
Abstract
Background
The migration of cadmium (Cd) from contaminated soil to rice is a cause for concern. However, the molecular mechanism underlying the response of rice roots to various Cd stresses remains to be clarified from the viewpoint of the co-expression network at a system-wide scale.
Results
We employed a comparative RNAseq-based approach to identify early Cd-responsive differentially expressed genes (DEGs) in rice ‘Nipponbare’ seedling roots after 1 h of high-Cd treatment. A multiplicity of the identified 1772 DEGs were implicated in hormone signaling and transcriptional regulation, particularly NACs and WRKYs were all upregulated under Cd stress. All of the 6 Cd-upregulated ABC transporters were pleiotropic drug resistance proteins (PDRs), whereas all of the 6 ZRT/IRT-like proteins (ZIPs) were consistently downregulated by Cd treatment.
To further confirm our results of this early transcriptomic response to Cd exposure, we then conducted weighted gene co-expression network analysis (WGCNA) to re-analyze our RNAseq data in combination with other 11 previously published RNAseq datasets for rice roots exposed to diverse concentrations of Cd for extended treatment periods. This integrative approach identified 271 transcripts as universal Cd-regulated DEGs that are key components of the Cd treatment coupled co-expression module. A global view of the 164 transcripts with annotated functions in pathway networks revealed several Cd-upregulated key functional genes, including transporter ABCG36/OsPDR9, 12-oxo-phytodienoic acid reductases (OPRs) for JA synthesis, and ZIM domain proteins JAZs in JA signaling, as well as OsWRKY10, NAC, and ZFP transcription factors. More importantly, 104 of these, including ABCG36/OsPDR9, OsNAC3, as well as several orthologs in group metalloendoproteinase, plastocyanin-like domain containing proteins and pectin methylesterase inhibitor, may respond specifically to various Cd pressures, after subtracting the 60 general stress-responsive genes reported to be commonly upregulated following multiple stresses.
Conclusion
An integrative approach was implemented to identify DEGs and co-expression network modules in response to various Cd pressures, and 104 of the 164 annotatable universal Cd-responsive DEGs may specifically respond to various Cd pressures. These results provide insight into the universal molecular mechanisms beneath the Cd response in rice roots, and suggest many promising targets for improving the rice acclimation process against Cd toxicity.
Similar content being viewed by others

Background
Cadmium (Cd) is a nonessential detrimental element that poses potential chronic toxicity to living organisms [1,2,3]. Cd is absorbed by the roots from the soil and transported to the shoot, so understanding the molecular mechanisms of plant roots coping with challenges imposed by unfavorable Cd element is one of crucial steps for improving acclimation of plants against Cd toxicity [4].
Like other nonessential elements, Cd enters cells as hitchhiker through some transporters specific for nutrient elements [5]. For rice, natural resistance-associated macrophage protein 5 (OsNramp5) is the major transporter for Mn and Cd transport into the inner root [6, 7]. The iron-regulated transporter 1 (OsIRT1) and OsNramp1 are potentially involved in this process [8], but their contributions appear small compared to that of OsNramp5 [3]. Under the adverse condition imposed by heavy metals, plants have evolved correspondingly a multiplicity of highly integrated adjustments to reduce the toxicity of absorbed heavy metals [9, 10]. In plant roots, Arabidopsis heavy metal ATPase AtHMA2 and AtHMA4 [11], as well as the ATP-binding cassette (ABC) transporter AtPDR8 [12] and OsHMA2 [13] localize to the plasma membrane and transport Cd out of the cell [14]. Another ABC-type transporter AtABCC3 [15], AtHMA3 [16] and OsHMA3 [17,18,19] play important roles in Cd detoxification by sequestering Cd to root vacuoles [3, 14].
Besides the strategies of heavy metal sequestration and detoxification, various signaling pathways are stimulated by Cd stress, and plant transcription factors (TFs) play vital roles in the regulation of signal transduction and protection of cells from stresses [20].
Although microarray-based transcriptomic analysis did provide highly valuable information on the plant responses to Cd pressure over the past decade [21], the complex Cd defense system in plants was still unexplored. Currently, ever-increasing RNAseq-based comparative transcriptomic studies have been conducted in several plant species, such as radish [22], Cd/zinc/Pb co-hyperaccumulating Crassulaceae [23], fast growing Cd-resistant tree [24], and maize [25, 26]. Taking the staple model crop rice into account, a few transcriptomic studies focusing on genes involved in the response to Cd stress have been conducted with the aid of RNAseq approach [1, 20, 27, 28]. However, there is still lack of an integrative investigation on these dispersed transcriptomes of rice roots under various Cd pressures over diverse treatment periods.
In the current omics era, co-expression networks and gene regulatory networks are widely used to predict functional roles of individual genes at a system-wide scale [29]. WGCNA is such a fascinating genome-wide approach focusing on elucidating biological networks instead of individual genes [29,30,31].
This powerful approach has been widely used on a range of systems for elucidating various processes, including dissecting flower development in Arabidopsis [32, 33] and strawberry [30], characterizing wheat endosperm development [34] and soybean domestication [35], revealing biotic stress responses in Arabidopsis [36], citrus [37] and Medicago truncatula [38], elucidating the holistic picture of drought response in grapevine [39], Cd response in maize [26] and the hyperaccumulating ecotype of Sedum alfredii [23]. Interestingly, WGCNA was also utilized to confirm the genome-wide association study (GWAS) inferred candidate genes associated with NaCl tolerance in Arabidopsis [40].
Through integrating transcriptome and metabolite data, gene co-expression networks related to inositol phosphate in maize [41] and fruit anthocyanin content in apple [42, 43] were constructed by WGCNA. Recently, three types of omics data including 31,447 mRNAs, 13,175 proteins and 4267 phosphoproteins were grouped individually by WGCNA into co-expression modules, and then integrated into omics networks in a developmental atlas of maize [29]. In addition to these integrations of different levels of omics data of the same sample, expression datasets generated by different groups under different experimental conditions and biological systems can be integrated by WGCNA to aid the annotation of rice genome [44]. It was also implemented to elucidate the genetic basis of drought tolerance in rice [45, 46] and discover the core biotic stress responsive genes in Arabidopsis [47]. However, there is no such a comprehensive resource aiming to elucidate the scaffolding mechanisms for Cd signaling pathways in rice from the viewpoint of co-expression network at a system-wide scale.
In this study, we implemented a comparative RNAseq-based approach to identify Cd-responsive DEGs in rice seedling roots under 1 h of high-Cd stress. Among the 1772 DEGs responsive to short-term high-Cd, 536 are novel Cd-responsive genes. To further clarify our findings of the early transcriptomic response to Cd exposure, we utilized WGCNA to re-analyze our RNAseq data in combination with other 11 published RNAseq datasets with different Cd concentrations and different time points. A universal Cd-responsive DEGs subset of 164 annotatable genes was extracted from the co-expression network modules generated by WGCNA, and 104 of them were Cd-specific regulated genes which were not reported to be general stress-responsive previously. Our results provide insight into the high-confidence universal molecular mechanisms beneath the Cd response in rice roots, and the universal Cd-regulated DEGs that may contribute to plant responses to Cd stress will be valuable for further improving the Cd response in rice using the genetic engineering approach.
Results
Global transcriptomic changes of rice roots in response to short-term high-Cd stress
Transcriptomic changes in roots response to 1 h of high-Cd stress were determined by comparing the control (ck1h) and 100 μM Cd-treated (Cd1h) rice seedlings (Additional file 1: Table S1). After using the stringent criteria (fold change ≥ 2 and q_value ≤ 0.05) to select transcripts, a total of 1772 gene isoforms were identified as being early Cd-responsive (Fig. 1). Since few genes had more than one Cd-regulated transcript isoform, differentially expressed transcripts were regarded as differentially expressed genes (DEGs) (Table 1, Additional file 2: Table S2).

A flowchart showing the comprehensive RNAseq data analysis procedure
To validate the expression pattern of DEGs that resulted from RNAseq, 8 DEGs were randomly selected for qRT-PCR assay (Additional file 3: Table S3). As anticipated, the qRT-PCR results were basically consistent with those from RNAseq (Additional file 4: Fig. S1), suggesting that DEGs resulted from RNAseq were credible for further analysis.
For the 1334 Cd-induced and 438 Cd-repressed transcript isoforms in rice ‘Nipponbare’ roots, 643 and 84 of them were also reported to be up- and down-regulated by 1 h of 50 μM medium Cd treatment (MCd1), correspondingly [27], whereas 575 up- and 44 down-regulated isoforms were also DEGs with same regulated pattern in the microarray-based analysis of early transcriptomic responses to 25 μM Cd treatment (lowCd) for 3 h [21]. Taking multiple Cd stresses commonly regulated genes into account, 298 up- and 1 down-regulated DEGs also displayed the same regulatory pattern in the previous reports, both with MCd1 and lowCd treatments [21, 27] (Additional file 5: Figure S2, Additional file 6: Dataset 1).
Excluding the 1070 DEGs also identified in two previous reports, the remaining 702 DEGs (433 up- and 269 down-regulated) are uniquely identified in this study (Additional file 5: Figure S2). Although 166 have other isoforms differentially expressed in previous two studies, 536 DEGs regulated by short-term high-Cd treatment are novel Cd-responsive genes (Table 1, Additional file 2: Table S2).
Functional characterization of early Cd-responsive DEGs in rice roots
To gain insights into the functionality of the 1772 DEGs that are likely to be associated with the Cd response, all of these Cd-responsive transcripts were visualized in the candidate pathway networks with MapMan.
Globally, the overrepresented biological functional pathway genes among Cd-regulated DEGs were involved in stress and hormone-signaling transduction, redox balance, regulation of transcription, ion transport, etc. (Fig. 2, Additional file 2: Table S2), which is consistent with previous studies [1, 21, 27]. Furthermore, the regulation patterns of JA signaling nodes, ABA-dependent pathway components, transporters, and TFs were essentially the same as those reported in previous Cd-treated rice transcriptomic studies (Fig. 2, Additional file 2: Table S2); therefore, these DEGs can be regarded as members of universal Cd-responsive genes.

Global view of 1772 DEGs involved in diverse metabolic pathways in rice roots under Cd stress. Among the 1759 data points of 1772 DEGs, 603 data points were mapped on the metabolic pathways (Arabidopsis seed-Molecular Networks) using MapMan software. The colored boxes indicate the Log2 ratio of Cd1h/ck1h
Among the DEGs within the ‘Stress’ group, all transcripts involved in cold stress response and metal binding, and the majority of heat shock protein transcripts, were upregulated in response to Cd. Of the transcripts mapped to ‘Hormones’ category, 9 DEGs in Jasmonate signaling, 8 in ABA signal transduction, and 10 of 11 in Auxin signal transduction, as well as the majority of isoforms mapped to ethylene signaling, were upregulated by Cd treatment.
Additionally, 6 transcripts that encode antioxidant enzymes and thioredoxin, and the majority of TFs, including 7 members of the HSF family, 4 NACs, and 22 WRKYs, were all upregulated under Cd stress (Table 1, Fig. 2, Additional file 2: Table S2).
Based on the transport overview, 3 groups of mapped transporters (including 6 ABC transporters mainly in the PDR category, 6 amino acid transporters, and 3 ammonium transporters) were all upregulated quickly following Cd treatment. Ten of 11 heavy metal-associated domain containing proteins (HMADs) and 4 members of multidrug and toxic compound extrusion (MATE) efflux family were all rapidly upregulated in Cd-treated rice roots. However, all 6 zinc or iron-regulated transporters (ZRT/IRT-like proteins, ZIPs) including OsIRT1 and OsZIP4 were downregulated by Cd stress. Similarly, 5 other transporters, including 1 chloride transporter and 1 malic acid transport protein were also inhibited by short-term high-Cd stress (Table 1, Additional file 7: Figure S3).
With regard to the novel Cd-responsive DEGs, CYP94C2b and OsJAZ9 were involved in Jasmonate signaling, one ethylene-responsive transcription factor (ERF110) and other 4 DEGs were involved in ethylene signaling, and the Cd-inhibited gibberellin-20 oxidase was responsible for gibberellin synthesis. Seven transporters are also putative novel Cd-responsive DEGs: these include 1 HMAD, OsZIP9, and OsIRT1, 2 MATEs, and 1 inorganic phosphate transporter OsPT8. Three TFs, including OsHsfB2c, 1 NAC, and WRKY108, are also among the novel Cd-responsive DEGs (Table 1, Additional file 2: Table S2).
Five DEGs, including one 12-oxophytodienoate reductase (OsOPR5), ABCG36/OsPDR9, stress responsive NAC (SNAC1) and WRKY71 transcription factors genes, were also reported to be upregulated by medium and low Cd stresses in rice seedling roots in 3 previous reports [21, 27, 48]. In the latter 2 previous high-throughput transcriptome studies, 30 DEGs were upregulated whereas OsZIP4 was downregulated by Cd treatment. Interestingly, OsNAC4 was also consistently upregulated in microarray analyses with 2 lowest Cd treatments, both at the 3-h time point. Transcription factors OsbZIP23 and OsNAC10 have also been previously shown to be responsive to medium Cd stress in RNAseq analysis [27], but in different gene isoforms, and mainly as the first isoforms (Table 1, Additional file 2: Table S2).
Identification of DEGs and co-expression network modules in response to various Cd pressures
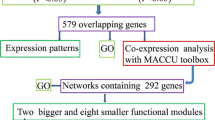
To investigate whether the early Cd-responsive genes identified in this study are involved in response to diverse concentrations of Cd treatments at different time points or not, 17 RNAseq samples covering those published and our RNAseq data of Cd-treated rice seedling roots (Additional file 1: Table S1) were processed by the new pipeline HISAT-StringTie-Ballgown. After filtering those transcripts with no more than 50% missing data, 22,080 transcripts across 17 RNAseq samples (Additional file 8: Dataset 2) were selected for analysis of DEGs and construction of gene co-expression network simultaneously.
For differential expression analysis, the stringent criteria (fold-change ≥ 2 and adj.P.Val ≤ 0.05) was used to select transcripts, thus 4 datasets containing DEGs [(1 h of Medium Cd treatment) MCd1 vs ck0, (1 d of Medium Cd) MCdD vs ck0, Cd24h vs ck24h and Cd1h vs ck1h)], representing various concentrations of Cd treatments across diverse time points, were obtained for further analysis (Additional file 9: Dataset 3). Meanwhile, the WGCNA package was employed to construct gene co-expression network of Cd response from the expression matrix of 22,080 transcripts across 17 RNAseq samples. This approach resulted in 22 distinct co-expressed modules (labeled by different colors), and the module ‘darkorange’ (with 1868 transcripts) is of particular interest (Fig. 3, Additional file 10: Table S4). This module has the highest level of module significance, and its module Eigengene expression profile suggests that it is correlated with Cd treatment, therefore, it is considered as the dominant Cd response-specific or Cd-coupled module.

Gene co-expression network analysis of 17 RNAseq datasets for rice roots exposed to diverse concentrations of Cd for extended treatment periods. a Barplot of module significance. b Eigengene expression profile for the Cd-coupled module in 17 RNAseq samples. The module significance is defined as the mean gene significance across all genes in the module. For the heatmap of the expression of eigengenes in the Cd-coupled module (rows) across 17 RNAseq samples (see Methods section), low values means a lot of module genes are under-expressed (green color in the heatmap) and high values for over-expressed (red in the heatmap) in this samples. The number of genes in the module is indicated in parenthesis
The comparison of transcriptomes across different time points and various dosages of Cd treatments might provide additional information about gene function, therefore the intersection of transcripts in Cd-coupled module and the 4 DEGs datasets was selected for functional analysis (Fig. 4).

A Venn diagram showing the intersections of transcripts in the Cd coupled module and DEGs in different concentrations of Cd treatments across diverse time points. Rice roots samples from diverse Cd treatments and their corresponding control were compared in couples to analyze DEGs using the limma software package
Functional characterization of universal Cd-responsive DEGs in co-expression network
After the comparison with the 4 DEGs datasets, 418 of the 1868 transcripts from Cd-coupled module were unveiled to be unrelated to Cd response, whereas 271 transcripts were universal Cd-responsive DEGs, constituting the central hub of Cd response-specific module subset (Fig. 4). The global function view showed that 164 of 271 DEGs from the Cd response-specific module across 17 samples can be annotated and located in MapMan pathway network (Fig. 5, Additional file 11: Table S5) and the remaining 107 unmapped were novel transcripts with “MSTRG” prefix (Additional file 10: Table S4) assigned by StringTie.

Global view of the 164 universal Cd-responsive DEGs involved in diverse metabolic pathways in rice roots under Cd stress. Among the 169 data points of 164 universal Cd-responsive DEGs, 51 data points were mapped on the metabolic pathways (Biotic stress) using MapMan software. The colored boxes indicate the Log2 ratio of Cd1h/ck1h
Taking the “Biotic stress” pathways as an example, all of the common upregulated transcripts were enriched in category ‘Hormone Signaling’, ‘proteolysis’, ‘Redox’, ‘Signaling’ and ‘transcription factors’. In detail, the expression of genes implicated in ‘Jasmonic acid’ and ‘Transcription factors’ (WRKYs and ERFs) were upregulated in Cd treated roots (Fig. 5). Among the 164 common data points, 6 of them involving in ethylene signaling, 8 in JA synthesis and signal transduction, 7 transcription factors, 7 zinc finger proteins (ZFPs), 7 glutathione-S-transferases (GSTs) and 4 transporters were all upregulated by Cd stress (Table 2).
Of particular interest is the identification of 1 key enzyme 9-cis-epoxycarotenoid dioxygenase (OsNCED4) for ABA synthesis, 2 key enzymes (OsOPR1 and OsOPR5) for JA synthesis, 6 transcription Factors including OsWRKY10 and SNAC1, 6 ZFPs, 3 GSTs and 3 transporters including ABCG36/OsPDR9, which were also uniformly upregulated by medium and low Cd stresses in rice seedling roots in at least 2 previous reports [21, 27, 48]. Additionally, one C2H2-type zinc finger protein (ZFP182) was significantly enhanced even at the lowest Cd concentration [48], and it was strongly induced from FPKM of 5 to 858 after the short-term high-Cd treatment (Table 2, Additional file 2: Table S2).
After subtracting the 60 general stress-responsive genes reported to be commonly upregulated following multiple stresses [27], 90 universal Cd-upregulated and 14 Cd-downregulated DEGs may respond specifically to various Cd pressures (Table 2; Additional file 11: Table S5). Regarding the 104 Cd-specific regulated genes, several DEGs could be categorized into orthologous groups of model plants (numbered as APK_ORTHOMCL). Among these DEGs, two metalloendoproteinases were in APK_ORTHOMCL1642, three plastocyanin-like domain containing proteins in APK_ORTHOMCL3089, while one pectin methylesterase inhibitor (Os12g18560, OsPMEI46) in APK_ORTHOMCL14694 (Table 2; Additional file 11: Table S5).
It is noteworthy that 14 of the 104 Cd-responsive members are also novel DEGs, only identified in this study through combing the two approaches. Among these 14 novel Cd-responsive DEGs, Os04g33390 (encoding prephenate dehydratase) is the ortholog of Arabidopsis arogenate dehydratases (Table 2, Additional file 2: Table S2, Additional file 11: Table S5).
Discussion
Early high-Cd responsive transporters in rice roots
To elucidate the rice early response to high-Cd stress, we performed pair-end RNAseq of rice roots with replicates treated with high Cd concentrations (100 μM) for 1 h, in parallel with control samples at the same time point. As expected, 702 of the 1772 DEGs were uniquely identified in our rice roots treated by short-term high-Cd treatment (Additional file 5: Figure S2, Additional file 2: Table S2), which differed from previous transcriptomic studies using short-term and moderate or low Cd treatment [21, 27].
Metal transporters play key roles in the acquisition, distribution, and homeostasis of Cd in plants [5]. Several Arabidopsis ABC transporters are suggested to mediate vacuolar sequestration of Cd-phytochelatin conjugates [15, 49]. In this study, all of the 6 Cd-upregulated ABCGs/PDRs were from one of the most highly represented subfamilies of ABC transporters (Table 1, Additional file 7: Figure S3).
In Arabidopsis, ABC transporter of the mitochondria AtATM3 [50] and AtABCC1–3 [15, 49] transported Cd into the vacuoles to increase Cd tolerance. AtABCC3 confers Cd tolerance by transporting phytochelatin (PC)-Cd complexes into the vacuoles to detoxify Cd [15]. In rice, the Cd-inducible OsABCG43 is likely to sequester Cd at the subcellular level [51], similar to vacuolar sequestration mediated by OsHMA3 [17].
Among the Cd-upregulated ABCG-type transporter genes, ABCG36/OsPDR9 is reported induced rapidly and markedly in rice roots by Cd and Zn [52]. Taking its homologs in Arabidopsis into consideration, AtPDR12 and AtPDR8 are all involved in heavy metal resistance, and the Cd-inducible AtPDR8 can export multiple functionally unrelated substrates, including auxinic compounds and Cd; thus, it confers heavy metal resistance as a Cd extrusion pump [12], strengthening the potential of employing ABCG36/OsPDR9 for genetic improvement of Cd tolerance in rice. Besides its mechanism as a pump to exclude lead, AtPDR12 functions as a plasma-membrane ABA-uptake transporter [53].
In addition to the upregulated PDR-type transporters, 4 members of the MATE efflux family were rapidly upregulated in 1-h Cd-treated rice roots (Table 1).
MATE proteins play a role in aluminum tolerance by mediating citric acid efflux from root cells to chelate Al [54]. Functional MATE homologs associated with Al tolerance were identified in Arabidopsis [55], Medicago truncatula [56], sorghum [57], barley [58, 59], wheat [60], maize [61], and rice (OsFRDL4, Os01g69010) [62].
In Arabidopsis, the MATE-related transporter DETOXIFICATION 1 (AtDTX1) has been described as an efflux transporter that detoxifies plant-derived or exogenous toxic compounds including Cd from the cytoplasm [63]. Interestingly, a recent study on mammalian MATEs has provided the first evidence that MATE transporters are involved in the cellular transport and detoxification of Cd [64]. Therefore, it would be worth investigating the role of the Cd-induced plant MATE transporters in combating Cd toxicity in planta.
Ten of 11 heavy metal-associated domain containing proteins (HMADs) were rapidly upregulated in Cd-treated rice roots (Table 1).
HMADs, such as AtHMAD1 [65] and heavy metal-associated isoprenylated plant proteins (HIPPs) metallochaperones [66], have been suggested play roles in metal binding and/or transport, thus regulating various processes under (a) biotic stress [65]. The main hypothesis for the mechanism of Cd homeostasis is that HIPPs protect the plant by trapping free Cd ion (via the CXXC core motif), thus preventing the ion from binding to a more essential protein [66, 67]. Whether these 10 Cd -upregulated rice HMADs are involved in Cd homeostasis and detoxification is of particular interesting.
With regard to the Cd-downregulated divalent cations transporters (Table 1, Additional file 7: Figure S3), 4 ZRT/IRT-like protein genes (OsZIP4, OsZIP5, OsZIP9, and OsZIP10) were consistently upregulated in the overexpression OsHMA3 line [19].
Overexpression of OsHMA3 enhanced the tolerance to Cd toxicity by increasing vacuolar sequestration of Cd in the roots, meanwhile, OsHMA3 is also responsible for vacuolar sequestration of Zn, but the translocation of Zn to the shoots is maintained by up-regulating these ZIP genes implicated in Zn uptake and translocation in the OsHMA3-overexpressed line [19].
Cd absorption from the soil is thought to occur mainly via ZIP family transporters in Zn/Cd-hyperaccumulators [10]. Among these, IRT1 protein is a broad-range metal ion transporter in plants. Arabidopsis AtIRT1 has previously been shown to transport Fe, Mn, Zn, Co, and Cd, whereas rice OsIRT1 and its homolog in barley HvIRT1 is able to transport Fe, Zn, and Cd [68,69,70]. However, OsIRT1 was neglected in 2 previous Cd-treated root transcriptomic studies, and it was found here to be suppressed by Cd treatment (Table 1, Additional file 2: Table S2). Moreover, the over-expression of OsIRT1 led to increased iron and zinc accumulation in rice, but Cd and other metal content in flag leaves and mature seeds from transgenic plants did not differ from those of WT plants, both growing in a paddy field not severely contaminated by Cd [69]. These findings again emphasize the potential use of these 4 Cd-suppressed ZIPs in Cd-contaminated soil to balance micronutrient fortification and toxicity reduction, which might be achieved by overexpressing them in rice.
Universal Cd-responsive genes in rice roots
To determine the universal Cd-responsive genes in rice ‘Nipponbare’ roots, we simultaneously performed a DEGs assay and gene co-expression network analysis. This combined approach produced 271 DEGs, which were putative universal Cd-responsive genes, and 164 DEGs of these with annotation information could be mapped to the MapMan pathway network (Table 2, Fig. 5, Additional file 11: Table S5).
Twelve mappable universal Cd-responsive DEGs were upregulated uniformly in previously published 3 reports including 2 microarray analyses, with the 2 lowest Cd treatments at the 3-h time point (Table 2, Additional file 11: Table S5) [21, 48], which demonstrated their high sensitivity to a wide range of Cd stresses across diverse time points. These included OsOPR5 and OsJAZ12 in JA signaling, the intensively studied SNAC1, 2 ZFPs, and ABCG36/OsPDR9. It is noteworthy that 14 universal Cd-responsive members are also novel DEGs only identified in this study through combining the 2 approaches (Table 2, Additional file 2: Table S2), and these include 2 experimentally verified genes, OsJAZ9 and CYP94C2b, involved in JA signaling [71,72,73].
From the perspective of global pathway distribution, 3 OPRs for JA biosynthesis and 4 OsJAZs for JA signaling were uniformly upregulated in all Cd-treated rice root samples (Fig. 5, Table 2). The universal Cd-responsive OsOPR1 identified here was also rapidly upregulated in leaves by heavy metals, Cu, Cd, and mercury [74]. Additionally, 2 maize zmOPRs were significantly upregulated in 2 maize genotypes following Cd stress [26]. Interestingly, CYP94C2b, in which overexpression can alleviate the JA response through inactivating JA and enhancing salt tolerance in rice [71], is also a universal Cd-responsive gene. Besides 3 OPRs and CYP94C2b for bioactive JA control, 4 OsJAZs for JA signaling were also shown to be universal Cd-responsive genes. As was expected, these 4 OsJAZs were also induced significantly by medium-term of medium Cd treatment (60 μM for 6 h) [28]. Among these, the stress-inducible OsJAZ9 acts as a transcriptional regulator in JA signaling and modulates salt stress tolerance in rice, and OsJAZ9-suppression rice plants exhibited increased sensitivity to JA, resulting in reduced salt tolerance [72, 73].
With regard to universal Cd-responsive TFs (Table 2, Fig. 5, Additional file 11: Table S5), SNAC1/OsNAC19 can be more strongly induced by exogenous application of JA than by ABA and ethylene [75], and OsWRKY10 can be strongly upregulated after JA treatment [76]. Collectively, the synchronized expression patterns of OsOPRs, OsJAZs, and JA-responsive TFs genes (SNAC1 and OsWRKY10) clearly indicate that the JA signaling pathway is one of the crucial elements in the plant response to Cd stress [21, 48]. Further identification of the upstream and downstream relationships between JA biosynthesis, and these TFs may contribute to elucidating the role of the JA-mediated signaling pathways underlying the responses of plants to Cd stress.
Beside SNAC1 and OsWRKY10, 7 ZFPs were also universally upregulated under 4 Cd exposure conditions (Table 2, Additional file 11: Table S5). Several ZFPs have been demonstrated to regulate plant tolerance to heavy metals. Among them, bean PvMTF-1 confers Cd tolerance in tobacco through activation of tryptophan biosynthesis, and the zinc finger-like motif is essential for the metal-responsive element binding of PvMTF-1 [77]. Maize zinc finger protein OXIDATIVE STRESS2 can enhance Cd tolerance in Arabidopsis [78], and Arabidopsis zinc-finger transcription factor ZAT6 positively regulates Cd tolerance through the glutathione-dependent pathway [79]. Arabidopsis STOP1 (sensitive to proton rhizotoxicity 1) operates in the signal transduction pathway controlling acid-soil tolerance [80]. Another zinc finger ART1 (for Al resistance transcription factor 1) regulates 31 genes implicated in Al tolerance in rice [81]. Regarding the 7 universal Cd-responsive ZFPs,ZFP182 was involved in ABA-induced antioxidant defense and its overexpression significantly enhanced salt, cold, and drought tolerance in rice [82, 83].
Since these transcription factors might trigger the expression of Cd detoxification genes and, thus, converge Cd stress signals, further investigation on them and their downstream targets involved in Cd response will aid to unveil ZFPs-mediated regulatory networks in rice under Cd stress.
Cd exposure somewhat perturbs the expression of genes in drought, high salinity, and low-temperature stress-signaling pathways; therefore, 9% of Cd-upregulated transcripts, including SNAC1 and ZFP252, were commonly upregulated among the 4 stresses [27]. Interestingly, 60 of the 164 universal Cd-responsive DEGs were Cd-upregulated and also commonly upregulated by the 4 stresses mentioned above (Table 2, Additional file 11: Table S5, Additional file 6: Dataset 1); therefore, these subset of 60 universal Cd-upregulated DEGs can be considered general stress-responsive genes. These include OsNCED4 for ABA synthesis, OsOPR1 for JA synthesis, OsJAZ12 and OsJAZ13 in JA signaling, 2 GSTs, and 7 TFs genes (SNAC1, OsBIERF3, 1 MYB, and 4 ZFPs) (Table 2). Regarding that overexpression of stress-inducible TFs can increase plant tolerance to multiple abiotic stresses, so upregulation of these general stress-responsive TFs could be useful for genetic improvement of crop tolerance to various abiotic stresses including diverse Cd pressure conditions.
Cd stress-specific responsive genes
Except the 60 general stress-responsive genes, 104 universal Cd-regulated were not general stress-responsive and 14 of them were not reported to be Cd-responsive previously (Table 2; Additional file 11: Table S5). Among these 14 novel Cd-responsive DEGs, Os04g33390 is the ortholog of Arabidopsis arogenate dehydratase 3 (ADT3). In plants, the arogenate pathway mediated by ADT isoforms is predominant in Phe biosynthesis [84], and ADT3 supply of Phe is required to control reactive oxygen species (ROS) concentration and distribution to protect cellular components [85]. These indicate the potential role of this rice ADT3 ortholog in buffering and restricting ROS under Cd pressure.
Regarding the 104 Cd-specific regulated genes, several DEGs could be categorized into orthologous groups of model plants [86] (Table 2; Additional file 11: Table S5).
Of the 49 rice OsPMEIs, OsPMEI46 and OsPMEI28 are in the same orthologous group APK_ORTHOMCL14694 [87], and these two PMEIs are all upregulated by Cd treatment, though OsPMEI28 (Os08g01670) is only in the list of short term High Cd-upregulated DEGs (Additional file 2: Table S2).
The activity of PME, responsible for catalyzing the demethylation of pectin in cell walls, is positively related to the binding of Cd and Al in plants [88]. Overexpression of one PME inhibitor (OsPMEI28) in rice resulted in dwarf phenotypes, and the cell wall thickness of culms of transgenic rice was greatly decreased [87]. Therefore, the roles of the two Cd-upregulated rice PMEIs in Cd stress tolerance and/or response need further investigation, especially the universal Cd-upregulated OsPMEI46 should gain more attention.
Plant MMPs (matrix metalloproteases) have been implicated in programmed cell death, and in response to biotic and abiotic stresses [89,90,91]. Tomato Sl2- and Sl3-MMP act upstream of P69B (an extracellular protease of the subtilase family) in an extracellular proteolytic cascade that contributes to the regulation of cell death [90]. In 4-week-old Arabidopsis plants, At2-MMP was induced in leaves by Cd or methyl JA and in roots by NaCl; however, Cd inhibited its expression in inflorescence and leaves of 10-week-old plants [90, 92].
As At2-MMP, the two rice MMPs in group APK_ORTHOMCL1642 are not general stress-responsive, but they are universal Cd-upregulated genes in rice roots (Table 2). Therefore, the identification of the substrates of these proteases will provide further insight into the mechanisms of cell death control under heavy metal stress.
Phytocyanins (PCs) are ancient blue copper binding proteins function as electron transporter, and plastocyanins as well as uclacyanins are typical family members of PCs [93, 94]. Moreover, PCs may also function in enhancing resistance to stress [93]. Overexpression of an Arabidopsis blue copper binding gene (AtBCB) could confer Al resistance in Arabidopsis [95]. Transgenic tobacco lines overexpressing an early nodulin-like protein gene from Boea crassifolia (BcBCP1) showed enhanced tolerance to osmotic stress [96]. Amplification of the PC genes in monocot plants (e.g. maize and rice) was suggested to be a mechanism of tolerance to harmful stresses [93]. Here, 3 members of the plastocyanin-type PCs in the orthologous group APK_ORTHOMCL3089 are universal Cd-upregulated genes (Table 2). Furthermore, the other member within this group (Os08g04340) is also upregulated by high-Cd stress (Additional file 2: Table S2). So, addressing whether these Cd-upregulated rice PCs play key roles in Cd tolerance is valuable.
Conclusion
To obtain more information from the RNAseq data on Cd-treated rice roots, the WGCNA package was employed to dissect the Cd-coupled co-expression gene modules from the filtered 22,080 transcripts across 17 RNAseq samples, including data from several published studies. Combined with the differential expression information, 164 universal Cd-responsive DEGs were identified as functioning under different concentrations of Cd across diverse time points.
More importantly, 104 of the 164 DEGs, including ABCG36/OsPDR9 and OsNAC3, might specifically respond to various Cd pressures, and 14 members of these are also novel DEGs, including OsJAZ9, OsNPR4 and Os04g33390 (encoding prephenate dehydratase), only identified in this study through integrative analysis. Few studies have been performed on these Cd-specific responsive DEGs; because they are promising novel candidate genes, more systemic investigation on them is needed to gain a better understanding of plant responses to Cd stress.
Methods
Rice seedlings cultivation, Cd treatment, and samples preparation
The seedlings of rice (Oryza sativa spp. japonica cv. Nipponbare) were cultivated in a hydroponic system (Kimura B nutrient solution) [97] in a growth chamber with a temperature of 28/22 °C (day/night), photosynthetic active radiation of 200 μmol m−2 s−1, and a photoperiod of 14/10 h (day/night). All hydroponic solutions were continuously aerated and renewed every 3 days. When the third leaves were fully expanded, the seedlings were transferred into fresh growing solutions containing 100 μM CdCl2 for 1 h. Following Cd treatment, roots were collected from rice seedlings of uniform size and separated into three groups: untreated 0 h, untreated 1 h, and Cd treated 1 h (labeled as ck0h, CK1h and Cd1h, respectively). All experiments were performed at least twice with 3 biological replicates each, and 3 replication samples for each treatment were mixed into one for RNAseq analysis, as described previously [1, 2, 27].
Rice roots RNA isolation, RNAseq library preparation and sequencing
Total RNA for RNAseq was extracted from rice seedling roots using a plant RNA kit (Omega, USA), and total RNA samples were then sent to Novogene Corporation (https://en.novogene.com/) for sequencing. Sequencing libraries were generated according to the manufacturer’s instructions (Illumina). Then the libraries were sequenced on an Illumina Hiseq 2000 platform and 100 bp paired-end reads were generated.
The process including RNA isolation, RNAseq library preparation and sequencing was repeated twice as 2 biological replicates. After the adaptor and low-quality sequences of pair-end reads were trimmed, a total of 32.63 Gb clean data from 6 cDNA libraries (labeled as ck0h, CK1h and Cd1h, each with 2 replicates) were obtained. Over 86% of the clean reads had scores at the Q30 level (Additional file 1: Table S1).
Mapping pair-end reads to the rice reference genome
A comprehensive analysis pipeline was formulated and our workflow for analysis of RNAseq data was illustrated in Fig. 1. In brief, two ‘Tuxedo’ packages, TopHat-Cufflinks [98] and HISAT-StringTie-Ballgown [99], were utilized in parallel to process the RNAseq data and output per kilobase transcript per million reads mapped (FPKM) matrix for differential expression analysis (Fig. 1). The rice reference genome and gene model annotation files (MSU6) were downloaded from Illumina’s iGenomes project (support.illumina.com/sequencing/sequencing_software/igenome.html) directly. For the 6 rice roots samples (ck0h, CK1h and Cd1h, each with replicates) prepared in our lab, the clean reads per sample were aligned to the rice reference genome (Nipponbare) using TopHat2 (v2.0.11). For each sample, the mapped reads were then assembled into transcripts with Cufflinks package (v2.2.1). Index of the reference genome was built using Bowtie (v2.2.2) and paired-end clean reads were aligned to the reference genome using TopHat2. After alignment, Cufflinks was employed to assemble the transcripts. Gene- and transcript-level expression values were represented by FPKM and were measured with Cufflinks.
Then the Cuffdiff was performed to evaluate the differential expression of genes and transcripts across various conditions (Fig. 1). A stringent criteria (fold-change ≥ 2 and q_value ≤ 0.05) was used to screen DEGs between each set of compared samples. MapMan (v3.6.0 RC1) was then used to annotate and subsequently visualize the DEGs on metabolic pathways [100].
Preprocessing public transcriptomic datasets of Cd-stressed rice roots
The RNAseq data (SRP053169, prepared in our lab) representing early transcriptomic response to high-Cd exposure (100 μM) were re-analyzed in combination with those published RNAseq data of Cd-treated rice seedling roots (DRP001141 and SRP058434, Additional file 1: Table S1), particularly from rice roots samples of medium Cd stress (50 μM) for 1 h and 24 h (designated MCd1 and MCdD, respectively) (single-end, each with 3 replicates, and only 0 h control ck0 available) [27], and high Cd pressure (100 μM) for 24 h (labeled as Cd24h) (pair-end, without replicate and the corresponding control ck24h available) [1]. In total, 17 datasets consisting of 3 time points and 3 concentrations of Cd were preprocessed by the ‘new Tuxedo’ RNAseq analysis package HISAT-StringTie-Ballgown (Fig. 1).
The RNAseq reads for each sample were mapped to the rice reference genome (MSU6) using HISAT2, and the output SAM files were sorted and converted to BAM files using SAMtools (version 0.1.19). Then the sorted alignments were assembled into transcripts and the expression levels of all genes and transcripts were estimated using StringTie. Similarly, StringTie merge function was performed after assembling each sample, then the merged transcripts are fed back to StringTie one more time so that it can re-estimate the transcript abundances using the merged structures, meanwhile create table counts for Ballgown [99].
Considering that some sample groups with less than three replicates or being single-end sequenced, the expression (FPKM values) matrix of all transcripts from Ballgown was extracted directly to analyze DEGs using the limma package [101]. The voom transformation was applied to the read counts. After this, the usual limma pipelines for differential expression was performed with the default stringent criteria (fold-change ≥ 2 and adj.P.Val ≤ 0.05).
Co-expression network analysis of Cd-stressed roots transcriptomes with WGCNA package
To reveal potential Cd-responsive modules in the public temporal transcriptome data of roots under various Cd pressures, the WGCNA software package in R was used to construct gene co-expression network of Cd-stressed rice roots from the normalized log2-transformed FPKM matrix from the Ballgown output mentioned above. To be included in the WGCNA workflow, transcript isoforms needed to have no more than 50% missing data [40]. Based on these criteria, there were 22,080 transcripts filtered for WGCNA unsigned co-expression network analysis (Fig. 1).
WGCNA network construction and module detection was conducted using an unsigned type of topological overlap matrix (TOM) with a soft-thresholding power β of 7 (R 2 > 0.7), a minimal module size of 30 and a branch merge cut height of 0.25, as described in previous reports [30, 33, 37, 43, 102].
Abbreviations
- ABC:
-
ATP-binding cassette transporters
- ADT:
-
Arogenate dehydratase
- ERF:
-
Ethylene-response factor
- GST:
-
glutathione-S-transferase
- HMA:
-
Heavy metal ATPase
- HMAD:
-
Heavy metal associated domain containing protein
- IRT1:
-
Iron-regulated transporter 1
- JA:
-
Jasmonic acid
- JAZ:
-
JA ZIM-domain family protein
- MATE:
-
Multidrug and toxic compound extrusion
- MMP:
-
Matrix metalloendoproteinase
- NCED:
-
9-cis-epoxycarotenoid dioxygenase
- OPR:
-
12-oxo-phytodienoic acid reductase
- PCs:
-
Phytocyanins
- PDR:
-
Pleiotropic drug resistance
- PMEI:
-
Pectin methylesterase inhibitor
- SNAC1:
-
Stress-responsive NAC 1
- WGCNA:
-
Weighted gene co-expression network analysis
- ZFP:
-
Zinc finger protein
- ZIPs:
-
ZRT- and IRT-like proteins
References
He F, Liu Q, Zheng L, Cui Y, Shen Z. RNA-seq analysis of rice roots reveals the involvement of post-transcriptional regulation in response to cadmium stress. Front Plant Sci. 2015;6:1136.
Kulik A, Anielska-Mazur A, Bucholc M, Koen E, Szymanska K, Zmienko A, Krzywinska E, Wawer I, McLoughlin F, Ruszkowski D, et al. SNF1-related protein kinases type 2 are involved in plant responses to cadmium stress. Plant Physiol. 2012;160(2):868–83.
Uraguchi S, Fujiwara T. Rice breaks ground for cadmium-free cereals. Curr Opin Plant Biol. 2013;16(3):328–34.
DalCorso G, Farinati S, Furini A. Regulatory networks of cadmium stress in plants. Plant Signal Behav. 2010;5(6):663–7.
Clemens S, Ma JF. Toxic heavy metal and metalloid accumulation in crop plants and foods. Annu Rev Plant Biol. 2016;67:489–512.
Ishikawa S, Ishimaru Y, Igura M, Kuramata M, Abe T, Senoura T, Hase Y, Arao T, Nishizawa NK, Nakanishi H. Ion-beam irradiation, gene identification, and marker-assisted breeding in the development of low-cadmium rice. Proc Natl Acad Sci U S A. 2012;109(47):19166–71.
Sasaki A, Yamaji N, Yokosho K, Ma JF. Nramp5 is a major transporter responsible for manganese and cadmium uptake in rice. Plant Cell. 2012;24(5):2155–67.
Takahashi R, Ishimaru Y, Senoura T, Shimo H, Ishikawa S, Arao T, Nakanishi H, Nishizawa NK. The OsNRAMP1 iron transporter is involved in Cd accumulation in rice. J Exp Bot. 2011;62(14):4843–50.
Lin YF, Aarts MG. The molecular mechanism of zinc and cadmium stress response in plants. Cell Mol Life Sci. 2012;69(19):3187–206.
Clemens S, Aarts MG, Thomine S, Verbruggen N. Plant science: the key to preventing slow cadmium poisoning. Trends Plant Sci. 2013;18(2):92–9.
Wong CK, Cobbett CS. HMA P-type ATPases are the major mechanism for root-to-shoot Cd translocation in Arabidopsis Thaliana. New Phytol. 2009;181(1):71–8.
Kim DY, Bovet L, Maeshima M, Martinoia E, Lee Y. The ABC transporter AtPDR8 is a cadmium extrusion pump conferring heavy metal resistance. Plant J. 2007;50(2):207–18.
Takahashi R, Ishimaru Y, Shimo H, Ogo Y, Senoura T, Nishizawa NK, Nakanishi H. The OsHMA2 transporter is involved in root-to-shoot translocation of Zn and Cd in rice. Plant Cell Environ. 2012;35(11):1948–57.
Takahashi R, Bashir K, Ishimaru Y, Nishizawa NK, Nakanishi H. The role of heavy-metal ATPases, HMAs, in zinc and cadmium transport in rice. Plant Signal Behav. 2012;7(12):1605–7.
Brunetti P, Zanella L, De Paolis A, Di Litta D, Cecchetti V, Falasca G, Barbieri M, Altamura MM, Costantino P, Cardarelli M. Cadmium-inducible expression of the ABC-type transporter AtABCC3 increases phytochelatin-mediated cadmium tolerance in Arabidopsis. J Exp Bot. 2015;66(13):3815–29.
Morel M, Crouzet J, Gravot A, Auroy P, Leonhardt N, Vavasseur A, Richaud P. AtHMA3, a P1B-ATPase allowing Cd/Zn/co/Pb vacuolar storage in Arabidopsis. Plant Physiol. 2009;149(2):894–904.
Miyadate H, Adachi S, Hiraizumi A, Tezuka K, Nakazawa N, Kawamoto T, Katou K, Kodama I, Sakurai K, Takahashi H, et al. OsHMA3, a P1B-type of ATPase affects root-to-shoot cadmium translocation in rice by mediating efflux into vacuoles. New Phytol. 2011;189(1):190–9.
Ueno D, Yamaji N, Kono I, Huang CF, Ando T, Yano M, Ma JF. Gene limiting cadmium accumulation in rice. Proc Natl Acad Sci U S A. 2010;107(38):16500–5.
Sasaki A, Yamaji N, Ma JF. Overexpression of OsHMA3 enhances Cd tolerance and expression of Zn transporter genes in rice. J Exp Bot. 2014;65(20):6013–21.
Kawahara Y, Oono Y, Wakimoto H, Ogata J, Kanamori H, Sasaki H, Mori S, Matsumoto T, Itoh T. TENOR: database for comprehensive mRNA-seq experiments in rice. Plant Cell Physiol. 2016;57(1):e7.
Lin CY, Trinh NN, Fu SF, Hsiung YC, Chia LC, Lin CW, Huang HJ. Comparison of early transcriptome responses to copper and cadmium in rice roots. Plant Mol Biol. 2013;81(4–5):507–22.
Xu L, Wang Y, Liu W, Wang J, Zhu X, Zhang K, Yu R, Wang R, Xie Y, Zhang W, et al. De novo sequencing of root transcriptome reveals complex cadmium-responsive regulatory networks in radish (Raphanus sativus L.). Plant Sci. 2015;236:313–23.
Han X, Yin H, Song X, Zhang Y, Liu M, Sang J, Jiang J, Li J, Zhuo R. Integration of small RNAs, degradome and transcriptome sequencing in hyperaccumulator Sedum alfredii uncovers a complex regulatory network and provides insights into cadmium phytoremediation. Plant Biotechnol J. 2016;14(6):1470–83.
Yang J, Li K, Zheng W, Zhang H, Cao X, Lan Y, Yang C, Li C. Characterization of early transcriptional responses to cadmium in the root and leaf of Cd-resistant Salix matsudana Koidz. BMC Genomics. 2015;16:705.
Yue R, Lu C, Qi J, Han X, Yan S, Guo S, Liu L, Fu X, Chen N, Yin H, et al. Transcriptome analysis of cadmium-treated roots in maize (Zea mays L.). Front Plant Sci. 2016;7:1298.
Peng H, He X, Gao J, Ma H, Zhang Z, Shen Y, Pan G, Lin H. Transcriptomic changes during maize roots development responsive to cadmium (Cd) pollution using comparative RNAseq-based approach. Biochem Biophys Res Commun. 2015;464(4):1040–7.
Oono Y, Yazawa T, Kawahara Y, Kanamori H, Kobayashi F, Sasaki H, Mori S, Wu J, Handa H, Itoh T, et al. Genome-wide transcriptome analysis reveals that cadmium stress signaling controls the expression of genes in drought stress signal pathways in rice. PLoS One. 2014;9(5):e96946.
Tang M, Mao D, Xu L, Li D, Song S, Chen C. Integrated analysis of miRNA and mRNA expression profiles in response to Cd exposure in rice seedlings. BMC Genomics. 2014;15:835.
Walley JW, Sartor RC, Shen Z, Schmitz RJ, Wu KJ, Urich MA, Nery JR, Smith LG, Schnable JC, Ecker JR, et al. Integration of omic networks in a developmental atlas of maize. Science. 2016;353(6301):814–8.
Hollender CA, Kang C, Darwish O, Geretz A, Matthews BF, Slovin J, Alkharouf N, Liu Z. Floral transcriptomes in woodland strawberry uncover developing receptacle and anther gene networks. Plant Physiol. 2014;165(3):1062–75.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559.
Bassel GW, Lan H, Glaab E, Gibbs DJ, Gerjets T, Krasnogor N, Bonner AJ, Holdsworth MJ, Provart NJ. Genome-wide network model capturing seed germination reveals coordinated regulation of plant cellular phase transitions. Proc Natl Acad Sci U S A. 2011;108(23):9709–14.
Xie W, Huang J, Liu Y, Rao J, Luo D, He M. Exploring potential new floral organ morphogenesis genes of Arabidopsis thaliana using systems biology approach. Front Plant Sci. 2015;6:829.
Pfeifer M, Kugler KG, Sandve SR, Zhan B, Rudi H, Hvidsten TR, Mayer KF, Olsen OA. Genome interplay in the grain transcriptome of hexaploid bread wheat. Science. 2014;345(6194):1250091.
Lu X, Li QT, Xiong Q, Li W, Bi YD, Lai YC, Liu XL, Man WQ, Zhang WK, Ma B, et al. The transcriptomic signature of developing soybean seeds reveals the genetic basis of seed trait adaptation during domestication. Plant J. 2016;86(6):530–44.
Appel HM, Fescemyer H, Ehlting J, Weston D, Rehrig E, Joshi T, Xu D, Bohlmann J, Schultz J. Transcriptional responses of Arabidopsis thaliana to chewing and sucking insect herbivores. Front Plant Sci. 2014;5:565.
Rawat N, Kiran SP, Du D, Gmitter FG Jr, Deng Z. Comprehensive meta-analysis, co-expression, and miRNA nested network analysis identifies gene candidates in citrus against Huanglongbing disease. BMC Plant Biol. 2015;15:184.
Formey D, Sallet E, Lelandais-Briere C, Ben C, Bustos-Sanmamed P, Niebel A, Frugier F, Combier JP, Debelle F, Hartmann C, et al. The small RNA diversity from Medicago truncatula roots under biotic interactions evidences the environmental plasticity of the miRNAome. Genome Biol. 2014;15(9):457.
Hopper DW, Ghan R, Schlauch KA, Cramer GR. Transcriptomic network analyses of leaf dehydration responses identify highly connected ABA and ethylene signaling hubs in three grapevine species differing in drought tolerance. BMC Plant Biol. 2016;16(1):118.
Kobayashi Y, Sadhukhan A, Tazib T, Nakano Y, Kusunoki K, Kamara M, Chaffai R, Iuchi S, Sahoo L, Kobayashi M, et al. Joint genetic and network analyses identify loci associated with root growth under NaCl stress in Arabidopsis thaliana. Plant Cell Environ. 2016;39(4):918–34.
Zhang S, Yang W, Zhao Q, Zhou X, Jiang L, Ma S, Liu X, Li Y, Zhang C, Fan Y, et al. Analysis of weighted co-regulatory networks in maize provides insights into new genes and regulatory mechanisms related to inositol phosphate metabolism. BMC Genomics. 2016;17:129.
Bai Y, Dougherty L, Cheng L, Zhong GY, Xu K. Uncovering co-expression gene network modules regulating fruit acidity in diverse apples. BMC Genomics. 2015;16:612.
El-Sharkawy I, Liang D, Xu K. Transcriptome analysis of an apple (Malus x domestica) yellow fruit somatic mutation identifies a gene network module highly associated with anthocyanin and epigenetic regulation. J Exp Bot. 2015;66(22):7359–76.
Childs KL, Davidson RM, Buell CR. Gene coexpression network analysis as a source of functional annotation for rice genes. PLoS One. 2011;6(7):e22196.
Sircar S, Parekh N. Functional characterization of drought-responsive modules and genes in Oryza sativa: a network-based approach. Front Genet. 2015;6:256.
Zhang L, Yu S, Zuo K, Luo L, Tang K. Identification of gene modules associated with drought response in rice by network-based analysis. PLoS One. 2012;7(5):e33748.
Amrine KC, Blanco-Ulate B, Cantu D. Discovery of core biotic stress responsive genes in Arabidopsis by weighted gene co-expression network analysis. PLoS One. 2015;10(3):e0118731.
Ogawa I, Nakanishi H, Mori S, Nishizawa NK. Time course analysis of gene regulation under cadmium stress in rice. Plant Soil. 2009;325(1–2):97–108.
Park J, Song WY, Ko D, Eom Y, Hansen TH, Schiller M, Lee TG, Martinoia E, Lee Y. The phytochelatin transporters AtABCC1 and AtABCC2 mediate tolerance to cadmium and mercury. Plant J. 2012;69(2):278–88.
Kim DY, Bovet L, Kushnir S, Noh EW, Martinoia E, Lee Y. AtATM3 is involved in heavy metal resistance in Arabidopsis. Plant Physiol. 2006;140(3):922–32.
Oda K, Otani M, Uraguchi S, Akihiro T, Fujiwara T. Rice ABCG43 is Cd inducible and confers Cd tolerance on yeast. Biosci Biotechnol Biochem. 2011;75(6):1211–3.
Moons A. Ospdr9, which encodes a PDR-type ABC transporter, is induced by heavy metals, hypoxic stress and redox perturbations in rice roots. FEBS Lett. 2003;553(3):370–6.
Kang J, Hwang JU, Lee M, Kim YY, Assmann SM, Martinoia E, Lee Y. PDR-type ABC transporter mediates cellular uptake of the phytohormone abscisic acid. Proc Natl Acad Sci U S A. 2010;107(5):2355–60.
Kochian LV, Pineros MA, Liu J, Magalhaes JV. Plant adaptation to acid soils: the molecular basis for crop aluminum resistance. Annu Rev Plant Biol. 2015;66:571–98.
Liu J, Magalhaes JV, Shaff J, Kochian LV. Aluminum-activated citrate and malate transporters from the MATE and ALMT families function independently to confer Arabidopsis aluminum tolerance. Plant J. 2009;57(3):389–99.
Wang J, Hou Q, Li P, Yang L, Sun X, Benedito VA, Wen J, Chen B, Mysore KS, Zhao J. Diverse functions of multidrug and toxin extrusion (MATE) transporters in citric acid efflux and metal homeostasis in Medicago truncatula. Plant J. 2017;90(1):79–95.
Magalhaes JV, Liu J, Guimaraes CT, Lana UG, Alves VM, Wang YH, Schaffert RE, Hoekenga OA, Pineros MA, Shaff JE, et al. A gene in the multidrug and toxic compound extrusion (MATE) family confers aluminum tolerance in sorghum. Nat Genet. 2007;39(9):1156–61.
Furukawa J, Yamaji N, Wang H, Mitani N, Murata Y, Sato K, Katsuhara M, Takeda K, Ma JF. An aluminum-activated citrate transporter in barley. Plant Cell Physiol. 2007;48(8):1081–91.
Zhou G, Pereira JF, Delhaize E, Zhou M, Magalhaes JV, Ryan PR. Enhancing the aluminium tolerance of barley by expressing the citrate transporter genes SbMATE and FRD3. J Exp Bot. 2014;65(9):2381–90.
Ryan PR, Raman H, Gupta S, Horst WJ, Delhaize E. A second mechanism for aluminum resistance in wheat relies on the constitutive efflux of citrate from roots. Plant Physiol. 2009;149(1):340–51.
Maron LG, Pineros MA, Guimaraes CT, Magalhaes JV, Pleiman JK, Mao C, Shaff J, Belicuas SN, Kochian LV. Two functionally distinct members of the MATE (multi-drug and toxic compound extrusion) family of transporters potentially underlie two major aluminum tolerance QTLs in maize. Plant J. 2010;61(5):728–40.
Yokosho K, Yamaji N, Ma JF. An al-inducible MATE gene is involved in external detoxification of al in rice. Plant J. 2011;68(6):1061–9.
Li L, He Z, Pandey GK, Tsuchiya T, Luan S. Functional cloning and characterization of a plant efflux carrier for multidrug and heavy metal detoxification. J Biol Chem. 2002;277(7):5360–8.
Yang H, Guo D, Obianom ON, Su T, Polli JE, Shu Y. Multidrug and toxin extrusion proteins mediate cellular transport of cadmium. Toxicol Appl Pharmacol. 2017;314:55–62.
Imran QM, Falak N, Hussain A, Mun BG, Sharma A, Lee SU, Kim KM, Yun BW. Nitric oxide responsive heavy metal-associated gene AtHMAD1 contributes to development and disease resistance in Arabidopsis thaliana. Front Plant Sci. 2016;7:1712.
de Abreu-Neto JB, Turchetto-Zolet AC, de Oliveira LF, Zanettini MH, Margis-Pinheiro M. Heavy metal-associated isoprenylated plant protein (HIPP): characterization of a family of proteins exclusive to plants. FEBS J. 2013;280(7):1604–16.
Tehseen M, Cairns N, Sherson S, Cobbett CS. Metallochaperone-like genes in Arabidopsis thaliana. Metallomics. 2010;2(8):556–64.
Korshunova YO, Eide D, Clark WG, Guerinot ML, Pakrasi HB. The IRT1 protein from Arabidopsis thaliana is a metal transporter with a broad substrate range. Plant Mol Biol. 1999;40(1):37–44.
Lee S, An G. Over-expression of OsIRT1 leads to increased iron and zinc accumulations in rice. Plant Cell Environ. 2009;32(4):408–16.
Pedas P, Ytting CK, Fuglsang AT, Jahn TP, Schjoerring JK, Husted S. Manganese efficiency in barley: identification and characterization of the metal ion transporter HvIRT1. Plant Physiol. 2008;148(1):455–66.
Kurotani K, Hayashi K, Hatanaka S, Toda Y, Ogawa D, Ichikawa H, Ishimaru Y, Tashita R, Suzuki T, Ueda M, et al. Elevated levels of CYP94 family gene expression alleviate the jasmonate response and enhance salt tolerance in rice. Plant Cell Physiol. 2015;56(4):779–89.
Wu H, Ye H, Yao R, Zhang T, Xiong L. OsJAZ9 acts as a transcriptional regulator in jasmonate signaling and modulates salt stress tolerance in rice. Plant Sci. 2015;232:1–12.
Ye H, Du H, Tang N, Li X, Xiong L. Identification and expression profiling analysis of TIFY family genes involved in stress and phytohormone responses in rice. Plant Mol Biol. 2009;71(3):291–305.
Agrawal GK, Jwa NS, Shibato J, Han O, Iwahashi H, Rakwal R. Diverse environmental cues transiently regulate OsOPR1 of the “octadecanoid pathway” revealing its importance in rice defense/stress and development. Biochem Biophys Res Commun. 2003;310(4):1073–82.
Lin R, Zhao W, Meng X, Wang M, Peng Y. Rice gene OsNAC19 encodes a novel NAC-domain transcription factor and responds to infection by Magnaporthe grisea. Plant Sci. 2007;172(1):120–30.
Ryu H-S, Han M, Lee S-K, Cho J-I, Ryoo N, Heu S, Lee Y-H, Bhoo SH, Wang G-L, Hahn T-R, et al. A comprehensive expression analysis of the WRKY gene superfamily in rice plants during defense response. Plant Cell Rep. 2006;25(8):836–47.
Sun N, Liu M, Zhang W, Yang W, Bei X, Ma H, Qiao F, Qi X. Bean metal-responsive element-binding transcription factor confers cadmium resistance in tobacco. Plant Physiol. 2015;167(3):1136–48.
He L, Ma X, Li Z, Jiao Z, Li Y, Ow DW. Maize OXIDATIVE STRESS2 homologs enhance cadmium tolerance in arabidopsis through activation of a putative SAM-dependent methyltransferase gene. Plant Physiol. 2016;171(3):1675–85.
Chen J, Yang L, Yan X, Liu Y, Wang R, Fan T, Ren Y, Tang X, Xiao F, Cao S. Zinc-finger transcription factor ZAT6 positively regulates cadmium tolerance through the glutathione-dependent pathway in arabidopsis. Plant Physiol. 2016;171(1):707–19.
Iuchi S, Koyama H, Iuchi A, Kobayashi Y, Kitabayashi S, Ikka T, Hirayama T, Shinozaki K, Kobayashi M. Zinc finger protein STOP1 is critical for proton tolerance in Arabidopsis and coregulates a key gene in aluminum tolerance. Proc Natl Acad Sci U S A. 2007;104(23):9900–5.
Yamaji N, Huang CF, Nagao S, Yano M, Sato Y, Nagamura Y, Ma JF. A zinc finger transcription factor ART1 regulates multiple genes implicated in aluminum tolerance in rice. Plant Cell. 2009;21(10):3339–49.
Huang J, Sun S, Xu D, Lan H, Sun H, Wang Z, Bao Y, Wang J, Tang H, Zhang H. A TFIIIA-type zinc finger protein confers multiple abiotic stress tolerances in transgenic rice (Oryza sativa L.). Plant Mol Biol. 2012;80(3):337–50.
Zhang H, Ni L, Liu Y, Wang Y, Zhang A, Tan M, Jiang M. The C2H2-type zinc finger protein ZFP182 is involved in abscisic acid-induced antioxidant defense in rice. J Integr Plant Biol. 2012;54(7):500–10.
Chen Q, Man C, Li D, Tan H, Xie Y, Huang J. Arogenate dehydratase isoforms differentially regulate anthocyanin biosynthesis in Arabidopsis thaliana. Mol Plant. 2016;9(12):1609–19.
Para A, Muhammad D, Orozco-Nunnelly DA, Memishi R, Alvarez S, Naldrett MJ, Warpeha KM. The dehydratase ADT3 affects ROS homeostasis and cotyledon development. Plant Physiol. 2016;172(2):1045–60.
Ouyang S, Zhu W, Hamilton J, Lin H, Campbell M, Childs K, Thibaud-Nissen F, Malek RL, Lee Y, Zheng L, et al. The TIGR Rice genome annotation resource: improvements and new features. Nucleic Acids Res. 2007;35(Database issue):D883–7.
Nguyen HP, Jeong HY, Jeon SH, Kim D, Lee C. Rice pectin methylesterase inhibitor28 (OsPMEI28) encodes a functional PMEI and its overexpression results in a dwarf phenotype through increased pectin methylesterification levels. J Plant Physiol. 2017;208:17–25.
Kan Q, Wu W, Yu W, Zhang J, Xu J, Rengel Z, Chen L, Cui X, Chen Q. Nitrate reductase-mediated NO production enhances Cd accumulation in Panax notoginseng roots by affecting root cell wall properties. J Plant Physiol. 2016;193:64–70.
Zimmermann D, Gomez-Barrera JA, Pasule C, Brack-Frick UB, Sieferer E, Nicholson TM, Pfannstiel J, Stintzi A, Schaller A. Cell death control by matrix metalloproteinases. Plant Physiol. 2016;171(2):1456–69.
Li D, Zhang H, Song Q, Wang L, Liu S, Hong Y, Huang L, Song F. Tomato Sl3-MMP, a member of the matrix metalloproteinase family, is required for disease resistance against Botrytis cinerea and Pseudomonas syringae pv. Tomato DC3000. BMC Plant Biol. 2015;15:143.
Marino G, Huesgen PF, Eckhard U, Overall CM, Schroder WP, Funk C. Family-wide characterization of matrix metalloproteinases from Arabidopsis thaliana reveals their distinct proteolytic activity and cleavage site specificity. Biochem J. 2014;457(2):335–46.
Golldack D, Popova OV, Dietz KJ. Mutation of the matrix metalloproteinase At2-MMP inhibits growth and causes late flowering and early senescence in Arabidopsis. J Biol Chem. 2002;277(7):5541–7.
Cao J, Li X, Lv Y, Ding L. Comparative analysis of the phytocyanin gene family in 10 plant species: a focus on Zea mays. Front Plant Sci. 2015;6:515.
Ma H, Zhao H, Liu Z, Zhao J. The phytocyanin gene family in rice (Oryza sativa L.): genome-wide identification, classification and transcriptional analysis. PLoS One. 2011;6(10):e25184.
Ezaki B, Sasaki K, Matsumoto H, Nakashima S. Functions of two genes in aluminium (al) stress resistance: repression of oxidative damage by the AtBCB gene and promotion of efflux of al ions by the NtGDI1gene. J Exp Bot. 2005;56(420):2661–71.
Wu H, Shen Y, Hu Y, Tan S, Lin Z. A phytocyanin-related early nodulin-like gene, BcBCP1, cloned from Boea crassifolia enhances osmotic tolerance in transgenic tobacco. J Plant Physiol. 2011;168(9):935–43.
Zheng L, Yamaji N, Yokosho K, Ma JF. YSL16 is a phloem-localized transporter of the copper-nicotianamine complex that is responsible for copper distribution in rice. Plant Cell. 2012;24(9):3767–82.
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and cufflinks. Nat Protoc. 2012;7(3):562–78.
Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc. 2016;11(9):1650–67.
Usadel B, Poree F, Nagel A, Lohse M, Czedik-Eysenberg A, Stitt M. A guide to using MapMan to visualize and compare Omics data in plants: a case study in the crop species, maize. Plant Cell Environ. 2009;32(9):1211–29.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47.
Wu J, Fu L, Yi H. Genome-wide identification of the transcription factors involved in citrus fruit ripening from the transcriptomes of a late-ripening sweet orange mutant and its wild type. PLoS One. 2016;11(4):e0154330.
Acknowledgments
We are grateful to Alyssa C Frazee (Center for Computational Biology, Johns Hopkins University) for suggestions on differential expression analysis, and Hanwei Mei and Shunwu Yu (Shanghai Agrobiological Gene Center, China) for providing the seeds of Nipponbare rice.
Funding
This work was supported by research grants from the Project of the National Key Research and Development Plan of China (No. 2016YFD0800700–3) and the National Natural Science Foundation of China (No. 31271421).
Availability of data and materials
All sequence reads analyzed during the current study have been deposited in the NCBI SRA datasets (www.ncbi.nlm.nih.gov/sra) under the accession number SRP053169.
Author information
Authors and Affiliations
Contributions
Research design, data interpretation and manuscript draft: MT, YC and MJ; Analyzing data: DC, JC, GZ and MT; Experiment: YY, GZ, MQ and JC; Conception and supervision of the research: MT, YC and MJ; and all authors approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable. The seeds of Nipponbare rice used in this study were kindly provided by Hanwei Mei and Shunwu Yu from Shanghai Agrobiological Gene Center. The samples collected from Nipponbare seedlings are for research use with permissions.
Consent for publication
Not applicable.
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1: Table S1.
RNAseq information of rice roots under various Cd stresses conditions. (XLSX 12 kb)
Additional file 2: Table S2.
Cd-responsive 1772 differentially expressed genes transcripts isoforms (DEGs) in rice roots exposed to Cd for 1 h. (XLSX 186 kb)
Additional file 3: Table S3.
Primers for quantitative RT-PCR. (XLSX 9 kb)
Additional file 4: Figure S1.
Quantitative RT-PCR of 8 randomly selected DEGs expression in in rice roots exposed to Cd for 1 h. Actin-1 (LOC4333919) was used to standardize transcript levels in each sample. The primers used in the qRT-PCR experiments are listed in additional Table S3. (PPT 75 kb)
Additional file 5: Figure S2.
A Venn diagram showing the intersections of Cd-responsive 1772 DEGs and those identified in previous two reports. Rice roots exposed to 1 h of Cd treatment (Cd1h) and its control (ck1h) are sampled in our lab. The published rice roots RNAseq data of medium Cd stress for 1 h (MCd1) [27], and the microarray analysis of low Cd stress for 3 h (lowCd) [21] are listed in Additional file 6: Dataset 1. (PPT 373 kb)
Additional file 6: Dataset 1.
Rice roots short-term Cd-responsive DEGs identified in previous three reports. (XLSX 621 kb)
Additional file 7: Figure S3.
Transport overview of 1772 DEGs in rice roots under Cd stress. DEGs were selected for the metabolic pathways analysis using the MapMan software (v3.6.0RC1). The colored boxes indicate the Log2 ratio of Cd1h/ck1h (1 h of Cd treated and untreated rice roots, respectively). (PPT 282 kb)
Additional file 8: Dataset 2.
The FPKM matrix of 22,080 rice genes transcripts across 17 RNAseq samples. (TXT 2098 kb)
Additional file 9: Dataset 3.
Four DEGs datasets (MCd1 vs ck0, MCdD vs ck0, Cd24h vs ck24h, Cd1h vs ck1h) output by limma package. (XLS 1903 kb)
Additional file 10: Table S4.
One thousand eight hundred sixty-eight rice genes and transcripts in the Cd response-specific module generated by WGCNA. (XLSX 47 kb)
Additional file 11: Table S5.
The expression of 164 universal Cd-responsive DEGs in Cd response-specific module across 4 DEGs datasets. (XLSX 38 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Tan, M., Cheng, D., Yang, Y. et al. Co-expression network analysis of the transcriptomes of rice roots exposed to various cadmium stresses reveals universal cadmium-responsive genes. BMC Plant Biol 17, 194 (2017). https://doi.org/10.1186/s12870-017-1143-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-017-1143-y