
Abstract
Background
Interaction between TERMINAL FLOWER 1 (TFL1) and LEAFY(LFY) seem to determine the inflorescence architecture inArabidopsis. In a parallel way, overexpression of VvTFL1A, agrapevine TFL1 homolog, causes delayed flowering and production of aramose cluster in the reiterated reproductive meristem (RRM) somatic variant ofcultivar Carignan. To analyze the possible contribution of this gene to clusterphenotypic variation in a diversity panel of cultivated grapevine (Vitisvinifera L. subsp. vinifera) its nucleotide diversity wascharacterized and association analyses among detected sequence polymorphisms andphenology and cluster traits was carried out.
Results
A total of 3.6 kb of the VvTFL1A gene, including its promoter, wassequenced in a core collection of 140 individuals designed to maximize phenotypicvariation at agronomical relevant traits. Nucleotide variation forVvTFL1A within this collection was higher in the promoter and intronsequences than in the exon regions; where few polymorphisms were located inagreement with a high conservation of coding sequence. Characterization of theVvTFL1A haplotype network identified three major haplogroups,consistent with the geographic origins and the use of the cultivars that couldcorrespond to three major ancestral alleles or evolutionary branches, based on theexistence of mutations in linkage disequilibrium. Genetic association studies withcluster traits revealed the presence of major INDEL polymorphisms, explaining 16%,13% and 25% of flowering time, cluster width and berry weight, respectively, andalso structuring the three haplogroups.
Conclusions
At least three major VvTFL1A haplogroups are present in cultivatedgrapevines, which are defined by the presence of three main polymorphism LD blocksand associated to characteristic phenotypic values for flowering time, clusterwidth and berry size. Phenotypic differences between haplogroups are consistentwith differences observed between Eastern and Western grapevine cultivars andcould result from the use of different genetic pools in the domestication processas well as different selection pressures on the development of table and winecultivars, respectively. Altogether, these results are coherent with previousclassifications of grapevine phenotypic diversity mainly based on cluster andberry morphotypes as well as with recent results on the structure of geneticdiversity in cultivated grapevine.
Similar content being viewed by others
Background
Grapevine (Vitis vinifera subsp. vinifera) was domesticated in theNeolithic period (ca. 8500–4000 BC) [1] from wild populations of Vitis vinifera subsp. sylvestris[2]. Archaeological data traced back the location of the earliest evidence forlarge-scale winemaking, likely linked to the use of domesticated plants, to the north ofZagros Mountains and in the Caucasian region [3] around 6000–5000 BC which supports that geographic area as the locationfor primo domestication events. From there, grapevine cuttings were widely spread: firstfrom North to South; and later from East to West around the Mediterranean basin pathway [3]. Vegetative propagation and dissemination, spontaneous events ofhybridization among cultivars, breeding with local wild plants and likely secondarydomestication events generated the pattern of admixture that is observed in currentcultivars [4]–[9]. The use of different genetic pools along the process of grapevinedomestication and human selection for different uses such as fresh consumption, raisinor wine production have resulted in large variation for cluster size, compactness andarchitecture among cultivars from different geographic locations [10].
The size and shape of grapevine clusters is determined by the development and growth ofinflorescences as well as the efficiency of pollination, fruit set and berry growth.Generally, wine grape cultivars present small (150-250 g) and compact clusters withsmall berries, while table grapes generally have large (300-400 g) and less compactclusters with large berries. Some of them can even be extremely big weighting up to1000-1500 g [11]. Negrul [12] distinguished different grape morphotypes based in part on cluster and berrytraits. Cluster architecture has implications on disease susceptibility, since cultivarswith compact clusters are more susceptible to rot by Botrytis cinerea thanthose of loose clusters [10],[13],[14]. In spite of the relevance of cluster structure and compactness, very littleis known about its genetic control probably due in part to the complexity of the trait,which depends on many different variables along the growth of the plant as well as theenvironmental interactions during its reproductive development. There is a need todefine cluster shape and size in terms of quantitative variables to understand itsgenetic determination. So far, only a few studies have tried to identify the mainvariables responsible for variation in bunch compactness in grapevine. In this sense,Vail and Marois [14] identified cluster weight as the main factor to explain its variation whileShavrukov et al. [15] proposed total cluster length and node number per rachis as two of the mainones. Recently, Tello and Ibañez [16] evaluated 19 indexes to estimate cluster compactness highlighting the role ofvarious cluster parameters such as branch length and number. The study proposed a fastand good estimator for cluster compactness based on cluster weight and length.
Genetic and molecular analyses in model plants, such as Arabidopsis thaliana,demonstrated the interaction between TERMINAL FLOWER 1 (TFL1) andLEAFY (LFY) [17],[18] in the establishment of inflorescence architecture. Their interactionssupported a simple model explaining the evolution of plants inflorescence architecture [19]. TFL1 belongs to a small gene family first identified in mammals asencoding phosphatidyl ethanolamine-binding proteins (PEBP) [20], which participates in a wide variety of biological functions in eukaryotes.In Arabidopsis, TFL1 has been shown to function in the transcriptionalrepression of flower meristem identity genes [21]. LFY encodes a plant specific transcription factor [22], which serves as a flower meristem identity regulator activating thetranscription of other flower meristem identity genes [23]. Recently, the existence of a common genetic pathway controllinginflorescence architecture in Arabidopsis and rice has been demonstratedindicating that this pathway could be highly conserved in angiosperms [24]. Following this report, four MADS-box genes are required to suppressTFL1 in emerging floral meristems; what seems to be indispensable toinitiate their differentiation.
In grapevine, the family of PEBP encoding genes includes at least five genes; three ofthem have deduced protein sequences related to Arabidopsis TFL1, beingVvTFL1A the closest homologous sequence [25]. In fact, over-expression of VvTFL1A in transgenic Arabidopsisplants generates phenotypes of large and late flowering inflorescences reminding thoseobserved when over-expressing the endogenous Arabidopsis gene [25]. Likewise, recent findings show that the extreme cluster proliferation anddelayed anthesis observed in the reiterated reproductive meristems (RRM) somatic variantof grapevine cultivar Carignan was caused by a single dominant mutation in theVvTFL1A gene. This dominant mutation was identified as the insertion of aclass II transposable element, Hatvine1-rrm, in the VvTFL1A promoter,triggering up-regulation of the corresponding VvTFL1A allele in reproductiveand vegetative organs of the shoot apex [26]. These results suggested a role for VvTFL1A in the determination ofinflorescence structure as well as on the branching pattern of the grapevine fruitclusters and the time of anthesis.
To further analyze the contribution of VvTFL1A to the phenotypic variationobserved for reproductive and inflorescence traits in grapevine, the nucleotidediversity shown by this gene in a core collection of grapevine accessions was analysedand a candidate gene association approach on the variation observed for fertility index,phenological variables as well as several inflorescence and berry related traits wascarried out. Herein the identification of VvTFL1A sequence polymorphismsassociated with flowering and cluster traits is reported, being the most relevant onescorresponding to several INDELs in two intron regions. These INDELs are in LD withadditional SNPs defining three LD blocks, which correspond to three major haplogroups.Interestingly, these haplogroups are characteristic of either wine or table cultivars inagreement with the cluster and flowering phenotype to which they are associated to.
Methods
Plant material
The plant material consisted of 140 grapevine cultivars corresponding to a corecollection of Vitis vinifera L. subsp. vinifera intended tomaximize agro-morphological diversity for 50 qualitative and quantitative traits [27]. All the cultivars are maintained at the INRA experimental station ofDomaine de Vassal, Marseillan-plage, France(http://www1.montpellier.inra.fr/vassal/). The list of cultivars,pedigree when available, classification according to use (wine, table or wine/table),geographical group according to Bacilieri et al. [6], Lacombe et al. [28] and available data of the Vitis International Variety Catalogue(http://www.vivc.de/) are shown in Additional file 1. Classification according to Eastern and Western origin was obtainedconsidering cultivars from the Iberian Peninsula (IBER), Western and Central Europe(WCEUR) and the Italian Peninsula (ITAP) as occidental cultivars; whereas cultivarsfrom the Balkans (BALK), Russia and Ukraine (RUUK), Eastern Mediterranean andCaucasus (EMCA), Middle and Far East (MFEAS) were considered as oriental cultivars.For newly bred grape varieties, their pedigree was used to assess Western or Easternorigin to classify them according to their genetic origin and not according tobreeding location. When genetic origin of pedigree was questionable, the cultivar wasconsidered to present mixed origin.
Phenotypic evaluation
Ten morphological traits related to the reproductive biology of grapevine wereconsidered in this study. Among them, four were related to phenology (budburst time,flowering time, veraison time and maturity time); one to yield (fertility index); andfive to berry and cluster features (berry weight and cluster length, width, weightand compactness). Cluster compactness was estimated from available data such as[cluster weight/(cluster length)2] [16]. Principal component analysis separated on axis 2 phenological traits fromcluster size traits being fertility index opposite to all other traits on axis 1(Additional file 2). Strongest correlations were foundbetween maturity and veraison time (Pearson’s r = 0.82) aswell as between cluster weight and width (Pearson’s r = 0.80). Alltraits were scored at the Domaine de Vassal and were expressed as the mean value forfive plants per accession analysed a maximum of three years following the recommendedOIV descriptors as shown in Table 1[11]. Details of phenotypic values obtained for each cultivar are given inAdditional file 3. The phenotype distribution for thesetraits within the core collection is provided in Additional file 4.
Genotyping
For each genotype, 3.6 kb of the VvTFL1A gene (GSVIVT01036145001,chr6_20199669-20203319, Genoscope 12X) were amplified and sequenced using primerslisted in Fernandez et al. [26]. DNA was extracted from young leaves of each genotype as described inAdam-Blondon et al. [29]. Amplifications were carried out using Taq DNA Polymerase (Qiagen) asrecommended by manufacturer. PCR products were treated with Exosap-IT reagent asrecommended by manufacturer and sequenced at the Genomic Service of the ParqueCientifico de Madrid in an ABI prism 3730 (Applied Biosystems) DNA sequencer. Basecalling, quality trimming and alignment of ABI chromatograms was performed usingSeqScape v2.5 (Applied Biosystems). Sequence polymorphisms were manually verified toestablish genotypes. The nomenclature system used to name polymorphisms correspondedto letters followed by numbers: single letter correspond to the involved nucleotidesubstitution using the IUB’s conventional nomenclature and “Ins” isused to designed INDEL; positive or negative numbers corresponded to polymorphismposition from the first base of the “ATG start codon”. Linkagedisequilibrium (LD) calculations between polymorphisms were carried out using the LDoption implemented in TASSEL v.2.1 [30].
Molecular diversity parameter estimates were calculated using DnaSP v4.50.2 [31]. Per site nucleotide diversity (π) [32], Watterson θ estimate [33] and Tajima’s D [34] were calculated for the whole haplotype set and separately for the threestructured sub-populations (K1, K2, K3).
Association tests
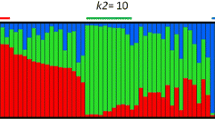
Knowing that population structure can bias association studies, the structuredassociation (SA) method [35] and the Mixed Linear Model MLM [36] were used to reduce false positives. Population structure of the corecollection was determined using 20 SSR markers well scattered throughout the 19 grapelinkage groups (LGs) [37] by a Bayesian clustering implemented in STRUCTURE v.2.3.4 [38]. The ADMIXTURE model was applied assuming that segregation of alleles wasindependent. A burn-in period of 100,000 followed by 150,000 Markov Chain Monte Carlo(MCMC) iterations with 5 replicate runs were carried out for each value of populationstructure tested (1 ≤ K ≤ 10). The optimalsub-population model was selected with the maximal likelihoodK = 3 according to the ΔK method [39] and later corrections for ΔK artefacts [40]. The corresponding Q-matrix was used for structured association tests. Anarbitrary cut-off value of 50% ancestry was set to assign each individual to one ofthe three sub-population clusters (Additional file 1).Individuals not assigned in one sub-population were considered as admixed. Thisstructure discriminates cultivars according to their use and geographic origin withK2 mainly constituted by western wine cultivars (78%), K3 by eastern table cultivars(78%) and K1 composed by eastern and western wine cultivars and table cultivars(Figure 1). The kinship matrix was calculated on thebasis of the same set of SSR markers [41] using TASSEL v.2.1.

Population structure of the V.vinifera core collection. Schematic representation of estimated membership wasobtained using STRUCTURE and classification of the individuals to one of thethree genetic groups (K1, K2 and K3) was obtained using 50% of ancestry.Geographic origin (A) and fruit use (B) of each cultivar areindicated using colour codes.
Comparison of the naïve General Linear Model (GLM) test, the structuredassociation test (GLM-Q) and the structured Mixed Linear Model (MLM-Q) using TASSELv.3 identified the last one as the most conservative model and was therefore selectedto perform the association tests. MLM-Q association tests were carried out using theR v.2.15 [42] and TASSEL v.3 software. Polymorphic sites carrying rare alleles(frequencies <5% within the total sample) and unbalanced genotypic classes(frequencies <5% within the total sample) were discarded to avoid biasedassociations. Rare genotypic classes were in this last case replaced by missing data.Polymorphisms were codified to test both additive and dominant effects using R to besimilar with marker model tested using TASSEL. For traits showing significantassociations after Bonferroni correction (P ≤ 0.05) usingeither TASSEL or R, multi-locus mixed-models using forward-backward stepwiseregression (MLMM) were implemented using the R software to identify majornon-redundant associated markers [43]. Population structure and kinship were both included in the multi-locusanalysis. Best models were selected according to the extended Bayesian informationcriteria (EBIC) and the multiple Bonferroni criteria (mBonf) according to Segura etal. [43].
Haplotype reconstruction and networks
As V. vinifera genotypes are generally highly heterozygous [37], the unphased genotypic dataset was analysed to identify the succession oflinked polymorphisms along the sequenced DNA region. Haplotypes were reconstructedusing a PLEM algorithm [44] implemented in PHASE v2.1 applying default values of the iterative scheme [45]. Reconstructed haplotypes were submitted separately and simultaneously tothree recombination detection tests implemented in the Recombination DetectionProgram v3beta41 [46]. Those were the MaxChi method with a window size of 12, 20, 25 or 30variables sites [47], the Chimaera method with a window size of 12, 20, 25 or 30 variablessites [48] and the 3SEQ method [49]. To ensure consistency, haplotypes showing a significant probability ofbeing the result of recombination (P ≤0.05) in at least two tests wereconsidered as recombinants and excluded from further analysis as previously done byFournier-Level et al. [50].
Network analysis was carried out using the median-joining method [51] implemented in Network v4.5.1.6 (Fluxus Technology, Sudbury, UK) andfixing a weight of 99 for the polymorphisms showing best associations with traits(Ins883, Ins422, K-737 and M-196). Three haplogroups HGA, HGB and HGC were definedaccording to the three LD blocks.
Results
VvTFL1Astructure and sequence polymorphisms
A total of 3646 bp of the VvTFL1A gene corresponding to 2442 bpand 1204 bp before and after ATG, respectively, were sequenced in all theindividuals of the core collection. Translation of coding sequences identified theannotated four exons in the V. vinifera PN40024 genome sequence [52] of 201, 62, 41 and 218 bp and three introns of 83, 467 and107 bp (Figure 2A). Nucleotide sequence analysesenabled the identification of 70 polymorphisms (64 SNP and 6 INDEL including 3microsatellites). Among them, 44 polymorphisms were located in the promoter sequence,4 in the 5’ untranslated region, and 5 and 17 polymorphisms were located inexonic and intronic regions, respectively. INDEL Ins-2054 and Ins-1389, located inthe promoter, and Ins883 in intron 3, involved 5, 21 and 1 nucleotides, respectively;whereas Ins-393 and Ins-3 in the promoter, and Ins422 in intron 2 representedmicrosatellite variations with Ins422 being biallelic and the other two multiallelic(Figure 2A). Out of the five exonic SNP, only three(W13, W1087, and M1094) caused non-synonymous amino acid substitutions and are shownin Figure 2A. Among the 70 polymorphisms, 32 (46%) wererepresented by a rare allele (frequency <5%), with 10 of them grouped betweenposition −1079 and −1430 before ATG. The complete genotypic data set isavailable in Additional file 5.

Sequence polymorphisms identified for the VvTFL1A gene sequence and their association to phenotypic traits. A.VvTFL1A polymorphisms found in 140 V. viniferaaccessions. Single-nucleotide substitutions are depicted as vertical bars,different colours denoting allele frequency found in the collection (bottomlegend box). INDEL are indicated as vertical arrows with similar colour codes.Amino acid changes are indicated. Polymorphisms are classified according tothree LD blocks using a colour code: B. Level of structured MLMassociation detected between phenotypic traits and markers alongVvTFL1A gene using TASSEL. P-value threshold 0.05 afterBonferroni correction is represented by the black line.
VvTFL1Anucleotide diversity
A total of 62 haplotypes, including 37 singletons, were identified based on phasereconstruction using the 70 segregating polymorphisms (Additional file 6). Genetic diversity of VvTFL1A gene was estimated interms of number of segregating sites (S) and polymorphism (π and θ) for allthe haplotypes and according to population structure. Comparison of genetic diversityindex among the 3 genetic groups inferred within the analysed core collection (seeMaterial and Methods) indicated that despite K3 sub-population included a smallernumber of haplotypes than the two other K1 and K2 sub-populations; this presented ahigh number of segregating sites and π and θ polymorphism indexes(Table 2). The Tajima’s D-tests showed ageneral neutral value considering all haplotypes and a slight but non-significantnegative value in K1 sub-population compared with K2 sub-population, which showed aslight positive value (Table 2). When Tajima’sD-test was estimated in sliding windows along VvTFL1A; the testrevealed a similar pattern of variation for K1 and K3 sub-populations with a generalnegative value along VvTFL1A promoter (Figure 3).In contrast, the value of D along the transcribed region in K3sub-population increased to reach a positive value. In K2 sub-population,Tajima’s D-test showed general positive value in both promoter andtranscribed regions. These patterns indicated different selection or demographicevents between haplotypes within the three genetic groups.

Pattern of Tajima’s D values along VvTFL1A gene. Neutral selection (D = 0) is represented by the greyline. Patterns have been obtained using sliding windows option from DnaSP(window length =500 and step size =350).
VvTFL1Ahaplotypes
In order to study relationship between VvTFL1A haplotypes, those likelyresulting from recombinant events were detected to avoid bias. Among the 62haplotypes originally identified, 26 were considered recombinants (Additional file6) and were removed for haplotype network analysis.Haplotype network was constructed using 36 non-recombinant haplotypes that included19 singletons. Haplotype network discriminated three groups of closely relatedhaplotypes or haplogroups (HGA, HGB and HGC), which were clearly structured inrelation with mutations in linkage disequilibrium (LD) (Figure 4). Within HGA major part of the haplotypes (44%) were present incultivars of the K1 sub-population including Eastern and Western cultivars used astable and wine grapes. The remaining haplotypes of HGA were similarly found incultivars of the K2 and K3 sub-population (25% and 24%, respectively). Regarding HGBand HGC 52% and 68% of their haplotypes were detected in cultivars of the K2sub-population mainly represented by Western wine cultivars (Figure 4).

Median joining networks derived from reconstructed DNA sequence haplotypesof VvTFL1A . Network analysis was carried for haplotypes identified for 70polymorphic sites in the VvTFL1A gene excluding recombined haplotypes.Haplotypes are represented by circles with circle size proportional tohaplotype frequency (circle size corresponding to haplotype numbers of 1, 5 and25 are illustrated). The haplotypes colours relate to the accessionsclassification according to according to structured genetic groups (Additionalfile 1). Black dots represent mutational steps.Colour lines represent the mutations in LD that separated the three haplogroupsidentified (HG).
Haplotypes 32 and 3 were the most frequent (frequency >0.15 on the total haplotypepool and >0.19 when excluding the recombinants) and belonged to HGA and HGB,respectively. Most of the cultivars of the core collection were heterozygous for twodifferent haplotypes (86%) with 20% of them being heterozygous for a combination ofHGA and HGB haplotypes (Additional file 1). Only 20cultivars were homozygous (14%) with eight and five varieties homozygous for HGA andHGB haplotypes, respectively. Two cultivars were homozygous for haplotype 54 of HGCand the remaining homozygous accessions presented putative recombinanthaplotypes.
Regarding the recombinant haplotypes, haplotype 18, which was the most frequent(frequency = 0.05), corresponded to a recombination between haplotypesfrom HGA and HGB (Additional file 6). Indeed, no allelespecifically assigned to HGC was present in this haplotype, which was always combinedwith alleles typical of both HGA and HGB haplotypes. Interestingly; haplotype 18 waspresent only in cultivars of K1 (40%) and K3 (60%) sub-populations classified mainlyas Eastern table grapes, with two cultivars being homozygous for this haplotype(Additional file 1). Furthermore, among the individualsthat presented at least one HGC haplotype mainly composed by cultivars of the K2sub-population, the only one Eastern table cultivar belonging to the K3sub-population was a combination with haplotype 18.
Certainly, LD pattern along VvTFL1A gene revealed three main blocks oflinked polymorphisms (Figure 5): linked polymorphismsspecific of HGA (Ins-2054, Y-1433, K-737, S-327, S-35, W270, Ins883) located in thepromoter, the first and the third introns of VvTFL1A gene; linkedpolymorphisms specific of HGB (Y-2237, K-2158, R-2124, R-2037, W-1991, R-1846, K-736,M224, R416, Y752 and M898) identified in the distal promoter region and in the threeintron regions; and linked polymorphisms specific of HGC (K-1704, R-1507, Ins-1389,Y-1362, Y-732, K-730, Y-19, R404, Ins422, R628) located in the promoter and thesecond intron of the VvTFL1A gene. Thus, haplogroups HGA, HGB, and HGC areconsistent with the existence of three ancestral alleles or evolutionary branchessupported by polymorphisms in the three LD blocks.

Linkage disequilibrium among polymorphisms in the gene VvTFL1A. LD plot based on R2 values for SNP and INDEL with frequency>5% were estimated according to Remington et al. [53]. The schematic representation of the VvTFL1A locusindicates ATG and stop codon position, exon regions represented by yellow boxesand UTR by grey boxes. Polymorphism classification into three LD blocks isrepresented by a colour code.
Candidate gene association
Considering the biological function established for the Arabidopsis TFL1gene as well as the phenological traits altered in the Carignan RRM somatic variant,the candidate gene association study was focused on those phenological and clustermorphological traits that could be related with its putative biological function ingrapevine. Association tests for VvTFL1A gene were run between the 38polymorphisms showing minor allele frequency ≥5% and each phenotypic trait.Rare genotyping classes (≤5%) were excluded from the association tests. Out ofthe 38 polymorphisms, only 8 presented significant associations (adjustedP-value ≤0.05) using either R or TASSEL with flowering time, berryweight and cluster width (Table 3, Figure 2B).
The strongest association was found between berry weight and SNP M-196(P = 1.4E−8) explaining 16% of the traitvariation. The highest association for flowering time and cluster width was foundwith Ins883 (P = 2.7E−4,P = 1.6E−5, respectively) that explained 10%and 13% of trait variation, respectively. Interestingly, Ins883 characteristic of HGAalso associated significantly (P ≤0.01) with berry weight(P = 5.7E−7). At a lesser extent, Ins422from HGC associated with the three traits explaining 9%, 6% and 7% of flowering time,berry weight and cluster width variations, respectively.
In order to determine whether the different associations detected were only due to LDor were the result of the particular effect of each polymorphism, the multi-locusmixed-model analysis was carried out. Flowering time showed the strongestassociations with polymorphisms characteristic of HGA and HGC under single-locusapproaches (Table 3). In the multi-locus analysis, thebest models to explain flowering time variation identified one and two polymorphismsbased on optimal mBonf and EBIC criteria, respectively (Table 4). The optimal models included Ins833 from HGA specific LD block andW1087 without LD with other polymorphisms and explained up to 16% of flowering timevariation. Association between W1087 and flowering time was not identified with thesingle-locus approach; the use of Ins883 as covariate in the model revealed W1087association. The remaining markers, not included in the model, had minor and/orredundant effects with those ones. Similarly, the best models explaining up to 25% ofberry weight variation included M-196 and Ins883 in agreement with the highestassociations detected with the single-locus approach. Instead, cluster widthvariation showing associations with polymorphisms within the three LD blocks undersingle-locus approaches was only explained by polymorphism Ins883 (HGA) after themulti-locus mixed-model analysis. The effect of polymorphisms characteristic of HGBwas minor and redundant with those of Ins883. Therefore, in addition to its highassociation (P ≤0.01) in single-locus analyses with three traits(flowering time, cluster width and berry weight), Ins883 was selected in the threebest multi-locus models explaining the variation of those traits. Besides, Ins883explains alone cluster width variation. These results highlight the major influenceof INDEL Ins883 in the possible role of VvTFL1A on phenology and clustertraits.
Phenotypic values related to major haplotypes
Based on haplotype network and association results, two molecular polymorphisms(Ins883 and Ins422) were selected that discriminated the three haplogroups (HGA:Ins883(G)6-Ins422(GA)7; HGB:Ins883(G)5-Ins422(GA)7; HGC:Ins883(G)5-Ins422(GA)8). The (G)6 allele ofIns883 was associated with late flowering time, high berry weight and large clusterwidth (Figure 6). All haplotypes in HGA presented the(G)6 allele, in contrast with haplotypes in HGB and HGC that containedthe (G)5 allele Regarding Ins422, the (GA)8 allele wasassociated with early flowering time, low berry weight and small cluster width(Figure 6). All haplotypes carrying the(GA)8 allele belonged to HGC, mainly represented by haplotype 60, whilehaplotypes in HGA and HGB contained the (GA)7 allele.

Effects of VvTFL1A INDEL Ins883 and Ins422 on flowering time, berry weight and clusterwidth. Box plots represent minimum and maximum (whisker), median (squaredot), and 25th and 75th percentiles (box) values.
As other polymorphisms appeared associated with these phenotypic traits, thephenotypic values were also analysed considering the three major haplotypes 32, 3 and60 from HGA, B and C, respectively. As an average, individuals presenting at leastone haplotype 32 exhibited late flowering, big berries and large cluster clearly incontrast to the phenotypic features of individuals containing at least one haplotype3 or 60 (Figure 7). This was more obvious when homozygousindividuals or heterozygous individuals for haplotypes 3 and 60 were taken intoaccount considering the absence of homozygous individuals for haplotype 60(Figure 7). The phenotypic value for the more frequentrecombinant haplotype 18 found in Eastern table cultivars was also analysed, whichpresented the (G)6 allele at the major INDEL Ins883 such as haplotypes ofHGA. Phenotypic values for flowering time, berry weight and cluster width forhaplotype 18 were slightly higher than those of individuals carrying haplotype 32(Figure 7).

Phenotypic value for flowering time, berry weight and cluster width relatedto major haplotypes. Phenotypic values of individuals containing themajor haplotypes H32, H3 and H60 and for the recombinant haplotype H18 atheterozygous or homozygous state. Box plots represent minimum and maximum(whisker), median (square dot), and 25th and 75th percentiles (box) values.
Discussion
Arabidopsis TFL1 plays a critical role in the specification of theinflorescence meristem and inflorescence architecture [54],[55]. This role seems to be conserved in other plant species [56] likely through a conserved regulatory pathway [24]. In grapevine, the previous identification of misexpression of theArabidopsis homolog VvTFL1A as the molecular cause of thereiteration of reproductive meristems (RRM) mutant [26], also supported the possible conservation of its biological function in thisspecies in agreement with previous results [57],[58]. Phenotypic characterization of the RRM plants showed that VvTFL1Aoverexpression was related to a delay in the time of anthesis and to an increase in thesize and branching pattern of the inflorescences [26], similar to the effects of TFL1 overexpression in transgenicArabidopsis [21]. To provide additional evidence on the involvement of VvTFL1A innatural variation for flowering time and inflorescence development and to identifynucleotide sequence polymorphisms that could be partially responsible for those traitsin grapevine, a genetic diversity analysis of this gene sequence and genetic associationstudies with those traits were carried out.
Nucleotide variations for VvTFL1A in the grapevine core collection analysed isrelatively high with an average of one polymorphic site every 50 nucleotides. However,only five out of the 70 polymorphisms detected are located in exonic regions and onlythree of them result in non-synonymous amino acid substitutions. This result is inagreement with the slight negative Tajima’s D values observed alongVvTFL1A coding sequences and suggests that the protein structure admits fewvariations. Reduction in overall level of nucleotide variation was also reported for theArabidopsis TFL1 gene when compared with other flowering genes [59]. Among the three non-synonymous polymorphisms identified in VvTFL1A,W13, located in first exon, had a very low frequency and was not considered for theassociation analyses. The two other, W1087 and M1094, are located in the fourth exon ina region of the protein responsible for the functional divergence between FT and TFL1 [60]; although the substituted amino acids do not correspond to conserved residues [60] and the SNP did not associate with inflorescence related traits undersingle-locus models. However, W1087 was selected by the multi-locus mixed-model analysisto explain part of flowering time variation together with Ins883 suggesting a possiblefunctional effect of this SNP in this trait. Regarding the 17 polymorphisms found inintron regions, two INDEL (Ins422 and Ins883) showed significant association withflowering and cluster trait variation. INDEL Ins422 is located in intron 2 andcorresponds to a microsatellite sequence of GA repetitions; while INDEL Ins883 islocated in intron 3 and corresponds to a G nucleotide repetition. The 48 remainingVvTFL1A polymorphisms identified in the core collection were locatedupstream of the translation start codon and included four INDEL. No traces of theHatvine1-rrm transposon were detected in the promoter of VvTFL1A inthe whole core collection which demonstrates the specificity of the insertion eventcausing the Carignan RRM mutant phenotype [26].
Nucleotide polymorphisms in VvTFL1A LD blocks that discriminate the threehaplogroups displayed differential association with cluster traits under linearregression models. Among all traits analysed, polymorphic sites characteristic of HGAand HGC haplogroups explained part of the phenotypic variation for flowering time, berryweight and cluster width. In the same way, polymorphisms specific of HGB associated withcluster width as well as polymorphisms from HGA. These results suggest that variation atVvTFL1A has an effect on flowering time, berry weight and cluster width withdifferent alleles having differential effects on the traits. Interestingly, bothflowering time delay and cluster width increase were observed in the phenotypiccharacterization of the RRM somatic variant related to VvTFL1A overexpression.Unfortunately, berry size was not measured in that study [26].
Among all the polymorphic sites tested, insertion Ins883 discriminating HGA from HGB andHGC explained alone part of flowering time, berry weight and cluster width variations.According to the multi-locus analysis, Ins833 explained up to 16%, 13% and 25% offlowering time, cluster width and berry weight variation in the best models, being theonly polymorphism contributing to berry weight variation. INDEL occurring infunctionally important regions of genes could affect gene function, through geneexpression modification [61] or RNA structure alterations [62]. However, a preliminary VvTLF1A RT qPCR expression analysis carriedout in young inflorescences of the cultivars of the core collection did not reveal anyassociation between gene expression variation and the VvTFL1A polymorphisms(data not shown). Likewise, no clear correlation (Pearson’s r <0.28) betweenVvTFL1A expression and phenotypic traits was identified (data not shown).Nevertheless, these negative results do not discard a possible role of this intronsequences in transcriptional or posttranscriptional processes given the difficulties incarrying out transcriptional comparisons among different genotypes with differentflowering behaviour. Analysis of maize TFL1 homologs expression in differenttissues and developmental stages showed the existence of differential transcriptprocessing [63]. In fact, in a preliminary study, the existence of alternatively spliced RNAforms was detected for the first and the second introns of VvTFL1A (data notshown). Further research will be required to demonstrate any functional role of thisalternative splicing as well as its relationship with the described VvTFL1AIns833 polymorphism. In any case, further association analyses using larger samples andspecific segregation analyses will be required to confirm the detected associations.
Together with Ins883, M-196 and W1087 without LD with other VvTFL1Apolymorphisms explain part of berry weight and flowering time variation according tomulti-locus analysis, respectively. In contrast to Ins883 that discriminates haplotypesof HGA from those of HGB and HGC, M-196 and W1087 corresponded to mutationsdifferentiating haplotypes within the HGA haplogroup (Figure 4). The M-196 base change located in the proximal promoter and the W1087non-synonymous substitution in the fourth exon of VvTFL1A might representrelevant structural modifications at the promoter and the protein sequence,respectively, likely affecting VvTFL1A function in a non-redundant way withIns883. Moreover, in silico analysis using SIFT program(http://sift.jcvi.org/) predicts that substitution of T by S at position144 of the VvTFL1A sequence affects protein function with a score of 0.04 based on thealignment of 240 closely related sequences.
VvTFL1A haplotype network differentiates three haplogroups of closely relatedhaplotypes. Each HG is represented by a high frequency haplotype, haplotypes 32 for HGA,3 for HGB and haplotype 60 from HGC. Consistent with the results of the associationanalyses, individuals containing haplotype 32 of HGA, exhibited late flowering, largecluster width and larger berries. Interestingly, most of the cultivars of K1 and K3classified to table or table/wine uses, characterized by these phenotypic features [6], present HGA haplotypes. This relationship is also true for accessionscarrying the recombinant haplotype 18. Indeed, haplotype 18 contains Ins883 insertionpresent in HGA haplotypes and mostly present in Eastern table cultivars belonging to K1and K3 genetic groups. The fact that cultivars carrying haplotype 18 display lateflowering, large cluster width and larger berries supports a clear relationship betweenIns883 polymorphism and the eastern table cluster characteristics. In contrast,individuals containing haplotype 60 of HGC with Ins422 insertion exhibited earlyflowering, shorter cluster width and smaller berries. Consistently, HGC haplotypes areenriched in Western wine grape cultivars mostly belonging to K2 genetic group, which areknown to display those cluster and berry features [6]. Finally, HGB haplotypes do not contain Ins422 or Ins883 insertions. Thephenotype of individuals containing haplotype 3 (most frequent within HGB) is similar tosome extent to that of cultivars carrying haplotype 60 (HGC). Consistently with thisphenotype, HGB haplotypes are mostly present in cultivars for wine use. Because nohomozygous individuals were observed for haplotype 60 in the core collection, thephenotypic effect of this haplotype is supposed to be much stronger in homozygous state,which suggests that haplotype 60, related to extreme phenology and clustercharacteristics, could be less favored in cultivars in homozygous state than haplotype3. Interestingly, partial sequencing of VvTFL1A in 20 V.v ssp.sylvestris plants from the Iberian Peninsula identified wild haplotypessimilar to haplotype 60 and belonging to HGC (data not shown). These data could indicatea western origin for haplotypes of HGC.
Conclusions
Three major VvTFL1A haplogroups were identified in cultivated grapevines basedon the presence of three main polymorphism LD blocks. These haplogroups are associatedto characteristic phenotypic values for flowering time, cluster width and berry size.Phenotypic differences between VvTFL1A haplogroups are consistent with theclassification of grapevine phenotypic diversity in three different morphotypes proposedby Negrul [12] and could result from the use of different genetic pools in grapevinedomestication and/or the existence of different selection pressures on the developmentof table and wine cultivars. Polymorphic markers identifying haplogroups can also berelevant in marker-assisted breeding programs addressing the improvement of clusterstructure and berry size.
Additional files
References
Mc Govern PE, Glusker DL, Exner LJ, Voigt MM: Neolithic resinated wine. Nature. 1996, 381: 480-481. 10.1038/381480a0.
Levadoux L: Les population sauvage et cultivées deVitis viniferaL. Annales de l' Amélioration des Plantes 1956, 6:59–118.,
Mc Govern PE: Ancient wine. The search for the origins of viniculture. 2003, Princeton University Press, Princeton, NJ
Aradhya MK, Dangl GS, Prins BH, Boursiquot JM, Walker MA, Meredith CP, Simon CJ: Genetic structure and differentiation in cultivated grape,Vitis viniferaL. Genet Res 2003, 81:179–192.,
Arroyo-Garcia R, Ruiz-Garcia L, Bolling L, Ocete R, Lopez MA, Arnold C, Ergul A, Soylemezoglu G, Uzun HI, Cabello F, Ibanez J, Aradhya MK, Atanassov A, Atanassov I, Balint S, Cenis JL, Costantini L, Goris-Lavets S, Grando MS, Klein BY, McGovern PE, Merdinoglu D, Pejic I, Pelsy F, Primikirios N, Risovannaya V, Roubelakis-Angelakis KA, Snoussi H, Sotiri P, Tamhankar S, et al: Multiple origins of cultivated grapevine (Vitis viniferaL. ssp. sativa) based on chloroplast DNA polymorphisms. Mol Ecol 2006, 15:3707–3714.,
Bacilieri R, Lacombe T, Le Cunff L, Di Vecchi-Staraz M, Laucou V, Genna B, Peros JP, This P, Boursiquot JM: Genetic structure in cultivated grapevines is linked to geography and humanselection. BMC Plant Biol. 2013, 13: 25-10.1186/1471-2229-13-25.
Emanuelli F, Lorenzi S, Grzeskowiak L, Catalano V, Stefanini M, Troggio M, Myles S, Martinez-Zapater JM, Zyprian E, Moreira FM, Grando MS: Genetic diversity and population structure assessed by SSR and SNP markers in alarge germplasm collection of grape. BMC Plant Biol. 2013, 13: 39-10.1186/1471-2229-13-39.
Myles S, Boyko AR, Owens CL, Brown PJ, Grassi F, Aradhya MK, Prins B, Reynolds A, Chia JM, Ware D, Bustamante CD, Buckler ES: Genetic structure and domestication history of the grape. Proc Natl Acad Sci U S A. 2011, 108: 3530-3535. 10.1073/pnas.1009363108.
Myles S, Chia JM, Hurwitz B, Simon C, Zhong GY, Buckler E, Ware D: Rapid genomic characterization of the genus vitis. PLoS One. 2010, 5: e8219-10.1371/journal.pone.0008219.
Branas J: Viticulture. 1974, Paul Dehan, Montpellier
Boursiquot JM, Dessup M, Rennes C: Distribution des principaux caractères phénologiques, agronomiques et technologiques chezVitis viniferaL. Vitis 1995, 34:31–35.,
Negrul AM: Origin and classification of cultured grape. The Ampelography of the USSR. Edited by: Baranov A, Kai YF, Lazarevski MA, Palibin TV, Prosmoserdov NN, Baranov A, KaiYF, Lazarevski MA, Palibin TV, Prosmoserdov NN. 1946, Pischepromizdat, Moscow, 159-216.
Molitor D, Rothmeier M, Behr M, Fischer S, Hoffman L, Evers D: Crop cultural and chemical methods to control grey mould on grapes. Vitis. 2011, 50: 81-87.
Vail ME, Marois JJ: Grape cluster architecture and the susceptibility of berries to Botrytiscinerea. Phytopatholog. 1991, 81: 188-191. 10.1094/Phyto-81-188.
Shavrukov YN, Dry IB, Thomas MR: Inflorescence and bunch architecture development inVitis viniferaL. Aust J Grape Wine Res 2003, 10:116–124.,
Tello J, Ibáñez J: Evaluation of indexes for the quantitative and objective estimation of grapevinebunch compactness. Vitis. 2014, 53: 9-16.
Bradley D, Ratcliffe O, Vincent C, Carpenter R, Coen E: Inflorescence commitment and architecture in Arabidopsis. Science 1997, 275:80–83.,
Shannon S, Meeks-Wagner DR: A mutation in the Arabidopsis TFL1 gene affects inflorescence meristem development. Plant Cell 1991, 3:877–892.
Prusinkiewicz P, Erasmus Y, Lane B, Harder LD, Coen E: Evolution and development of inflorescence architectures. Science. 2007, 316: 1452-1456. 10.1126/science.1140429.
Schoentgen F, Saccoccio F, Jolles J, Bernier I, Jolles P: Complete amino acid sequence of a basic 21-kDa protein from bovine braincytosol. Eur J Biochem. 1987, 166: 333-338. 10.1111/j.1432-1033.1987.tb13519.x.
Hanano S, Goto K: Arabidopsis TERMINAL FLOWER1 is involved in the regulation of flowering time andinflorescence development through transcriptional repression. Plant Cell. 2011, 23: 3172-3184. 10.1105/tpc.111.088641.
Weigel D, Alvarez J, Smyth DR, Yanofsky MF, Meyerowitz EM: LEAFY controls floral meristem identity in Arabidopsis. Cell 1992, 69:843–859.,
Parcy F, Nilsson O, Busch MA, Lee I, Weigel D: A genetic framework for floral patterning. Nature. 1998, 395: 561-566. 10.1038/26903.
Liu C, Teo ZW, Bi Y, Song S, Xi W, Yang X, Yin Z, Yu H: A conserved genetic pathway determines inflorescence architecture in Arabidopsisand rice. Dev Cell. 2013, 24: 612-622. 10.1016/j.devcel.2013.02.013.
Carmona MJ, Cubas P, Calonje M, Martinez-Zapater JM: Flowering transition in grapevine (Vitis vinifera L.). Can J Bot 2007, 85:701–711.,
Fernandez L, Torregrosa L, Segura V, Bouquet A, Martinez-Zapater JM: Transposon-induced gene activation as a mechanism generating cluster shape somaticvariation in grapevine. Plant J. 2010, 61: 545-557. 10.1111/j.1365-313X.2009.04090.x.
Barnaud A, Lacombe T, Doligez A: Linkage disequilibrium in cultivated grapevine,Vitis vinifera L. Theor Appl Genet 2006, 112:708–716.,
Lacombe T, Boursiquot JM, Laucou V, Di Vecchi-Staraz M, Peros JP, This P: Large-scale parentage analysis in an extended set of grapevine cultivars (Vitis vinifera L.). Theor Appl Genet 2013, 126:401–414.,
Adam-Blondon AF, Roux C, Claux D, Butterlin G, Merdinoglu D, This P: Mapping 245 SSR markers on the Vitis vinifera genome: a tool for grapegenetics. Theor Appl Genet. 2004, 109: 1017-1027. 10.1007/s00122-004-1704-y.
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES: TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 2007, 23: 2633-2635. 10.1093/bioinformatics/btm308.
Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R: DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003, 19: 2496-2497. 10.1093/bioinformatics/btg359.
Nei M: Molecular Evolutionary Genetics. 1987, Columbia University Press, New York, NY
Watterson GA: On the number of segregating sites in genetical models without recombination. Theor Pop Biol. 1975, 7: 256-276. 10.1016/0040-5809(75)90020-9.
Tajima F: Statistical method for testing the neutral mutation hypothesis by DNApolymorphism. Genetics. 1989, 123: 585-595.
Thornsberry JM, Goodman MM, Doebley J, Kresovich S, Nielsen D, Buckler ES: Dwarf8 polymorphisms associate with variation in flowering time. Nat Genet. 2001, 28: 286-289. 10.1038/90135.
Yu J, Pressoir G, Briggs WH, Vroh Bi I, Yamasaki M, Doebley JF, McMullen MD, Gaut BS, Nielsen DM, Holland JB, Kresovich S, Buckler ES: A unified mixed-model method for association mapping that accounts for multiplelevels of relatedness. Nat Genet. 2006, 38: 203-208. 10.1038/ng1702.
Laucou V, Lacombe T, Dechesne F, Siret R, Bruno JP, Dessup M, Dessup T, Ortigosa P, Parra P, Roux C, Santoni S, Vares D, Peros JP, Boursiquot JM, This P: High throughput analysis of grape genetic diversity as a tool for germplasmcollection management. Theor Appl Genet. 2011, 122: 1233-1245. 10.1007/s00122-010-1527-y.
Pritchard JK, Stephens M, Donnely P: Inference of population structure using multilocus genotype data. Genetics. 2000, 155: 945-959.
Evanno G, Regnaut S, Goudet J: Detecting the number of clusters of individuals using the software STRUCTURE: asimulation study. Mol Ecol. 2005, 14: 2611-2620. 10.1111/j.1365-294X.2005.02553.x.
Vigouroux Y, Glaubitz JC, Matsuoka Y, Goodman MM, Sanchez GJ, Doebley J: Population structure and genetic diversity of New World maize races assessed byDNA microsatellites. Am J Bot. 2008, 95: 1240-1253. 10.3732/ajb.0800097.
Fournier-Level A, Le Cunff L, Gomez C, Doligez A, Ageorges A, Roux C, Bertrand Y, Souquet JM, Cheynier V, This P: Quantitative genetic bases of anthocyanin variation in grape (Vitis vinifera L.ssp. sativa) berry: a quantitative trait locus to quantitative trait nucleotideintegrated study. Genetics. 2009, 183: 1127-1139. 10.1534/genetics.109.103929.
Kang HM, Zaitlen NA, Wade CM, Kirby A, Heckerman D, Daly MJ, Eskin E: Efficient control of population structure in model organism associationmapping. Genetics. 2008, 178: 1709-1723. 10.1534/genetics.107.080101.
Segura V, Vilhjalmsson BJ, Platt A, Korte A, Seren U, Long Q, Nordborg M: An efficient multi-locus mixed-model approach for genome-wide association studiesin structured populations. Nat Genet. 2012, 44: 825-830. 10.1038/ng.2314.
Qin ZS, Niu T, Liu JS: Partition-ligation-expectation-maximization algorithm for haplotype inference withsingle-nucleotide polymorphisms. Am J Hum Genet. 2002, 71: 1242-1247. 10.1086/344207.
Stephens M, Donnelly P: A comparison of bayesian methods for haplotype reconstruction from populationgenotype data. Am J Hum Genet. 2003, 73: 1162-1169. 10.1086/379378.
Martin DP, Williamson C, Posada D: RDP2: recombination detection and analysis from sequence alignments. Bioinformatics. 2005, 21: 260-262. 10.1093/bioinformatics/bth490.
Maynard Smith J: Analyzing the mosaic structure of genes. J Mol Evol. 1992, 34: 126-129.
Posada D, Crandall KA: Evaluation of methods for detecting recombination from DNA sequences: computersimulations. Proc Natl Acad Sci U S A. 2001, 98: 13757-13762. 10.1073/pnas.241370698.
Boni MF, Posada D, Feldman MW: An exact nonparametric method for inferring mosaic structure in sequencetriplets. Genetics. 2007, 176: 1035-1047.
Fournier-Level A, Lacombe T, Le Cunff L, Boursiquot JM, This P: Evolution of the VvMybA gene family, the major determinant of berry colour incultivated grapevine (Vitis vinifera L.). Heredity (Edinb). 2010, 104: 351-362. 10.1038/hdy.2009.148.
Bandelt HJ, Forster P, Rohl A: Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999, 16: 37-48. 10.1093/oxfordjournals.molbev.a026036.
Jaillon O, Aury JM, Noel B, Policriti A, Clepet C, Casagrande A, Choisne N, Aubourg S, Vitulo N, Jubin C, Vezzi A, Legeai F, Hugueney P, Dasilva C, Horner D, Mica E, Jublot D, Poulain J, Bruyère C, Billault A, Segurens B, Gouyvenoux M, Ugarte E, Cattonaro F, Anthouard V, Vico V, Del Fabbro C, Alaux M, Di Gaspero G, Dumas V, et al: The grapevine genome sequence suggests ancestral hexaploidization in majorangiosperm phyla. Nature. 2007, 449: 463-467. 10.1038/nature06148.
Remington DL, Thornsberry JM, Matsuoka Y, Wilson LM, Whitt SR, Doebley J, Kresovich S, Goodman MM, Buckler ES: Structure of linkage disequilibrium and phenotypic associations in the maizegenome. Proc Natl Acad Sci U S A. 2001, 98: 11479-11484. 10.1073/pnas.201394398.
Jack T: Molecular and genetic mechanisms of floral control. Plant Cell. 2004, 16 (Suppl 1): S1-S17.
Ratcliffe OJ, Amaya I, Vincent CA, Rothstein S, Carpenter R, Coen ES, Bradley DJ: A common mechanism controls the life cycle and architecture of plants. Development. 1998, 125: 1609-1615.
McGarry RC, Ayre BG: Manipulating plant architecture with members of the CETS gene family. Plant Sci. 2012, 188–189: 71-81.
Boss PK, Sreekantan L, Thomas MR: A grapevine TFL1 homologue can delay flowering and alter floral development whenoverexpressed in heterologous species. Funct Plant Biol. 2006, 33: 31-41. 10.1071/FP05191.
Carmona MJ, Calonje M, Martinez-Zapater JM: The FT/TFL1 gene family in grapevine. Plant Mol Biol. 2007, 63: 637-650. 10.1007/s11103-006-9113-z.
Olsen KM, Womack A, Garrett AR, Suddith JI, Purugganan MD: Contrasting evolutionary forces in the Arabidopsis thaliana floral developmentalpathway. Genetics. 2002, 160: 1641-1650.
Ahn JH, Miller D, Winter VJ, Banfield MJ, Lee JH, Yoo SY, Henz SR, Brady RL, Weigel D: A divergent external loop confers antagonistic activity on floral regulators FTand TFL1. Embo J. 2006, 25: 605-614. 10.1038/sj.emboj.7600950.
Warren ST, Zhang F, Licameli GR, Peters JF: The fragile X site in somatic cell hybrids: an approach for molecular cloning offragile sites. Science. 1987, 237: 420-423. 10.1126/science.3603029.
Collins FS, Drumm ML, Cole JL, Lockwood WK, Vande Woude GF, Iannuzzi MC: Construction of a general human chromosome jumping library, with application tocystic fibrosis. Science. 1987, 235: 1046-1049. 10.1126/science.2950591.
Danilevskaya ON, Meng X, Hou Z, Ananiev EV, Simmons CR: A genomic and expression compendium of the expanded PEBP gene family frommaize. Plant Physiol. 2008, 146: 250-264.
Acknowledgments
We would like to thank Dr Javier Ibanez and Dr Stéphanie Mariette for helpfuldiscussions; Virginia Rodriguez for her assistance for DNA extraction; Dr CarlosAlonso Blanco for his helpful comments and critical reading of the manuscript; and DrVincent Segura is acknowledged for assistance using MLMM software and criticalreading of the manuscript. This work was funded by grant BIO2008-03892 andBIO2011-026229 from the MICINN and MINECO of Spain to JMM-Z. Networking activitieswere also funded by COST Action FA1003 “East–west Collaboration forGrapevine Diversity Exploration and Mobilization of Adaptive Traits forBreeding”. JT was recipient of a fellowship from the MICINN.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
LF, PT, LT and JMM-Z conceived the study. LLC, TL, JMB, SL and PT created andcharacterized the core collection. LF and GB generated and characterized the sequencedata. LF, LLC, JT, AFL analysed the data. LF and JMM-Z wrote the paper with the input ofall authors. All authors read and approved the final manuscript.
Electronic supplementary material
12870_2014_209_MOESM1_ESM.xls
Additional file 1: List of the 140 individuals of the core collection used in this study.Variety name, pedigrees when available, bred cultivars and classificationaccording to origin and use are indicated. Classification of individualsaccording to K1, K2, K3 and admixed groups is described (see materials andmethods section) as well as haplotype combination and correspondinghaplogroups. [N.D. not determined]. (XLS 64 KB)
12870_2014_209_MOESM3_ESM.xls
Additional file 3: Phenotypic value presented by the 140 individuals of the core collection forthe 10 traits analysed in this study.(XLS 48 KB)
12870_2014_209_MOESM4_ESM.jpeg
Additional file 4: Distributions of the ten phenotypic traits analysed in the corecollection. Red line corresponds to the expected normaldistribution. (JPEG 840 KB)
12870_2014_209_MOESM5_ESM.xls
Additional file 5: Genotype of the 140 core collection accessions for the 70 polymorphismsof VvTFL1A. The standart IUB’s nomenclature for nucleic acid has been used tocode SNPs. For INDEL A = deletion, C = insertion,M = heterozygous, and for triallelic microsatellites third allelehas been codify as T. [n/a = not available]. (XLS 169 KB)
12870_2014_209_MOESM6_ESM.xls
Additional file 6: Sequences of the 53 haplotypes found for the VvTFL1A gene. Only polymorphic sites are indicated. Frequency and recombinanthaplotypes are indicated. (XLS 90 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Fernandez, L., Le Cunff, L., Tello, J. et al. Haplotype diversity of VvTFL1A gene and association with cluster traits ingrapevine (V. vinifera). BMC Plant Biol 14, 209 (2014). https://doi.org/10.1186/s12870-014-0209-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-014-0209-3