
Abstract
Background
Red clover (Trifolium pratense L.) is a diploid perennial temperate legume with 14 chromosomes (2n = 14) native to Europe and West Asia, with high nutritional and economic value. It is a very important forage grass and is widely grown in marine climates, such as the United States and Sweden. Genetic research and molecular breeding are limited by the lack of high-quality reference genomes. In this study, we used Illumina, PacBio HiFi, and Hi-C to obtain a high-quality chromosome-scale red clover genome and used genome annotation results to analyze evolutionary relationships among related species.
Results
The red clover genome obtained by PacBio HiFi assembly sequencing was 423 M. The assembly quality was the highest among legume genome assemblies published to date. The contig N50 was 13 Mb, scaffold N50 was 55 Mb, and BUSCO completeness was 97.9%, accounting for 92.8% of the predicted genome. Genome annotation revealed 44,588 gene models with high confidence and 52.81% repetitive elements in red clover genome. Based on a comparison of genome annotation results, red clover was closely related to Trifolium medium and distantly related to Glycine max, Vigna radiata, Medicago truncatula, and Cicer arietinum among legumes. Analyses of gene family expansions and contractions and forward gene selection revealed gene families and genes related to environmental stress resistance and energy metabolism.
Conclusions
We report a high-quality de novo genome assembly for the red clover at the chromosome level, with a substantial improvement in assembly quality over those of previously published red clover genomes. These annotated gene models can provide an important resource for molecular genetic breeding and legume evolution studies. Furthermore, we analyzed the evolutionary relationships among red clover and closely related species, providing a basis for evolutionary studies of clover leaf and legumes, genomics analyses of forage grass, the improvement of agronomic traits.
Similar content being viewed by others


Background
Red clover (Trifolium pratense L.) (Fig. 1a) is a temperate legume (Leguminosae) native to Europe and West Asia. It is not only inexpensive and rich in nutritional value but also contains a crude protein content of about 20%, with dry matter digestibility of about 70%, making it highly palatable and easily eaten by animals [1, 2]. Like alfalfa, red clover is self-incompatible and can be crossed with other subspecies [3, 4]. Its rich genetic diversity enables the production of many excellent agronomic traits. Red clover not only serves as a high-quality livestock feed but also has environmental and medicinal value [5,6,7,8]. As an important feed for dairy and beef cattle, red clover has important implications for the dairy and beef industries [9,10,11]. Although the world is affected by the new crown rot epidemic, the number of beef and dairy cattle is increasing, with corresponding increases in demand for high-quality forage [4, 12]. Red clover also has implications for environmental protection as well as medicinal value. It can co-produce nitrogen with Rhizobium, reducing the need for nitrogen fertilizers [13, 14]. Its developed root system promotes the reproduction of soil microorganisms and the growth of surrounding trees [7, 15]. Red clover contains high amounts of anthocyanins and is a potential candidate for reducing inflammation and oxidative stress [16]. Despite these advantages of red clover, its disadvantages include flatulence in ruminants, cold tolerance, and root sensitivity to soil pH. Accordingly, the development of improved varieties by plant breeding is an important goal.

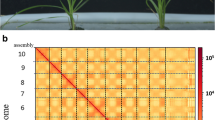
Plant morphology and Hi-C-assisted genome assembly of red clover. a Phenotype of the sequenced red clover plant. b Hi-C interaction heatmap showing 100-kb resolution super scaffolds
As a non-model legume, the structure of the red clover genome and important genetic information have not been fully determined, substantially limiting breeding and crop improvement. Relative to closely related species (e.g., white clover, alfalfa, and soybean) [17,18,19], progress in genomic research on red clover has been slow. The genome structure was initially explored by fluorescence in situ hybridization [20, 21]. Subsequently, simple repeat sequence (SSR) and single nucleotide polymorphism (SNP) markers were used to construct a red clover linkage genetic map [22]; however, few genes were mined, and the impact on variety improvement was limited. Subsequently, the red clover genome was constructed using the Illumina HiSeq 2000 platform; however, the final assembly results were insufficient for data mining in terms of continuity and completeness [23]. Therefore, it is necessary to construct a high-quality red clover genome to accelerate genetic and genomic research on red clover and future breeding.
In this study, we obtained a high-quality chromosome-level red clover genome by using Illumina, PacBio HiFi, and Hi-C data. The assembly contained 96 scaffolds (~ 423 Mb), with N50 = 55 Mb, accounting for 92.8% of the estimated genome size (456 Mb). Compared with that of the previously reported red clover genome assembly [23], the quality was greatly improved. From the genome, we annotated 44,588 genes. Furthermore, we evaluated gene family expansion and contraction and positive selection and performed phylogenetic analyses. In addition, analyses of the collinear relationship between the chromosomes of red clover and closely related species supported the accuracy and sensitivity of the data for studies of genomic evolution and for predictions of genomic patterns [24, 25]. Complete genomic information for the species and closely related species provides a basis for studying the differentiation and evolution of the species [26, 27]. This study provides a new starting point for evolutionary genomics research and a new research direction for analyzing the evolutionary relationships between red clover and related species.
Results
Genome-survey, sequencing, and assembly. The size, duplication rate, heterozygosity, and other parameters of the red clover genome were assessed by Jellyfish [28]. After quality control, Illumina sequencing yielded 26 Gb of data. We randomly selected 10,000 clean reads for comparison with the NT, and the rate of successful alignment against plant genomes (Medicago truncatula, Trifolium repens, Cicer arietinum and Trifolium meduseum) was 98.63%. A K-mer analysis further predicted a genome size of 456 Mb, with heterozygosity accounting for 1.57% and repeat sequences accounting for 54% of the genome.
We used traditional next-generation sequencing (NGS) data assembly methods to predict the genome size and third-generation HiFi sequencing (TGS) developed by PacBio for assembly of the red clover genome. In addition to making up for shortcomings of NGS in assembly applications, TGS does not require PCR amplification, produces ultra-long reads, GC preference and genome assembly by directly using high quality HiFi reads for splicing, which simplifies the process of sequencing and assembly, and improves the accuracy of assembly results [29,30,31]. High-quality HiFi reads were obtained after correcting subreads for ccs processing. In total, 1,764,862 HiFi reads was generated, and the N50 length was 18 kbp.
Contigs were then generated according to the phased string graph [32]. The assembled genome (423 Mb) contained 194 contigs, with an N50 size of 13 Mbp, and the largest contig was 34 Mbp. The average GC content of the assembled genome was 33.6%, which was slightly lower than those of the previously reported genomes of White lupin (33.7%) [33] and Pisum sativum (37.6%) [34]. The Illumina reads were then aligned to the assembled contigs to assess the integrity and quality of the assembly. The concordant paired mapped alignment rate was 91.47%. Furthermore, the single-copy homologous gene pool used to assess genetic spatial integrity showed a BUSCO [35] of 97.9% for the assembled genome, highlighting its good completeness.
Scaffold construction and curation
Hi-C is a high-throughput chromosome conformation capture technology. Taking the entire nucleus as the research object, this approach fixes and captures the interacting parts of the chromosome, followed by high-throughput sequencing to evaluate the spatial distribution of chromatin DNA throughout the genome and to obtain high-resolution chromosomal regulatory elements from positional relationships [36, 37]. In this study, we used 50 Gb of Hi-C data (100× coverage) to generate chromosome-level super-scaffolds. Subsequent analysis of the Hi-C library results showed that the genome was 423 Mbp with a scaffold N50 of 55 Mb. Compared with those of previous red clover sequence data (scaffold N50 = 223 kb), the quality and integrity were substantially better. After Hi-C-assisted assembly, 412 Mbp of genome sequence was mapped to seven chromosomes, accounting for 97.32% of the total sequence. Inter- and intra-chromosomal linkages were calculated to further verify the accuracy of the assembly. The linkages within chromosomes were much stronger than those between chromosomes, as revealed by the Hi-C heatmap. Furthermore, interactions were stronger for chromosomes at closer physical locations than at more distant physical locations (Fig. 1b). These results demonstrate the high accuracy of the assembled genome. Table 1 summarizes the assembly information.
Genome annotation
Gene functions in the genome are inferred by computational homology-based alignments and the prediction of repeat sequences. In this study, we identified miniature inverted-repeat transposable elements (MITEs) and long terminal repeat (LTR) transposable elements. Structural features were used for prediction, and these elements accounted for 52.81 and 30.01% of the total sequences, respectively. LTR-retrotransposons classified as Copia and Gypsy accounted for 11.26 and 7.70%, respectively, and 7605 simple repeats were found in the assembled genome. In addition, 16,227 ncRNAs were found and included 13 types.
Similarly, 44,588 high-confidence gene models and 46,089 transcripts were obtained using RNA-seq and de novo prediction strategies after removing the gene models containing early stop codons and frameshifts [38]. The gene models were unevenly distributed across the seven chromosomes.
The average lengths of genes and transcripts were 3620 bp and 1730 bp, respectively. Each gene contained an average of 1 transcript, and each transcript contained an average of 5 exons. The average lengths of CDS, exons, and introns were 1585 bp, 348 bp, and 495 bp, respectively. In addition, we compared the genome with those of five closely related species, including Medicago truncatula (MtrunA17r5.0-ANR from NCBI), Trifolium medium (ASM349008v1 from NCBI), Vigna radiate (ver6 from NCBI), Cicer arietinum (ASM33114v1 from NCBI), and Glycine max (v4.0 from NCBI). Among these, soybean had the largest genome (1012 Mb) [39], which was 2.4 times larger than the red clover genome. The T. meduium assembly had the largest number of genes (119102), C. arietinum the fewest (28772), and the other three closely related species had similar gene counts and average CDS lengths (Table 2). Using the NR, SwissProt, KEGG, GO, and eggNOG databases, 44,018, 30,158, 13,365, 26,349, and 38,110 genes were annotated, and gene functions and counts were obtained. A dataset of 10,805 common genes was visualized by a Venn diagram (Fig. 2, Table S1).

Venn analysis of five major databases (NR, Swiss-Prot, eggNOG, GO, KEGG) containing gene function annotation information
Gene family and evolution analysis
Comparing the genome of red clover with those of six representative species, a gene family analysis revealed that 44,588 genes clustered in 29,508 gene families. Among species, T. medium had the most gene families (5230). As visualized by a Venn diagram, 6602 gene families were shared by all species (Fig. 3a). Furthermore, over 11.8 million years of differentiation between red clover and T. medium, 619 gene families in red clover expanded and 3 gene families contracted (Fig. 3b). A GO enrichment analysis showed that the expanded gene families were enriched in terms such as ADP binding, oxidoreductase activity and defense response (Table S2). A GO enrichment analysis of genes with the signature of positive selection, as detected by the branch-site test, was also performed [40]. Most of the genes in red clover under positive selection were related to biosynthetic reactions (Table S3). We speculate that the expanded gene families and genes under positive selection may be involved in stress resistance and energy metabolism, which could help to increase the environmental adaptability of plants.

Gene family and phylogenetic tree analyses of red clover and other representative plant genomes. a Venn diagram of the number of shared gene families. b A phylogenetic tree based on shared single-copy gene families (left), gene family expansions and contractions among red clover and seven other species (middle), and Gene family clustering in red clover and seven other plant genomes (right). c Genome-wide replication Ks distribution map of red clover and its related species. d Genome-wide replication Ks analysis of red clover
A phylogenetic tree was constructed based on 754 single-copy homologous genes, with Arabidopsis (TAIR10.1 from NCBI) as the outgroup [41]. Red clover clustered with G. max, V. radiata, M. truncatula, T. medium, and C. arietinum to form a monophyletic group. Red clover was most closely related to T. medium, with an estimated divergence time of about 12 million years ago. The collinearity of several closely related species was then evaluated, revealing that red clover and M. truncatula show a certain degree of synteny (Fig. S1). The seven chromosomes of red clover and eight chromosomes of M. truncatula had good collinearity (Fig. 4), indicating high conservation after species divergence.

Features of the red clover and M.truncatula genome. a Length of each pseudochromosome (Mb). b Distribution of repetitive sequence. c Distribution of gene density. d Distribution of the GC content (e) Red clover and M.truncatula synteny analysis; the beginning of NC represents the chromosome of M.truncatula, while the beginning of CHR represents the chromosome of red clover
Whole genome duplication (WGD) events are important indicators of plant evolution and are considered to be the driving force behind plant adaptation to various environments [42]. Using differences in synonymous substitution rates to detect gene duplication and loss in the red clover genome. The results showed that the divergence of red clover occurred prior to the WGD event shared by M. truncatula and V. radiata. Red clover experienced a common WGD event with M. truncatula and V. radiata with KS values of 0.28 and 0.67, respectively (Fig. 3c). In addition, a WGD event in red clover also corresponded to a KS peak of 0.75 (Fig. 3d).
Discussion
Genetic and genomic data for leguminous plants with excellent agronomic traits are important resources for the study of genetics, breeding, and functional omics. In this study, we assembled a high-quality diploid genome for a widely important forage crop, the autodiploid red clover. A highly contiguous and complete reference genome is necessary for detailed population genetics analyses and experimental studies. The new genomic data contribute to the legume genome resource bank and provide a good reference for further research on other crops in the clover genus. By combining state-of-the-art technologies (including PacBio HiFi, Hi-C, and high-quality genome assemblers), we generated a representative red clover diploid genome. Compared with the previously reported red clover reference genome, the continuity and completeness of the newly assembled red clover genome showed a high quality: 13 Mbp contig N50, 55 Mb scaffold N50, and 97.9% BUSCO score. The assembly result was high-quality in comparison with legume genomes published to date. Notably, after combining high-throughput sequencing and Hi-C scaffolding, the total sequence length across all seven chromosomes was 412 Mbp, which was very similar to the size of the assembled genome (423 Mb). Genome annotation revealed 44,588 genes with high confidence models, providing an important resource for molecular breeding and evolutionary studies.
Red clover belongs to the legume family, and its evolutionary history is not well-understood. Our analyses of gene family expansion and contraction and positive selection revealed the molecular basis underlying survival under geographical environment and climate change as well as its excellent agronomic traits. The genetic information provides very important resources for future research on legume improvement and stress-resistant species. According to a phylogenetic analysis, Trifolium species arose after the differentiation of G. max, V. radiata, M. truncatula, and C. arietinum, followed by the divergence of red clover and T. medium. Red clover and Medicago truncatula showed good genomic collinearity and were closely related based on the phylogeny and sequence similarity. The genome of red clover will help to clarify the evolutionary processes shaping legume species.
Conclusion
We report a high-quality chromosomal-level red clover genome assembled using third-generation HiFi sequencing. The newly generated genome has the highest coverage and integrity among published legume genomes. This study provides an excellent genetic resource for molecular breeding and the improvement of legumes and provides an important basis for research on the evolution of legumes.
Experimental procedures
For genomic DNA sequencing, the leaves samples of well-grown from single red clover were collected by a professional graduate student and were collected into vacutainer tubes. Red clover (2n = 14) was grown in a light incubator at the Grassland Agri-husbandry Research Center. The study followed ethics norms and was in compliance with Chinese and international regulations.
DNA isolation and sequencing
The red clover Emerson was chosen as the test plant. Plants were grown in an incubator at the Qingdao Agricultural University in Shandong, China. After the leaves were treated with liquid nitrogen, DNA was extracted using the Tiangen DNA Secure Kit for Genome Sequencing (Beijing, China). Genome sequencing was performed by Berry Hekang (Beijing, China) using the third-generation PacBio Sequel II assembly sequencing platform. The extracted DNA was used for library preparation and paired-end (PE) sequencing of the library using Illumina NovaSeq [30, 43, 44]. Sequence reads containing adapters, repeats, and low-quality reads (quality scores < 20) were first filtered out. Then, 10,000 reads were randomly selected for NT comparison using the BLAST tool [45]. No apparent external contamination was detected. A K-mer analysis was used to estimate the genome size, duplication rate, and heterozygosity. 17-mer frequency distribution analysis of quality-filtered reads by Jellyfish [28]. The genome size of red clover was estimated using the following formula: G = Knum/Kdepth, where Knum is the number of k-mers, Kdepth is the expected depth of k-mers.
The quality of genomic DNA was checked using a NanoDrop 2000 spectrophotometer. The purified genome was used to construct the SMRTbell library and sequenced using PacBio SMRT technology [46, 47]. The library size was determined using an Agilent 2100 Bioanalyzer (Santa Clara, CA, USA). The acquired data were filtered and then processed using smrtlink for CCS processing.
Genome assembly and quality evaluation
HiFi reads were assembled using hifiasm, and purge-dups was used to remove hybridized fragments in the contig sequence [48, 49]. A single-copy orthologous gene library using a combination of tblastn, augustus, and hummer was finally used to assess the integrity of the assembled genome [35].
Hi-C data analysis and chromosome construction
The leaf tissue (100 mg) of red clover which was the same individual used in HiFi sequencing. It was soaked in paraformaldehyde, a cell cross-linking agent, for 15 minutes. Glycine was then added to the mixture to stop the chromatin cross-linking reaction, and the treated tissue was collected and frozen in liquid nitrogen. These tissues were then ground into a powder to extract DNA. Biotin-labeled oligonucleotide ends were added during end-repair, and the extracted DNA was subsequently fragmented into 350 bp fragments using a Covaris breaker [36, 50]. The biotin-conjugated DNA was captured and purified using avidin magnetic beads, and the library was constructed and sequenced using the Illumina PE150 platform [37, 51]. The raw reads were filtered, and 10,000 randomly selected sequencing reads were aligned with the NT library using the BLAST tool. The Hi-C data and draft genome were then compared using JUICER [52]. The Hi-C library results were subsequently analyzed using 3D-DNA alignments to obtain valid Hi-C data and generate the chromosome-level scaffold of the red clover genome [53].
Functional annotation
Repeat sequences were analyzed and predicted using RepeatMasker, MITE Hunter, LTRharvest, LTR Finder, LTR retriever, and RepeatModeler, and MITES and LTR transposable elements were identified using structure-based prediction methods [54,55,56,57,58,59]. The software parameters for LTRharvest and LTR Finder were -similar 90 -vic 10 -seed 20 -seqids yes -minlenltr 100 -maxlenltr 7000 -mintsd 4 -maxtsd 6 -motif TGCA -motifmis And -D 15000 -d 1000 -L 7000 -l 100 -p 20 -C -M 0.9 [60]. The parameters settings for RepeatModeler for the de novo identification of repeat sequences in masked genomes were -engine ncbi -pa 60. Similarly, the parameters for RepeatMasker for masking repeat sequences in the genome were -s -nolow -norna -gff -engine ncbi -parallel 20 [56]. tRNAscan-SE was used to predict tRNA ab initio rRNA [61]. The Rfam database was used to search for other types of ncRNAs [62, 63]. Specific information was obtained by a similarity analysis.
Except for tandem repeats, all repeat regions were soft-masked and subsequently used for the annotation of protein-coding genes [64]. The coding sequences of G. max, V. radiata, M. truncatula, T. medium, and C. arietinum were downloaded. These coding sequences were subjected to Blast (v. 2.2.20) searches against the red clover genome [45]. Homologs containing premature stop codons and frameshifts were discarded. Red clover RNA-seq data from different tissues were aligned to red clover contigs using GeMoMa-1.6.1 and a comprehensive transcriptome database was built using PASA (v. 2.0.1) [65, 66]. Open reading frames were predicted using PASA (v. 2.0.1) and the resulting database was used to train parameters for the following four de novo gene prediction software packages: AUGUSTUS (v. 3.2.2) [67], GeneMarker-ET (v. 4.57) [68], GlimmerHMM (v. 3.0.2) [69], and SNAP [70]. Predictions obtained using these packages were then combined using EVM [71, 72], and 44,588 genes were retrieved and functionally annotated by blast searches against databases, including NR, Swiss-Pro, eggNOG, GO, and KEGG [73,74,75]. A Venn diagram of the genes obtained from the five major databases was then generated to obtain more accurate annotation information.
Comparative analysis
A collinearity analysis of red clover and closely related species was performed using MCscan with parameters -g 1000 -c 90 -l 200 [76]. Notably, the OrthoMCL cluster analysis was used to identify seven gene protein families (Red clover, G. max, V. radiata, M. truncatula, T. medium, A. thaliana, and C. arietinum) [77]. First, an all-vs.-all BLAST alignment of the protein-coding sequences of all of the above species was generated, and the similarity between the sequences was calculated [78]. The Markov clustering algorithm (expansion coefficient of 1.5) was used for protein family identification [42, 79, 80]. Single-copy genes in each species were selected as reference markers. Four-fold degenerate sites were selected to construct supergenes [81]. Multiple sequence comparisons of supergenes were subsequently performed using Mafft [82, 83]. An appropriate base substitution model was selected and a species-based maximum likelihood (ML) phylogenetic tree was constructed using RAxML [84,85,86]. The mcmctree tool in the PAML software package was used to estimate the differentiation times based on 754 single-copy homologous genes [87, 88]. The single-copy homologous genes were identified by OrthoMCL, a correlated molecular clock model and a REV substitution model [89]. After a burn-in of 5,000,000 iterations, the MCMC process was repeated 1,000,000 times with a sample frequency of 50. The times were further calibrated using the predicted divergence times of M. truncatula-T. pratense (17.23–32.3 Mya), M. truncatula- V. radiata (44.0–74.5 Mya) and G. max-V. radiata (20.7–29.5 Mya) based on available TimeTree (http://timetree.org) [90]. Then, the variation in gene family sizes among species was analyzed using Café, followed by a GO functional enrichment analysis of the expanded gene families [91]. The branch-site model was used to detect positive selection on specific branches and affecting a portion of sites by selecting one-to-one orthologous proteins from red clover and its closely related species and using PRANK [40]. After aligning homologous protein sequences, Gblocks was used to filter the alignment results [92]. CODEML in PAML was used to evaluate positive selection in specific clades and affecting only certain loci with correction for multiple hypothesis testing using Chi2 [88]. The duplicate age distribution was used to detect WGD events, and blastp was used to align the longest protein sequences for genes in the red clover genome [93]. The results were then filtered using MCScanX and synonymous substitution rates were calculated using the Yn00 tool in the PAML software package [94]. A density distribution map based on the Ks values for all paralogous gene pairs and Ks values of ortholog gene pairs between the genomes of red clover, M. truncatula, and V. radiata were then drawn using Matlab [95].
Availability of data and materials
All data generated and analyzed during this current study are available in the Grassland Agri-husbandry Research Center, Qingdao Agricultural University with permission from the Competent Authority. All sequencing data were submitted in NCBI Database having BioProject ID PRJNA765108 and details of software used are in Table S4. Biological materials used in this study available from the corresponding author.
Abbreviations
- NT:
-
Nucleotide Sequence Database
- NGS:
-
Next-Generation Sequencing
- CCS:
-
Circular Consensus Sequencing
- BUSCO:
-
Benchmarking Universal Single-Copy Orthologs
- Hi-C:
-
High-through chromosome conformation capture
- MITEs:
-
miniature inverted repeat transposable elements
- LTR:
-
Long terminal repeat
- LTR-RT:
-
Long terminal repeat retrotransposons
- ncRNA:
-
Non-coding RNA
- NR:
-
NCBI nucleotide sequences
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- WGD:
-
Whole genome duplications
References
Niderkorn V, Martin C, Rochette Y, Julien S, Baumont R. Associative effects between orchardgrass and red clover silages on voluntary intake and digestion in sheep: Evidence of a synergy on digestible dry matter intake. J Anim Sci. 2015;93(10):4967–76.
Farghaly MM, Youssef IMI, Radwan MA, Hamdon HA. Effect of feeding Sesbania sesban and reed grass on growth performance, blood parameters, and meat quality of growing lambs. Trop Anim Health Pro. 2022;54(1):3.
Jones C, De Vega J, Lloyd D, Hegarty M, Ayling S, Powell W, et al. Population structure and genetic diversity in red clover (Trifolium pratense L.) germplasm. Sci Rep. 2020;10(1):8364.
Riday H, Krohn AL. Genetic map-based location of the red clover (Trifolium pratense L.) gametophytic self-incompatibility locus. TAG Theor Appl Genet. 2010;121(4):761–7.
Akbaribazm M, Khazaei F, Naseri L, Pazhouhi M, Zamanian M, Khazaei M. Pharmacological and therapeutic properties of the Red Clover (Trifolium pratense L.): an overview of the new findings. J Tradit Chin Med. 2021;41(4):642–9.
Harlow BE, Flythe MD, Kagan IA, Goodman JP, Klotz JL, Aiken GE. Isoflavone supplementation, via red clover hay, alters the rumen microbial community and promotes weight gain of steers grazing mixed grass pastures. PLoS One. 2020;15(3):e0229200.
Nazarova EA, Nazarov AV, Egorova DO, Anan'ina LN. Influence of destructive bacteria and red clover (trifolium pratense L.) on the pesticides degradation in the soil. Environ Geochem Health. 2022;44(2):399–408.
Wahdan SFM, Tanunchai B, Wu YT, Sansupa C, Schadler M, Dawoud TM, et al. Deciphering Trifolium pratense L. holobiont reveals a microbiome resilient to future climate changes. MicrobiologyOpen. 2021;10(4):e1217.
Moorby JM, Ellis NM, Davies DR. Assessment of dietary ratios of red clover and corn silages on milk production and milk quality in dairy cows. J Dairy Sci. 2016;99(10):7982–92.
Bertilsson J, Aerlind M, Eriksson T. The effects of high-sugar ryegrass/red clover silage diets on intake, production, digestibility, and N utilization in dairy cows, as measured in vivo and predicted by the NorFor model. J Dairy Sci. 2017;100(10):7990–8003.
Hart EH, Onime LA, Davies TE, Morphew RM, Kingston-Smith AH. The effects of PPO activity on the proteome of ingested red clover and implications for improving the nutrition of grazing cattle. J Proteome. 2016;141:67–76.
Greenwood PL. Review: An overview of beef production from pasture and feedlot globally, as demand for beef and the need for sustainable practices increase. Animal. 2021;15(Suppl 1):100295.
Stefan A, Van Cauwenberghe J, Rosu CM, Stedel C, Labrou NE, Flemetakis E, et al. Genetic diversity and structure of Rhizobium leguminosarum populations associated with clover plants are influenced by local environmental variables. Syst Appl Microbiol. 2018;41(3):251–9.
Janczarek M, Urbanik-Sypniewska T. Expression of the Rhizobium leguminosarum bv. trifolii pssA gene, involved in exopolysaccharide synthesis, is regulated by RosR, phosphate, and the carbon source. J Bacteriol. 2013;195(15):3412–23.
Duodu S, Carlsson G, Huss-Danell K, Svenning MM. Large genotypic variation but small variation in N2 fixation among rhizobia nodulating red clover in soils of northern Scandinavia. J Appl Microbiol. 2007;102(6):1625–35.
Lee SG, Brownmiller CR, Lee SO, Kang HW. Anti-Inflammatory and Antioxidant Effects of Anthocyanins of Trifolium pratense (Red Clover) in Lipopolysaccharide-Stimulated RAW-267.4 Macrophages. Nutrients. 2020;12(4):1089.
Griffiths AG, Moraga R, Tausen M, Gupta V, Bilton TP, Campbell MA, et al. Breaking Free: The Genomics of Allopolyploidy-Facilitated Niche Expansion in White Clover. Plant Cell. 2019;31(7):1466–87.
Chen H, Zeng Y, Yang Y, Huang L, Tang B, Zhang H, et al. Allele-aware chromosome-level genome assembly and efficient transgene-free genome editing for the autotetraploid cultivated alfalfa. Nat Commun. 2020;11(1):2494.
Li YH, Zhou G, Ma J, Jiang W, Jin LG, Zhang Z, et al. De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits. Nat Biotechnol. 2014;32(10):1045–52.
Sato S, Isobe S, Asamizu E, Ohmido N, Kataoka R, Nakamura Y, et al. Comprehensive structural analysis of the genome of red clover (Trifolium pratense L.). DNA Res Int J Rapid Publ Rep Genes Genomes. 2005;12(5):301–64.
Kataoka R, Hara M, Kato S, Isobe S, Sato S, Tabata S, et al. Integration of linkage and chromosome maps of red clover (Trifolium pratense L.). Cytogenet Genome Res. 2012;137(1):60–9.
Lopez-Maestre H, Brinza L, Marchet C, Kielbassa J, Bastien S, Boutigny M, et al. SNP calling from RNA-seq data without a reference genome: identification, quantification, differential analysis and impact on the protein sequence. Nucleic Acids Res. 2016;44(19):e148.
De Vega JJ, Ayling S, Hegarty M, Kudrna D, Goicoechea JL, Ergon A, et al. Red clover (Trifolium pratense L.) draft genome provides a platform for trait improvement. Sci Rep. 2015;5:17394.
Cui F, Taier G, Li M, Dai X, Hang N, Zhang X, et al. The genome of the warm-season turfgrass African bermudagrass (Cynodon transvaalensis). Hortic Res. 2021;8(1):93.
International Brachypodium I. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature. 2010;463(7282):763–8.
Gao L, Gonda I, Sun H, Ma Q, Bao K, Tieman DM, et al. The tomato pan-genome uncovers new genes and a rare allele regulating fruit flavor. Nat Genet. 2019;51(6):1044–51.
Hubner S, Bercovich N, Todesco M, Mandel JR, Odenheimer J, Ziegler E, et al. Sunflower pan-genome analysis shows that hybridization altered gene content and disease resistance. Nat Plants. 2019;5(1):54–62.
Marcais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27(6):764–70.
Nurk S, Walenz BP, Rhie A, Vollger MR, Logsdon GA, Grothe R, et al. HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome Res. 2020;30(9):1291–305.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60.
Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9(11):e112963.
Cheng H, Concepcion GT, Feng X, Zhang H, Li H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods. 2021;18(2):170–5.
Hufnagel B, Marques A, Soriano A, Marques L, Divol F, Doumas P, et al. High-quality genome sequence of white lupin provides insight into soil exploration and seed quality. Nat Commun. 2020;11(1):492.
Kreplak J, Madoui MA, Capal P, Novak P, Labadie K, Aubert G, et al. A reference genome for pea provides insight into legume genome evolution. Nat Genet. 2019;51(9):1411–22.
Seppey M, Manni M, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness. Methods Mol Biol. 2019;1962:227–45.
Dudchenko O, Batra SS, Omer AD, Nyquist SK, Hoeger M, Durand NC, et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science. 2017;356(6333):92–5.
Mascher M, Gundlach H, Himmelbach A, Beier S, Twardziok SO, Wicker T, et al. A chromosome conformation capture ordered sequence of the barley genome. Nature. 2017;544(7651):426.
Ter-Hovhannisyan V, Lomsadze A, Chernoff YO, Borodovsky M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008;18(12):1979–90.
Liu YC, Du HL, Li PC, Shen YT, Peng H, Liu SL, et al. Pan-Genome of Wild and Cultivated Soybeans. Cell. 2020;182(1):162.
Yang JZNRZ. Evaluation of an Improved Branch-Site Likelihood Method for Detecting Positive Selection at the Molecular Level. Mol Biol Evolu. 2005;12:2472–9.
Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290(5494):1151–5.
Berthelot C, Brunet F, Chalopin D, Juanchich A, Bernard M, Noel B, et al. The rainbow trout genome provides novel insights into evolution after whole-genome duplication in vertebrates. Nat Commun. 2014;5:3657.
Vurture GW, Sedlazeck FJ, Nattestad M, Underwood CJ, Fang H, Gurtowski J, et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics. 2017;33(14):2202–4.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–9.
McGinnis S, Madden TL. BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 2004;32(Web Server issue):W20–5.
Koren S, Rhie A, Walenz BP, Dilthey AT, Bickhart DM, Kingan SB, et al. De novo assembly of haplotype-resolved genomes with trio binning. Nat Biotechnol. 2018.
Quinlan AR. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr Protoc Bioinforma. 2014;47:11–2.11–34.
Roach MJ, Schmidt SA, Borneman AR. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinformatics. 2018;19(1):460.
Jiao Y, Peluso P, Shi J, Liang T, Stitzer MC, Wang B, et al. Improved maize reference genome with single-molecule technologies. Nature. 2017;546(7659):524–7.
Jarvis DE, Ho YS, Lightfoot DJ, Schmockel SM, Li B, Borm TJA, et al. Corrigendum: The genome of Chenopodium quinoa. Nature. 2017;545(7655):510.
Teh BT, Lim K, Yong CH, Ng CCY, Rao SR, Rajasegaran V, et al. The draft genome of tropical fruit durian (Durio zibethinus). Nat Genet. 2017;49(11):1633–41.
Durand NC, Shamim MS, Machol I, Rao SS, Huntley MH, Lander ES, et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 2016;3(1):95–8.
Bickhart DM, Rosen BD, Koren S, Sayre BL, Hastie AR, Chan S, et al. Single-molecule sequencing and chromatin conformation capture enable de novo reference assembly of the domestic goat genome. Nat Genet. 2017;49(4):643–50.
Gao L, McCarthy EM, Ganko EW, McDonald JF. Evolutionary history of Oryza sativa LTR retrotransposons: a preliminary survey of the rice genome sequences. BMC Genomics. 2004;5(1):18.
Price AL, Jones NC, Pevzner PA. De novo identification of repeat families in large genomes. Bioinformatics. 2005;21(Suppl 1):i351–8.
Tarailo-Graovac M, Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinforma. 2009:10 Chapter 4:Unit 4.
Ouyang S, Buell CR. The TIGR Plant Repeat Databases: a collective resource for the identification of repetitive sequences in plants. Nucleic Acids Res. 2004;32(Database issue):D360–3.
Ou SJ, Jiang N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 2018;176(2):1410–22.
Han Y, Wessler SR. MITE-Hunter: a program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res. 2010;38(22):e199.
Xu Z, Wang H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007;35(Web Server issue):W265–8.
Storz G. An expanding universe of noncoding RNAs. Science. 2002;296(5571):1260–3.
Nawrocki EP, Eddy SR. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013;29(22):2933–5.
Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR. Rfam: an RNA family database. Nucleic Acids Res. 2003;31(1):439–41.
Korf I. Gene finding in novel genomes. BMC bioinformatics. 2004;5:59.
Keilwagen J, Hartung F, Grau J. GeMoMa: Homology-Based Gene Prediction Utilizing Intron Position Conservation and RNA-seq Data. Methods Mol Biol. 2019;1962:161–77.
Avram O, Kigel A, Vaisman-Mentesh A, Kligsberg S, Rosenstein S, Dror Y, et al. PASA: Proteomic analysis of serum antibodies web server. PLoS Comput Biol. 2021;17(1):e1008607.
Stanke M, Steinkamp R, Waack S, Morgenstern B. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 2004;32(Web Server issue):W309–12.
Holland MM, Parson W. GeneMarker(R) HID: A reliable software tool for the analysis of forensic STR data. J Forensic Sci. 2011;56(1):29–35.
Majoros WH, Pertea M, Salzberg SL. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics. 2004;20(16):2878–9.
Puma JE, Young M, Foerster S, Keller K, Bruno P, Franck K, et al. The SNAP-Ed Evaluation Framework: Nationwide Uptake and Implications for Nutrition Education Practice, Policy, and Research. J Nutr Educ Behav. 2021;53(4):336–42.
Haas BJ, Salzberg SL, Wei Z, Pertea M. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008;9(1):R7.
Minematsu K. Evidence-based medicine (EVM) for thrombolytic therapy during the ultra-acute stage of cerebrovascular diseases and its current status in Japan. Nihon Naika Gakkai Zasshi J Jpn Soc Intern Med. 2004;93(9):1821–6.
Xie C, Mao X, Huang J, Ding Y, Wu J, Dong S, et al. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011;39(Web Server issue):W316–22.
Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, Apweiler R, et al. InterProScan: protein domains identifier. Nucleic Acids Res. 2005;33(Web Server issue):W116–20.
Syed A, Upton C. Java GUI for InterProScan (JIPS): a tool to help process multiple InterProScans and perform ortholog analysis. BMC Bioinformatics. 2006;7:462.
Tang H, Bowers JE, Wang X, Ming R, Alam M, Paterson AH. Synteny and collinearity in plant genomes. Science. 2008;320(5875):486–8.
Li L, Stoeckert CJ Jr, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13(9):2178–89.
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19(9):1639–45.
Jiao Y, Wickett NJ, Ayyampalayam S, Chanderbali AS, Landherr L, Ralph PE, et al. Ancestral polyploidy in seed plants and angiosperms. Nature. 2011;473(7345):97–100.
Chen B, Silvestri GA, Dahne J, Lee K, Carpenter MJ. The Cost-Effectiveness of Nicotine Replacement Therapy Sampling in Primary Care: a Markov Cohort Simulation Model. J Gen Intern Med. 2022.
Vanneste K, Van de Peer Y, Maere S. Inference of genome duplications from age distributions revisited. Mol Biol Evol. 2013;30(1):177–90.
Nakamura T, Yamada KD, Tomii K, Katoh K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics. 2018;34(14):2490–2.
Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30(14):3059–66.
Hohler D, Pfeiffer W, Ioannidis V, Stockinger H, Stamatakis A. RAxML Grove: an empirical phylogenetic tree database. Bioinformatics. 2022;38(6):1741–2.
Kozlov AM, Darriba D, Flouri T, Morel B, Stamatakis A. RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics. 2019;35(21):4453–5.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–3.
Yang C, Su XP, Liu DP, Guo ZW, Wang F, Lu YL. A New Method of Aquatic Animal Personality Analysis Based on Machine Learning (PAML): Taking Swimming Crab Portunus trituberculatus as an Example. Front Mar Sci. 2020;7.
Xu B, Yang ZH. pamlX: A Graphical User Interface for PAML. Mol Biol Evol. 2013;30(12):2723–4.
Fischer S, Brunk BP, Chen F, Gao X, Harb OS, Iodice JB, et al. Using OrthoMCL to assign proteins to OrthoMCL-DB groups or to cluster proteomes into new ortholog groups. Curr Protoc Bioinforma. 2011:11–9 Chapter 6:Unit 6 12.
Kumar S, Stecher G, Suleski M, Hedges SB. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol Biol Evol. 2017;34(7):1812–9.
Panda S. The CaFe project: Optical <monospace>Fe II</monospace> and near-infrared <monospace>Ca II</monospace> triplet emission in active galaxies: simulated EWs and the co-dependence of cloud size and metal content. Astron Astrophys. 2021;650.
Van der Straeten J, De Brouwer W, Kabongo E, Dresse MF, Fostier K, Schots R, et al. Validation of a PCR-Based Next-Generation Sequencing Approach for the Detection and Quantification of Minimal Residual Disease in Acute Lymphoblastic Leukemia and Multiple Myeloma Using gBlocks as Calibrators. J Mol Diagn. 2021;23(5):599–611.
Mahram A, Herbordt MC. NCBI BLASTP on high-performance reconfigurable computing systems. Acm T Reconfig Techn. 2015;7(4).
Wang YP, Li JP, Paterson AH. MCScanX-transposed: detecting transposed gene duplications based on multiple colinearity scans. Bioinformatics. 2013;29(11):1458–60.
Nemati M, Tabari MMR, Hosseini SA, Javadi S. A novel approach using hybrid fuzzy vertex method-MATLAB framework based on GMS model for quantifying predictive uncertainty associated with groundwater flow and transport models. Water Resour Manag. 2021;35(12):4189–215.
Acknowledgements
The author would like to thank Professor Guofeng Yang, Professor Zengyu Wang, and Professor Juan Sun (Professor of Grassland Science, Qingdao Agricultural University) for their help in data analysis and article writing. Thank you for the scientific research funding provided by the College of Grassland Science of Qingdao Agricultural University. Thanks for the experimental help provided by Beijing Berry and Kang.
Funding
This study was supported by the National Nature Science Foundation of China (U1906201), Shandong Forage Research System (SDAIT-23-01), China Agriculture Research System (CARS-34) and the First Class Grassland Science Discipline Program of Shandong Province China (1619002).
Author information
Authors and Affiliations
Contributions
ZY, ZW and GY conceived and designed this research. ZY analyzed data and wrote the manuscript. ZY, LS, YM, JW and JS executed the data analyses. JS participated in the discussion of the results. LM, YH, FM and XZ collected samples. GY contributed to the evaluation and discussion of the results and manuscript revisions. All authors have read and approved the final version.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Red clover is not endangered or a protected species in China, and it was purchased from BEST grass industry and planted in a light incubator. The seeds are collected by Professor Guofeng Yang in BEST grass industry. All the study procedures were carried out in accordance with relevant guidelines.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
GO, eggNOG, NR, KEGG and SP annotation results.
Additional file 2: Table S2.
The gene families described (including their GO terms) and their numbers between red clover and the gene family expansion in red clover.
Additional file 3: Table S3.
The genes in red clover under positive selection were described (including their GO terms) and their numbers.
Additional file 4: Table S4.
URLs and code of the software.
Additional file 5: Figure S1.
Red clover and its closely related species synteny analysis. (a) The beginning of NC represents the chromosome of M.truncatula, while the beginning of CHR represents the chromosome of red clover. (b) The beginning of NC represents the chromosome of C.arietinum, while the beginning of CHR represents the chromosome of red clover. (c) The beginning of NC represents the chromosome of G.max, while the beginning of CHR represents the chromosome of red clover. (d) The beginning of NC represents the chromosome of V.radiata, while the beginning of CHR represents the chromosome of red clover.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yan, Z., Sang, L., Ma, Y. et al. A de novo assembled high-quality chromosome-scale Trifolium pratense genome and fine-scale phylogenetic analysis. BMC Plant Biol 22, 332 (2022). https://doi.org/10.1186/s12870-022-03707-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-022-03707-5