapevine MATE-type proteins act as vacuolar H+-dependent acylated anthocyanin transporters. Plant Physiol. 2009;150:402–15.
Chen L, Liu Y, Liu H, Kang L, Geng J, Gai Y, Ding Y, Sun H, Li Y. Identification and expression analysis of MATE genes involved in flavonoid transport in blueberry plants. PLoS ONE. 2015;10:e0118578.
Chai YR, Lei B, Huang HL, Li JN, Yin JM, Tang ZL, Wang R, Chen L. TRANSPARENT TESTA 12 genes from Brassica napus and parental species: cloning, evolution, and differential involvement in yellow seed trait. Mol Genet Genomics. 2009;281:109–23.
Dossa K, Mmadi MA, Zhou R, Zhou Q, Yang M, Cisse N, Diouf D, Wang L, Zhang X. The contrasting response to drought and waterlogging is underpinned by divergent DNA methylation programs associated with transcript accumulation in sesame. Plant Sci. 2018;277:207–17.
Dossa K, Mmadi MA, Zhou R, Zhang T, Su R, Zhang Y, Wang L, You J, Zhang X. Depicting the core transcriptome modulating multiple abiotic stresses responses in sesame (Sesamum indicum L.). Int J Mol Sci. 2019;20:3930.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real time quantitative PCR and the 2(-Delta Delta C (T)) method. Methods. 2001;25:402–8.
Acknowledgements
We thank Zhongjie Cai, Lianwei Qu, Rongxiao Liu, and Xiaofei Zhang from Liaoning Academy of Agricultural Sciences, Shenyang (110161), China, for their assistance in the experiments.
Funding
This work was supported by the Province Key R & D Program of Liaoning (2020JH2/10200016) and the National Key R & D Program of China (2019YFD1001001). The Funder has no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Conceptualization, J.L. and H.S.; methodology, Q.Z.; software, C.J.; validation, J.L. and Q.Z.; formal analysis, J.L., Q.Z. and L.L.; investigation, X.W.; resources, L.L.; writing—original draft preparation, J.L.; writing—review and editing, J.L. and H.S. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1
. (a) Multi-peak detection plot of metabolites in the multiple reaction monitoring mode; (b) Total ions current overlaps of the quality control samples by mass spectrometry detection; (c) Total ions current of one quality control sample by mass spectrometry.
Additional file 2: Figure S2
. Differential gene volcano figure.
Additional file 3: Figure S3
. qRT-PCR validation of the selected differentially expressed genes.
Additional file 4: Table S1
. Types and contents of flavonoid compounds in H and S. Table S2: Summary of the sequencing. Table S3: List of novel genes identified in rose petals. Table S4: List of the uniquely expressed genes in rose petals. Table S5: A total of genes were assigned to the three pathways. Table S6: List of the differentially expressed MATE genes. Table S7: Primer sequences of genes used for qRT-PCR verification.
Additional file 5: Table S8-1
. Gene-metabolite pairs involved in Ko00941. S8-2: Gene-metabolite pairs involved in Ko00942. S8-3: Gene-metabolite pairs involved in Ko00943. S8-4: Gene-metabolite pairs involved in Ko00944.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
Cite this article
Lu, J., Zhang, Q., Lang, L. et al. Integrated metabolome and transcriptome analysis of the anthocyanin biosynthetic pathway in relation to color mutation in miniature roses. BMC Plant Biol 21, 257 (2021). https://doi.org/10.1186/s12870-021-03063-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-021-03063-w