
Abstract
Background
LTR Retrotransposons transpose through reverse transcription of an RNA intermediate and are ubiquitous components of all eukaryotic genomes thus far examined. Plant genomes, in particular, have been found to be comprised of a remarkably high number of LTR retrotransposons. There is a significant body of direct and indirect evidence that LTR retrotransposons have contributed to gene and genome evolution in plants.
Results
To explore the evolutionary history of long terminal repeat (LTR) retrotransposons and their impact on the genome of Oryza sativa, we have extended an earlier computer-based survey to include all identifiable full-length, fragmented and solo LTR elements in the rice genome database as of April 2002. A total of 1,219 retroelement sequences were identified, including 217 full-length elements, 822 fragmented elements, and 180 solo LTRs. In order to gain insight into the chromosomal distribution of LTR-retrotransposons in the rice genome, a detailed examination of LTR-retrotransposon sequences on Chromosome 10 was carried out. An average of 22.3 LTR-retrotransposons per Mb were detected in Chromosome 10.
Conclusions
Gypsy-like elements were found to be >4 × more abundant than copia-like elements. Eleven of the thirty-eight investigated LTR-retrotransposon families displayed significant subfamily structure. We estimate that at least 46.5% of LTR-retrotransposons in the rice genome are older than the age of the species (< 680,000 years). LTR-retrotransposons present in the rice genome range in age from those just recently inserted up to nearly 10 million years old. Approximately 20% of LTR retrotransposon sequences lie within putative genes. The distribution of elements across chromosome 10 is non-random with the highest density (48 elements per Mb) being present in the pericentric region.
Similar content being viewed by others


Background
LTR Retrotransposons transpose through reverse transcription of an RNA intermediate and are ubiquitous components of all eukaryotic genomes thus far examined [1]. Plant genomes, in particular, have been found to be comprised of a remarkably high number of LTR retrotransposons [2–4]. For example, more than half of the maize [5] and over 90% of the wheat [6] genomes are comprised of LTR retrotransposons. There is a significant body of direct and indirect evidence that LTR retrotransposons have contributed to gene and genome evolution in both animals and plants [4, 7–13].
Rice (Oryza sativa L.) is a staple food for over half of the world's population. Unlike most other cereal grasses, rice has a relatively small genome estimated at 430 Mb [14, 15]. Comparative genetic maps within the grass family indicate extensive regions of conserved gene content and orders [16]. The development of many important tools for genetic analysis, including excellent genetic and physical maps [17, 18], efficient genetic transformation techniques [19], an ever increasing dataset of expressed sequence tags (ESTs) [20], and a large collection of diverse germplasm [21] has established rice as an ideal model for the study of mechanisms underlying plant evolution. The recent release of draft genome sequences of two rice subspecies, indica [22] and japonica [23], and ongoing efforts to compile a complete japonica rice genome sequence publicly available by International Rice Genome Sequencing Project (IRGSP) [15], promises to greatly facilitate the study of rice genome evolution.
Most TEs were earlier discovered by chance or by limited assays using conserved regions of retrotransposons in rice [24–26]. For example, based on the screening of genomic libraries with a conserved retrotransposon probe, Hirochika and his colleagues [24] made an estimate of ~1000 retrotransposons totally in the rice genome, which fall into 32 families. By hybridizing a rice BAC library with a variety of RT probes, however, Wang et al. [27] estimated that the genome contains only about 100 copia-like elements in the entire haploid genome. Computer based sequence similarity searches of the nucleic acid databases are considered to be a more accurate method to identify retrotransposon families and estimate TE content and distribution in rice genome [28–31]. Using a new data-mining program (LTR retrotransposon structure program), we previously mined the GeneBank rice database (GBRD) as well as a more extensive Monsanto rice data set (MRD) (259 Mb) of LTR retrotransposons [32]. Including numerous LTR retrotransposon families that have not been previously reported, our comprehensive survey indicated that there are at least 59 distinct O. sativa LTR retrotransposon families comprising ≈17% of the rice genome. Although the majority of rice retrotransposon families have been identified and their phylogenetic relationships have been inferred based on the sequence similarity of the RT domains, the evolutionary dynamics, substructure, and relative integration time or age of rice retrotransposon elements remain poorly understood.
Our laboratory has long been interested in the evolution of LTR retrotransposons and their contribution to genome evolution. The current availability of a considerable part of the rice sequences at NCBI database is providing an excellent and timely opportunity to analyze the evolutionary history of rice LTR-retrotransposons. The present study was undertaken (i) to assess retrotransposon diversity and timing of insertion events by identifying all distinguishable rice LTR-retrotransposon sequences in the published sequence database, and(ii) to further infer, based on the establishment of substructure of all characterized families using the sequence divergence among the more rapidly evolving LTRs, possible retrotransposon evolution. In order to gain insight into the chromosomal distribution of LTR-retrotransposons in the rice genome, we also selected chromosome 10 to conduct a detailed examination of the intra-chromosomal distribution of LTR-retrotransposons. Finally, we have made a preliminary attempt to assess the potential contribution of LTR-retrotransposons to the evolution of gene structure and function in rice by identifying elements located in or near putative genes.
Results and discussion
Characterization of O. sativa LTR retrotransposons
LTR retrotransposons displaying ≥ 90% reverse transcriptase (RTs) pairwise identity at the amino acid level were, by earlier convention, assigned to the same family [33]. Using this criterion, McCarthy et al. [32] previously identified and characterized 55 families of LTR retrotransposons in the rice genome. Four additional groups of non-autonomous LTR retrotransposons identified in this previous study did not display a RT sequence homology but were, nevertheless, designated as families based on their distinct structures [32]. Only 38 retrotransposon families for which both LTRs have been identified [32] were included in this study.
In this study, we have extended the earlier survey of full-length LTR retrotransposons in the rice genome to include all identifiable fragments of LTR retrotransposon sequences. In accord with previously established criteria [34], we classified rice LTR retrotransposon sequences into three major groups: 1) full-length elements: a) autonomous full-length elements contain all of the characteristic features of LTR retrotransposons including putative gag, pol and, in some cases, env genes flanked by LTRs; b) non-autonomous full-length elements are a recently identified [32, 39] sub-class of full-length elements that have two LTRs flanking a series of repeating motifs of various lengths. Although numerous, relatively small and, in some cases, recently transposed, these non-autonomous elements encode none of the typical LTR retrotransposon ORFs and must acquire essential reverse transcription and intergration functions in trans; 2) solo LTRs are solitary LTR elements believed to be the products of recombination events between the flanking LTRs of full-length elements [1]; 3) fragmented elements are defined as partially deleted or truncated LTR retrotransposon sequence. This third category is a "catch all" grouping that includes all LTR retrotransposon sequences that are neither full-length elements or solo LTRs. For information purposes, we report in this paper both the total number of fragmented elements, as well as, the number of these that are fragmented solo LTRs.
We identified a total of 1219 LTR retrotransposon sequences in the portion of the rice genome surveyed in this study distributed over 38 families (Tables 1, see Additional file: 1; Table 2). Gypsy-like elements are • 4 × more numerous than copia-like elements (990 vs. 229). The number of element sequences within families was found to vary considerably, ranging from 1(Osr 19) to 255 (Osr 34) (Table 2). We identified 217 full-length elements representing 17.8 % of all LTR retrotransposon sequences identified. Osr 27 contains the largest number of full-length elements (61) in the genome, while 12 families (Osr 5–7, 14, 19–20, 22–23, 28, 42–44) have only 1 full-length element. The most abundant copia-like family was Osr 8 (59 sequences) while Osr 34 is the most abundant gypsy-like family (255 sequences).
Of the total LTR retrotransposon sequences identified, 822 or 67.4% were found to be fragmented or truncated elements. Fragmented elements were detected in 31 families varying in number from 1 (Osr 4, 20, 22, and 23) to 214 (Osr 34) per family. Seven families of LTR retrotransposons (Osr 2, 12, 16, 19, 24, 35, and 36) were found to contain no fragmented elements. We identified 179 solo LTRs representing 14.8 % of all LTR retrotransposon sequences identified. The solo LTRs are distributed over 21 families (Osr 1, 4, 8, 13–17, 25–27, 29–34, 37, and 40–41), and range in number from 1 (Osr 15 and Osr 16) to 39 (Osr 27) per family. The Osr 19 family consists of a single full-length element without any distinguishable fragmented elements or solo LTRs.
Other distinguishing characteristics of the LTR retrotransposon sequences identified in this study are presented in Table 1 (see Additional file: 1), including clone accession numbers, chromosomal location, sequence length, target site repeats (TSRs), LTR pairwise identities and estimated element age. TSRs result from a duplication of the unoccupied insertion site following element insertion [35]. The TSRs of all rice LTR retrotransposon are five bp long except for Osr 26.1 which has a TSR of seven bp. While members of O. sativa families share >90% RT sequence identity [32], sequence identity values among the more rapidly evolving LTRs are highly variable within families, ranging from 75.505% to 100%.
Phylogenetic substructure of O. sativa LTR retrotransposons
The slowly evolving RT encoding region of LTR retrotransposons is ideal for calculating evolutionary distances among even distantly related families of retroelements [6, 36]. However, sequence analyses of the more rapidly evolving LTRs are better suited for the characterization of phylogenetic substructure within families of LTR retrotransposons. We used LTR sequence divergence among elements within each family to identify sub-structure within the O. sativa LTR retrotransposon (Osr) phylogeny.
Significant sub-structure was detected in eleven families by neighbor-joining and parsimony criteria (Osr 4, 10, 12, 25, 26, 27, 30, 34, 35, 43, and 44).
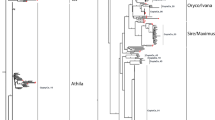
The Osr 26 family, for example, is comprised of 32 elements falling into at least 5 distinct clades (Fig. 1). The Osr 27 family, which is composed of 134 elements, displayed complicated substructure, consisting of at least 10 divergent clades with strong bootstrap support (Fig. 2). The remaining 27 families are closely related and displayed no significant intra-family substructure.

Phylogenetic trees of subfamily structure based on LTR nucleotide sequence data. The 32 elements of Osr 26 family fall into at least 5 clades, with Osr 25 as the outgroup. To better exhibit tree structure, all Osr 27 elements were removed. Insertions/deletions were ignored while performing phylogenetic analyses. Values on individual branches are bootstrap percentages using 1000 bootstrap repetitions. Each LTR in the tree is named by the genomic clone in which it was found. For elements with two LTRs, the 3' LTR is labeled by a lower case "b" while the 5' LTR is labeled by a lower case "a". Each tree is exhibited with a scale bar determined by the number of nucleotide substitutions per site between two sequences. The tight clustering seen in both families represents a high degree of nucleotide identity between elements within a subfamily.

Phylogenetic trees of subfamily structure based on LTR nucleotide sequence data. The Osr 27 family forms at least 10 divergent clades with strong bootstrap support, with Osr 41 as the outgroup. To better exhibit tree structure, all Osr 27 elements were removed. Insertions/deletions were ignored while performing phylogenetic analyses. Values on individual branches are bootstrap percentages using 1000 bootstrap repetitions. Each LTR in the tree is named by the genomic clone in which it was found. For elements with two LTRs, the 3' LTR is labeled by a lower case "b" while the 5' LTR is labeled by a lower case "a". Each tree is exhibited with a scale bar determined by the number of nucleotide substitutions per site between two sequences. The tight clustering seen in both families represents a high degree of nucleotide identity between elements within a subfamily.
Aging of the LTR-retrotransposons of O. sativa
LTR nucleotide identity can be used to estimate the time of LTR retrotransposon integration [37–39]. Of the 217 full-length elements that we have identified in O. sativa, 116 (53.5%) have >99% LTR similarity with 34 of these (15.7%) being identical. The remaining 101(46.5%)have relatively low levels of nucleotide divergence (<99%). Among them, 31(14%), 24(11%), 30(14%), and 15(7%) fell into different ranges of LTR similarities of 98–99%, 97–98%, 95–97%, and <95%, respectively. Identical LTRs imply that the elements have inserted recently and have not had time to accumulate mutations between LTRs. Using the average neutral substitution rate for grasses [37, 39, 40] of 0.065 substitutions per site per million years and the intra-element LTR divergence calculated here, we have computed the estimated integration time of the 181 elements with LTR nucleotide divergence (Table 1, see Additional file: 1). Based on the full-length elements available in this study, we estimate over half of rice retroelements (53.5%) with >99% LTR identity have inserted within the last 770,000 years while the older elements (46.5%) have integrated over a span of 770,000–7,700,000 years ago. Figure 3, 4 and 5 show estimated ages of all the full-length elements found in our study.

O. sativa copia-like LTR-retrotransposon age calculated using intraelement LTR nucleotide similarities. Only are those elements that contain LTR nucleotide divergence values other than zero included.

O. sativa gypsy-like LTR-retrotransposon age calculated using intraelements LTR Nucleotide Similarities (A). Only are those elements that contain LTR nucleotide divergence values other than zero included.

O. sativa gypsy-like LTR-retrotransposon age calculated using intraelements LTR Nucleotide Similarities (B). Only are those elements that contain LTR nucleotide divergence values other than zero included.
The LTRs of one gypsy-like (Osr 27.2) and one copia-like (Osr 13.8) element displayed atypically high levels of sequence divergence indicating that these elements are exceptionally old or possibly that these elements are, in fact, hybrid elements generated by homologous recombination or some other recombination process. Indeed, such inter-element recombination events have been previously documented in yeast [41, 42]. However, in this case, recombination is unlikely since the target site duplications of these (and indeed all full-length elements identified in this study) are identical. Thus, we have no direct evidence that any of the full-length rice LTR retrotransposons analyzed in this study were generated by recombination.
Distribution of Rice LTR-retrotransposons on chromosome 10
At the time of this study, about 48% rice genomic sequences were available in the public database, which included the almost entirely sequenced chromosome 1 and 10. In an initial effort to gain insight into the intra-chromosomal distribution of O. sativa LTR retrotransposons, we selected Chromosome 10 for a more detailed analysis. Tests were conducted to determine whether LTR retrotransposon sequences were randomly distributed on Chromosome 10. We found that the average density of elements on Chromosome 10 is 22.321/Mb (Table 3). There is a nonrandom clustering of both copia-like and gypsy-like LTR retrotransposons along the chromosome (Figure 6, 7 and 8).

Distribution of full-length, fragmented and solo LTR element sequences on the Chromosome 10 in the O. sativa genome. The figure shows the distribution of copia-like, gypsy-like and all elements on the chromosome 10. A genomic position for an element was estimated based on a PAC or BAC clone which contains the element. All elements were plotted to their respective chromosome location. Both copia-like and gypsy-like LTR retrotransposons exhibit nonrandom clustering along the chromosome. More LTR retrotransposons reside in the pericentric regions of the chromosome than in the arms, and short arm of Chromosome 10 also displays a higher density of the elements than long arm. The number of gypsy-like elements are more than that of copia-like elements on chromosome 10.

The distribution of full-length, solo and fragmented LTR elements along the chromosome 10. The borders of the pericentromeric regions of chromosome 10 were assigned as being 5 cM from the center of the centromere on each arm. The remainders of the chromosome were designated as arms. Both copia-like and gypsy-like LTR retrotransposons exhibit nonrandom clustering along the chromosome. More LTR retrotransposons reside in the pericentric regions of the chromosome than in the arms, and short arm of Chromosome 10 also displays a higher density of the elements than long arm. The number of gypsy-like elements are more than that of copia-like elements on chromosome 10.

Insertion frequencies of full-length, solo and fragmented LTR elements along the chromosome 10. DNA density for chromosomal arms and pericentromeric regions was calculated from the total DNA of three contigs (~10.4 cM and ~62.8 cM in arms, ~10.5 cM in pericentromeric regions) on Chromosome 10 (see Methods). Both copia-like and gypsy-like LTR retrotransposons exhibit nonrandom clustering along the chromosome. More LTR retrotransposons reside in the pericentric regions of the chromosome than in the arms, and short arm of Chromosome 10 also displays a higher density of the elements than long arm. The number of gypsy-like elements are more than that of copia-like elements on chromosome 10.
There are • 3 × more LTR retrotransposon sequences in the pericentric regions of the chromosome than in the arms (48.069 vs. 16.019 elements per Mb DNA, p < 0.005). Moreover, the short arm of Chromosome 10 displays a higher density of the elements than long arm (58.163 vs. 11.183 elements per Mb DNA). Consistent with what was observed on the whole genome level, there are proportionally more gypsy-like than copia-like elements on chromosome 10 (17 vs. 5 elements per Mb DNA). Comparing total number of retrotransposons in rice genome (Table 2) and retrotransposons in Chromosome 10), we noticed that two families (Osr 12 and 33) have more elements in Chromosome 10 than in the whole sequenced genome. This is not unexpected because many sequences from Chromosome 10 were released after we stopped searching rice genomic sequences in GenBank as of January 1, 2002 (see methods).
O. sativa LTR retroelement sequences are associated with putative genes
In an initial effort to determine whether and how frequently LTR retrotransposon sequences are associated with putative genes in the rice genome, we examined elements from all 19 families of copia-like elements and 5 representative families gypsy-like elements present in O. sativa. Our results indicate that 111/510 or 22% of LTR retrotransposon sequences lie within putative or established rice genes over the region of the genome analyzed in this study (Table 4, Figure 9 and 10). Fragmented elements are more frequently associated with genes (16%), followed by solo LTRs (3%) and full-length elements (2%). While these numbers are likely to change somewhat as the rice genome is better annotated, these preliminary estimates indicate that the potential contribution of LTR retrotranspson sequences to the evolution of gene structure and function in rice may be significant.

Five cases of association between retroelements with O. sativa putative genes observed in this study. Red arrows indicate positions of LTR retroelements with the direction of transcription. Green bars represent NCBI database-predicted gene regions with their orientation of transcription. All the five associations (from A to E) are located in the following genomic clones: AP003103, AP003105, H0423H10, AC018727 and H0806H05. (A) An entire full-length Osr 43 retrotransposon is completely a part of a putative rice gene (B1085F09.5) of unknown function. (B) An Osr 13 solo LTR is entirely a part of a putative rice gene (P0489A05) of unknown function. (C) An Osr 15 fragmented LTR is completely a part of an unknown gene (H0423H10.8) and overlaps two introns (No. 1 and 2) and No. 2 extron of the gene. (D) An Osr 17 full-length retrotransposon overlaps two putative genes nearby (OSJNBa0056G17.33 and OSJNBa0056G17.22) of unknown function. (E) An Osr 34 fragmented LTR is associated with two putative genes nearby (H0806H05.13 and H0806H05.14) of unknown function. It constitutes extrons and introns of these two genes.

Four cases of association between retroelements with O. sativa putative genes observed in this study. Red arrows indicate positions of LTR retroelements with the direction of transcription. Green bars represent NCBI database-predicted gene regions with their orientation of transcription. All the four associations (from F to I) are located in the following genomic clones: AP003627, AC079685, AP003021 and AP003144. (F) An Osr 34 solo LTR is associated with two putative genes nearby (P0459B04.22 and P0459B04.23) of unknown function. (G) An Osr 8 fragmented LTR is partly associated with a putative LeoOPT1 (oligopeptide transport) gene (OSJNBb0012A20.5). (H) An Osr 13 solo LTR constitutes almost half of a putative gene (P0503E05.17) of unknown function; (I) An Osr 13 full-length retrotransposon is part of a putative rice gene (P0507H05.22) of unknown function.
Conclusions
We have previously determined that two classes (gypsy-like and copia-like) of full-length LTR retrotransposons comprise ~17% of the Oryza sativa genome[32]. In this study, we have extended the earlier survey to include all identifiable fragments of LTR retrotransposon sequences. We have classified rice LTR retrotransposon sequences into three groups: full-length elements, solo LTRs, and fragmented elements. We have identified a total of 1219 LTR retrotransposon sequences in the region of the O. sativa genome analyzed in this study distributed over 38 families. Gypsy- like elements are >4 × more abundant than copia-like elements. Eleven of the thirty-eight investigated LTR-retrotransposon families display significant subfamily structure. We estimate that at least 46% of LTR-retrotransposons in the Oryza sativa genome are older than the age of the species (<680,000 years). A detailed examination of chromosome 10 revealed that LTR retrotransposon sequences are not randomly distributed across this chromosome but are more dense in the pericentric region. We found that aproximately 20% of Oryza sativa LTR retrotransposon sequences lie within putative genes and thus may play a significant role in gene evolution.
Methods
Sequence identification and retrieval
Whole genome analysis
LTRs representing previously identified families of rice LTR retrotransposons [32] were used as queries in BLASTN searches against rice genomic sequences present in GenBank as of January 1, 2002 http://www.ncbi.nlm.nih.gov/. About ~17% of rice genomic sequences was represented in this release. Subsequent sequence similarity searches against 29,285,477 bp of the complete chromosome 10 sequences in the GeneBank were performed up to April 25, 2002. Only 38 retrotransposon families for which both LTRs have been identified [32] were included in our study. To be considered an LTR sequence in this study, a BLAST "hit" had to display • 60% sequence homology to the LTR query sequence in a pair-wise comparison test [43] and have a size • 40% of that of the LTR query sequence. Each LTR identified by these criteria was given the name of the Osr (O. sativa retrotransposon) to which it was most homologous.
Multiple sequence alignments and phylogenetic analyses
Using the clone coordinates from the BLAST searches, the rice LTR sequences were copied and placed into individual files. Alignments were created using ClustalW and edited with MacVector 7.0 http://www.gcg.com/. ClustalX 1.8 [44] was used to generate neighbor-joining (NJ) trees with bootstrap values and visualized with TreeView 1.5.3 [45].
Age determination of elements
Full-length elements were aged by comparing their 5' and 3' LTR sequences [37]. Kimura-2 parameter distances (K) between 5' and 3' LTRs of individual elements were calculated by MEGA-2 [46]. The average substitution rate (r) of 6.5 × 10-9 substitutions per synonymous site per year for grasses [40] was used to calibrate the ages of the rice LTR-retrotransposons. The time (T) since element insertion was estimated using the formula T = K /2r, where T = time of divergence. K = divergence, and r = substitution rate [47].
Location of LTR-retrotransposon sequences on chromosome 10
Rice LTR retrotransposons were used as queries in BLASTN searches against chromosome 10 sequences present in GenBank as of April 25, 2002. The distribution of LTR retrotransposon sequences on Chromosome 10 of Nipponbare was estimated based on the position of LTR retrotransposon sequences in PACs and BACs http://rgp.dna.affrc.go.jp. The term "insertion frequency" is defined here as the number of LTR sequences identified in a PAC or BAC clone divided by the amount of DNA in the clone. DNA density for chromosomal arms and pericentromeric regions was calculated from the total DNA of three contigs (~10.4 cM and ~62.8 cM in arms, ~10.5 cM in pericentromeric regions) on Chromosome 10. According to the estimates of Harushima et al. [17] and Cheng et al. [48], we assigned the borders of the pericentromeric regions of chromosome 10 as being 5 cM from the center of the centromere on each arm. The remainder of the chromosome was designated as arms.
Gene annotation
The O. sativa expressed sequence tag database http://www.ncbi.nih.gov/dbEST/index.html was BLASTed (in April, 2002) for homology to all (228) copia-like LTR sequences and 278 gypsy-like LTR sequences of 5 representative families. About ~19% of rice genomic sequences was represented in this release. TBLASTN searches (default parameters) of these LTR associated genes were then ran against the NCBI O. sativa genome database to search for homology to previously characterized genes. GeneFinder http://ftp.genome.washington.edu/cgi-bin/Genefinder was used to delineate the exon boundaries of the putative genes.
References
Berg DE, Howe MM: Mobile DNA. 1989, Washington, DC: American Society for Microbiology
Voytas DF, Boeke JD: Yeast retrotransposon revealed. Nature. 1992, 358: 717-10.1038/358717a0.
Suoniemi A, Tanskanen J, Pentikainen O, Johnson MS, Schulman AH: The core domain of retrotransposon integrase in Hordeum: predicted structure and evolution. Mol Biol Evol. 1998, 15: 1135-44.
Kumar A, Bennetzen JL: Plant retrotransposons. Annu Rev Genet. 1999, 33: 479-532. 10.1146/annurev.genet.33.1.479.
SanMiguel P, Tikhonov A, Jin YK, Motchoulskaia N, Zakharov D, Melake-Berhan A, Springer PS, Edwards KJ, Lee M, Avramova Z, Bennetzen JL: Nested retrotransposons in the intergenic regions of the maize genome. Science. 1996, 274: 765-768. 10.1126/science.274.5288.765.
Flavell RB: Repetitive DNA and chromosome evolution in plants. Philos Trans R Soc Lond B Biol Sci. 1986, 312: 227-242.
McDonald JF: Evolution and consequences of transposable elements. Curr Opin Genet Dev. 1993, 3: 855-864.
McDonald JF: Transposable elements: Possible catalysts of organismic evolution. Trends Ecol Evol. 1995, 10: 123-126. 10.1016/S0169-5347(00)89012-6.
McDonald JF: Transposable elements and genome evolution. 1995, Boston, MA: Kluewer Academic Press
Britten RJ: Mobile elements inserted in the distant past have taken on important functions. Gene. 1997, 205: 177-182. 10.1016/S0378-1119(97)00399-5.
Brosius J: RNAs from all categories generate retrosequences that may be exapted as novel genes or regulatory elements. Gene. 1999, 238: 115-134. 10.1016/S0378-1119(99)00227-9.
Kidwell MG, Lisch DR: Transposable elements and host genome evolution. Trends Ecol Evol. 2000, 15: 95-99. 10.1016/S0169-5347(99)01817-0.
Feschotte C, Jiang N, Wessler SR: Plant transposable elements: where genetics meets gemomics. Nature Reviews Genetics. 2002, 3: 329-341. 10.1038/nrg793.
Arumuganathan K, Earle ED: Nuclear DNA content of some important plant species. Plant Mol Biol Rep. 1991, 9: 208-218.
Sasaki T, Burr B: International rice genome sequencing project: The effort to completely sequence the rice genome. Curr Opin Plant Biol. 2000, 3: 138-141. 10.1016/S1369-5266(99)00047-3.
Gale MD, Devos KM: Comparative genetics in the grasses. Proc Natl Acad Sci. 1998, 95: 1971-1974. 10.1073/pnas.95.5.1971.
Harushima Y, Yano M, Shomura A, Sato M, Shimano T, Kuboki Y, Yamamoto T, Lin SY, Antonio BA, Parco A: A high-density rice genetic linkage map with 2275 markers using a single F2 population. Genetics. 1998, 148: 479-494.
Cheng ZK., Buell CR, Wing RA, Gu MH, Jiang JM: Toward a Cytological Characterization of the Rice Genome. Genome Res. 2001, 11: 2133-2141. 10.1101/gr.194601.
Hiei Y, Ohta T, Komari T, Kumashiru T: Efficient transformation of rice (Oryza sativa L.) mediated by Agrobacterium and sequence analysis of the boundaries of the T-DNA. Plant J. 1994, 6: 271-282. 10.1046/j.1365-313X.1994.6020271.x.
Yamamoto K, Sasaki T: Large-scale EST Sequencing in Rice. Plant Mol Biol. 1997, 35: 135-144. 10.1023/A:1005735322577.
Jackson MT: Conservation of rice genetic resources: the role of the International Rice Genebank at IRRI. Plant Mol Biol. 1997, 35: 61-67. 10.1023/A:1005709332130.
Yu J, Hu S, Wang J, Wong GK-S, Li S, Liu B, Deng Y, Dai L, Zhou Y, Zhang X, Cao M, Liu J, Sun J, Tang J, Chen Y, Huang X, Lin W, Ye C, Tong W, Cong L, Geng J, Han Y, Li L, Li W, Hu G, Huang X, Li W, Li J, Liu Z, Li L, Liu J, Qi Q, Liu J, Li L, Li T, Wang X, Lu H, Wu T, Zhu M, Ni P, Han H., Dong W, Ren X, Feng X, Cui P, Li X, Wang H, Xu X, Zhai W, Xu Z, Zhang J, He S, Zhang J, Xu J, Zhang K, Zheng X, Dong J, Zeng W, Tao L, Ye J, Tan J, Ren X, Chen X, He J, Liu D, Tian W, Tian C, Xia H, Bao Q, Li G, Gao H, Cao T, Wang J, Zhao W, Li P, Chen W, Wang X, Zhang Y, Hu J, Wang J, Liu S, Yang J, Zhang G, Xiong Y, Li Z, Mao L, Zhou C, Zhu Z, Chen R, Hao B, Zheng W, Chen S, Guo W, Li G, Liu S, Tao M, Wang J, Zhu L, Yuan L, Yang H: A Draft Sequence of the Rice Genome (Oryza sativa L. ssp. indica). Science. 2002, 296: 79-92. 10.1126/science.1068037.
Goff SA, Ricke D, Lan TH, Presting G, Wang R, Dunn M, Glazebrook J, Sessions A, Oeller P, Varma H: A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science. 2002, 296: 92-100. 10.1126/science.1068275.
Hirochika H, Fukuchi A, Kikuchi F: Retrotransposon families in rice. Mol Gen Genet. 1992, 233: 209-216.
Kumekawa N, Ohtsubo H, Horiuchi T, Ohtsubo E: Identification and characterisation of novel retrotransposons of the gypsy type in rice. Mol Gen Genet. 1999, 260: 593-602. 10.1007/s004380050933.
Motohashi R, Mochizuiki K, Ohtsubo H, Ohtsubo E: Structures and distribution of p-SINE1 members in rice genomes. Theor Appl Genet. 1997, 95: 359-68. 10.1007/s001220050571.
Wang SP, Liu N, Peng KM, Zhang QF: The distribution and copy number of copia-like retrotransposons in rice (Oryza sativa) and their implications in the organization and evolution of the rice genome. Proc Natl Acad Sci USA. 1999, 96: 6824-6828. 10.1073/pnas.96.12.6824.
Tarchini R, Biddle P, Wineland R, Tingey S, Rafalski A: The complete sequence of 340 kb of DNA around the rice Adh1-Adh2 region reveals interrupted colinearity with maize chromosome 4. Plant Cell. 2000, 12: 381-391. 10.1105/tpc.12.3.381.
Turcotte K, Srinivasan S, Bureau T: Survey of transposable elements from rice genomic sequences. Plant J. 2001, 25: 169-179. 10.1046/j.1365-313x.2001.00945.x.
Mao L, Wood TC, Yu Y, Budiman MA, Tomkins J, Woo S, Sasinowski M, Presting G, Frisch D, Goff S: Rice transposable elements: a survey of 73,000 sequence-tagged-connectors. Genome Res. 2000, 10: 982-990. 10.1101/gr.10.7.982.
Vicient CM, Schulman AH: Copia-Like retrotransposons in the rice genome:few and assorted. Genome Letters. 2002, 1: 35-47. 10.1166/gl.2002.002.
McCarthy EM, Liu JD, GAO LZ, McDonald JF: LTR retrotransposons of Oryza sativa. Genome Biol. 2002, 3: research0053.1-0053.11. 10.1186/gb-2002-3-10-research0053.
Bowen NJ, McDonald JF: Genomic analysis of Caenorhabditis elegans reveals ancient families of retroviral-like elements. Genome Res. 1999, 9: 924-935. 10.1101/gr.9.10.924.
Ganko EW, Fielman KT, McDonald JF: Evolutionary history of Cer elements and their impact on the C. elegans Genome. Genome Res. 2001, 11: 2066-2074. 10.1101/gr.196201.
Coffin JM, Hughes SH, Varmus HE: Retroviruses. 1997, New York: Cold Spring Harbor Laboratory Press
Xiong Y, Eickbush TH: Origin and evolution of retroelements based upon their reverse transcriptase sequences. EMBO J. 1990, 9: 3353-3362.
SanMiguel P, Gaut BS, Tikhonov A, Nakajima Y, Bennetzen JL: The paleontology of intergene retrotransposons of maize. Nature Genet. 1998, 20: 43-45. 10.1038/1695.
Bowen NJ, McDonald JF: Drosophila euchromatic LTR retrotransposons are much younger than the host species in which they reside. Genome Res. 2001, 11: 1527-1540. 10.1101/gr.164201.
Jiang N, Bao Z, Temnykh S, Cheng Z, Jiang J, Wing RA, McCouch SR, Wessler SR: Dasheng: a recently amplified nonautonomous long terminal repeat element that is a major component of pericentromeric regions in rice. Genetics. 2002, 161: 1293-1305.
Gaut BS, Morton BR, Mccaig BC, Clegg MT: Substitution rate comparisons between grasses and palms: synonymous rate differences at the nuclear gene Adh parallel rate differences at the plastid gene rbcL. Proc Natl Acad Sci USA. 1996, 93: 10274-10279. 10.1073/pnas.93.19.10274.
Jordan IK, McDonald JF: Evidence for the role of recombination in the regulatory evolution of Saccharomyces cerevisiae Ty elements. J Mol Evol. 1998, 47: 14-20.
Jordan IK, McDonald JF: Phylogenetic perspective reveals abundant Ty1/Ty2 hybrid elements in the Saccharomyces cerevisiae genome. Mol Biol Evol. 1999, 16: 419-422.
Tatusova TA, Madden TL: BLAST 2 Sequences, a new tool for comparing protein and nucleotide sequences. FEMS Microbiol Lett. 1999, 174: 247-250. 10.1016/S0378-1097(99)00149-4.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG: The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25: 4876-4882. 10.1093/nar/25.24.4876.
Page RD: TreeView: An application to display phylogenetic trees on personal computers. Comput Appl Biosci. 1996, 12: 357-358.
Kumar S, Tamura K, Jakobsen IB, Nei M: MEGA (Arizona State Univ., Tempe), Version 2.1. 2001
Li WH: Molecular Evolution. 1997, Sunderland, MA: Sinauer
Cheng ZK, Presting GG, Buell CR, Wing RA, Jiang JM: High resolution pachytene chromosome mapping of bacterial artificial chromosomes anchored by genetic markers reveals the centromere location and the distribution of genetic recombination along chromosome 10 of rice. Genetics. 2001, 157: 1749-1757.
Acknowledgements
L.G. is grateful to other members of the McDonald lab and Dr. Susan R. Wessler's group for their friendly help. This work was supported by NSF grant DBI-0077709.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contribution
L.G. carried out all data collection, completed sequence analyses, and drafted the manuscript. J.M. participated in the design, and coordination of the study and greatly contributed to the revision of the manuscript. E.M. provided all LTR sequences of rice retrotransposon families, and technically helped data collection in earlier stage of this study. E.G. provided technical assistance during sequence collection and data analyses, and gave valuable comments to improve the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Gao, L., McCarthy, E.M., Ganko, E.W. et al. Evolutionary history of Oryza sativa LTR retrotransposons: a preliminary survey of the rice genome sequences. BMC Genomics 5, 18 (2004). https://doi.org/10.1186/1471-2164-5-18
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-5-18