
Abstract
Background
Seed dimorphism has been thought to be a bet-hedging strategy that helps plants survive in the disturbed environment and has been widely studied for its ecological adaptation mechanism. Many studies showed that seed-associated microorganisms play an important role in enhancing plant fitness, but information regarding endophytic bacteria associated with dimorphic seeds is limited. This study explores the influence of seed coat structure and seed phytochemical properties on the community composition and diversity of endophytic bacteria of dimorphic seeds of Suaeda glauca. In this study, we used 16S rRNA high-throughput gene sequencing method to compare the community composition and bacterial diversity between brown and black seeds of Suaeda glauca.
Results
A significant difference was observed in seed coat structure and phytochemical properties between brown and black seeds of S. glauca. Total 9 phyla, 13 classes, 31 orders, 53 families, 102 genera were identified in the dimorphic seeds. The dominant phyla were Proteobacteria, Firmicutes, and Actinobacteria. The results showed that seed dimorphism had little impact on the diversity and richness of endophytic bacterial communities but significantly differs in the relative abundance of the bacterial community between brown and black seeds. At the phylum level, Actinobacteria tend to be enriched significantly in brown seeds. At the genus level, Rhodococcus, Ralstonia, Pelomonas and Bradyrhizobium tend to be enriched significantly in brown seeds, while Marinilactibacillus was mainly found in black seeds. Besides, brown seeds harbored a large number of bacteria with plant-growth-promoting traits, whereas black seeds presented bacteria with enzyme activities (i.e., pectinase, cellulolytic and xylanolytic activities).
Conclusion
The endophytic bacterial community compositions were significantly different between dimorphic seeds of Suaeda glauca, and play an important role in the ecological adaptation of dimorphic seeds by performing different biological function roles. The endophytic bacterial communities of the dimorphic seeds may be influenced mainly by the seed coat structureand partly by the seed phytochemical characteristics. These findings provide valuable information for better understanding of the ecological adaptation strategy of dimorphic seeds in the disturbed environment.
Similar content being viewed by others

Background
Seed dimorphism is thought to be a bet-hedging strategy where plant species produce two distinct types of seed within the same plant [1, 2], and usually associated with differences in seed size, shape, color and absence/presence of seed appendages [3,4,5]. Seed dimorphism is a common phenomenon in the halophyte such as Suaeda spp., observed in S. glauca [6], S. salsa [7,8,9,10], S. acuminate [11], S. aralocaspica [12, 13], S. corniculata sp. mongolica [14], S. splendens [2] and S. moquinii [15].
In the past few years, researchers have studied the ecological adaptation mechanism of Suaeda spp. related to seed dimorphism, which primarily focused on seed ecological behaviors including seed germination/dormancy traits [7, 11, 12, 15], competitive abilities [8] and phenotypic plasticity [10, 14]; seed physiological properties including seed coat structure [16] and seed phytochemical characteristics (ion content, nutrient composition) [16,17,18]; as well as transcriptome analysis of dimorphic seeds during germination [13]. However, with the emergence of the concept of the “holobiont” [19, 20], plants are no longer viewed as monogenetic individuals but as polygenetic entities, where the microbiota plays an important role in the ecological adaptation of plants [21]. Seed endophytic bacteria have been reported to influence seed germination [22, 23], seed preservation [24], seedling establishment and development [23, 25,26,27], as well as play an important role in enhancing plant fitness [28]. Besides enhancing plant fitness, they also help the plant to tolerate stress conditions [29].
Numerous studies have shown that the composition of seed endophytic microbiota not only influenced by soil factors [30] and plant genotype [31,32,33], but also by seed phytochemical traits (including antioxidants content, starch content and nutrition component) [34, 35] and seed physiological characteristics (such as salt tolerance) [33]. Interestingly, previous studies showed that the dimorphic seeds of Suaeda spp. exhibit significant differences in seed phytochemical properties including fatty acid composition [18], total unsaturated fatty acids content [36], total phenols, flavonoids, carotenoids content [17], soluble sugar, soluble protein, total nitrogen, total phosphorus, inorganic ion content [16], seed coat structure [16] and seed salt-tolerance [7, 9, 37,38,39]. So, we hypothesized that differential endophytic bacterial communities can be detected between two distinct types of seeds of Suaeda spp., and may perform different bacterial function roles.
Suaeda glauca Bunge, a common annual halophyte, produces two distinct types of fruit (large utricles vs small utricles) and exhibits different germination behavior [6]. The present study aims to explore as follows:
(1) observe the morphological structure difference between the dimorphic seeds of S. glauca, (2) investigate the difference in the seed phytochemical characteristics (soluble protein, soluble starch, soluble sugar, fat content, and total phenols) between the dimorphic seeds of S. glauca, (3) compare the difference in the composition of the endophytic bacterial communities between the two distinct seed types, (4) provide useful information to understand the ecological role of the seed-associated endophytic bacterial community and to understand the ecological adaptation strategy for dimorphic seeds.
Results
Seed morphology and phytochemical properties
The spatial site distribution pattern of dimorphic utricles (seeds) of S. glauca was observed in the mother plant grown in the same natural environment. Glomerules of S. glauca, usually inserted near the base of the leaves, usually consisted of 1–3 flowers. The glomerules at the top of the branches were one flower, which forms a large utricle, while the glomerules at the middle and lower axils of the branches usually consisted of three flowers, which produces three utricles. The two large utricles were usually located on the two lateral sides of the glomerules and the small utricles in the middle (Fig. 1a).

Suaeda glauca. a Positions of large utricles and small utricles of S. glauca on a branch in fruiting stage. b Fruit and seed morphological characteristics of large utricles in mature stage. c Fruit and seed morphological characteristics of small utricles in mature stage. d Morphological characteristics of seed coat of brown seed. e Morphological characteristics of exotesta of black seed. f Morphological characteristics of endotesta of black seed
Indoor observations showed that S. glauca produced two types of utricles: large utricles, pentagram-shaped, with five expanded tepals in the fruiting stage, which surround and protect brown seeds (Fig. 1b); whereas small utricles, spheroid-shaped, with non-expanded tepals in the fruiting stage, which protect black seeds (Fig. 1c). At maturity, the brown seed had only a soft and membranous seed coat (Fig. 1d). On the contrary, the black seed had a rigid cuticle exotesta (Fig. 1e) and a soft membranous endotesta (Fig. 1f). The hard shell of the exotesta resists strong inward pressure at maturity. The results showed significant differences in fruit size, seed size, seed coat structure between brown and black seeds.
The phytochemical properties of the seeds were tested and the results showed a very significant difference in protein, soluble starch, soluble sugar and total phenolic content between brown and black seeds. As shown in Fig. 2, protein, soluble starch, soluble sugar and total phenolic content in brown seeds were higher than those in black seeds. In contrast, the fat content of brown seeds was lower than that of black seeds.

Comparison analysis of five seed phytochemical properties between brown seeds and black seeds. (a) Protein content; (b) Soluble starch content; (c) Soluble sugar content; (d) Fat content; (e) Total phenolic content. Results are presented as means of three replicates and vertical bars indicate standard deviations of the means. Different letters indicate significant differences between two types seed according to Student’s T-test at p < 0.05. Br: brown seeds; Bl: black seeds
Characteristics of 16S rRNA gene sequencing and alpha-diversity
The surface-sterilized seeds placed on TSA agar plates showed no microbial growth (Additional file 1: Fig. S1). It was therefore assumed that the bacteria identified from all seed samples were endophytic, or very closely associated with seed epidermal tissue.
The bacterial 16S rRNA gene (V5-V6 regions) was sequenced to characterize the endophytic bacterial community composition between brown and black seeds. After the quality filtration of raw data, a total of 114,770 high-quality sequences were obtained from 6 samples. The mean sequence number per sample was 19,128 ± 5604, ranging from11,433 to 24,822 (Table 1) (each sample = 0.20 g seeds weight and each group = 3 samples). The sequence numbers, coverage, the number of operational taxonomic units (OTUs), richness, and diversity indices for each sample were presented in Table 1. The high-quality sequences were clustered into 175 OTUs (at 97% sequence identity) and each library contains different phylogenetic OTUs ranging from 29 to 120.
The diversity and richness indices of all samples were calculated to illustrate the complexity of each sample (Table 1). The diversity of each sample was obtained by using Shannon index and Simpson index. The Shannon index ranged from 1.125 to 2.596, while the Simpson index ranged from 0.157 to 0.409. The Chao index and ACE index were usually used to express the richness of each sample. Chao index ranged from 55.250 to 112.500, while ACE index ranged from 55.233 to 121.687. In total, the values of ACE, Chao, Simpson and Shannon varied among six samples. However, no significant differences in all alpha-diversity estimators were observed between brown and black seed populations (p > 0.05, student’s t-test; Fig. 3). The Good’s coverage value per sample was > 0.99 (from 0.997 to 0.999), indicating that the information was sufficient to reveal most of the bacterial communities in each sample.

Comparison of the richness and diversities of bacterial OUT level between brown seeds and black seeds. (a) ACE index; (b) Chao index; (c) Shannon index; (d) Simpson index. Results are presented as means of three replicates and vertical bars indicate standard deviations of the means. Different letters indicate significant differences between two types seed according to Student’s T-test at p < 0.05. Br: brown seeds; Bl: black seeds
Taxonomic composition of endophytic bacterial community
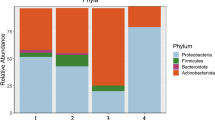
The 16S rRNA gene sequence results showed that the microbial communities of all seed samples covered 9 phyla, 13 classes, 31 orders, 53 families, 102 genera and 137 species. Sequences that were less than 1.0% in abundance were merged into “others”. The relative abundant phyla in all samples were Proteobacteria (58.0%), Firmicutes (34.1%), Actinobacteria (6.6%), Bacteroidetes (1.1%) and others (0.03%) (Fig. 4a).

The bacterial community in all seed samples at phylum level (a), genus level (b). The comparison (c) of the endophytic bacterial communities at genus level between brown seeds and black seeds. The community composition of endophytic bacteria of brown seeds (d), and black seeds (e) at genus level, respectively. 3 samples in each group. Each sample = 0.20 g seeds
There were 19 genera with a relative abundance of > 1.0% in at least one of the six samples (Additional file 3: Table S1). Of all 19 genera, 8 classified genera (average relative abundance more than 1.0% at genus level) were Kushneria (27.4%), Halomonas (17.2%), Bacillus (16.5%), Marinilactibacillus (13.6%), Rhodococcus (6.1%), Ralstonia (5.9%), Pelomonas (2.3%) and Bradyrhizobium (1.0%) (Fig. 4b). The Venn diagram (Fig. 4c) at the genus level was also constructed to further identify the shared genus present in brown and black seeds. The results suggest that 44 genera were shared between the two groups. The core genera present in the dimorphic seeds of S. glauca were Kushneria (24.0–30.8%), Bacillus (13.1–19.9%) and Holomonas (1.0–33.5%) (Fig. 4d and e). In addition, more bacterial taxa were found in brown seeds (53 genera), compared to black seeds (5 genera) (Fig. 4c).
Community analysis of endophytic bacterial compositions
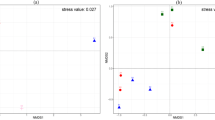
Principal coordinate analysis (PCoA) was used to determine the similarity of the endophytic bacterial communities between brown and black seeds. The PCoA biplot revealed that all seed samples show a clear separation across PC1 axis, except for Br_2 sample, which coincides with the different seed types (Fig. 5a). The results showed that all black seed samples were grouped in the left of the biplot and presented better similarity in their endophytic bacterial communities, however, there were considerable differences in endophytic bacterial communities of brown seeds. In total, black and brown seed samples were separated along the first axis (PC1), explaining 34.8% and the second axis (PC2) of explaining 19.9%.

The principal co-ordinates analysis (PCoA) (a) and hierarchical clustering tree (b) of the bacterial community at OTU level in the two groups. 3 samples in each group. Each sample = 0.20 g seeds. The hierarchical clustering tree was calculated using the unweighted unifrac method, and the relationship between samples was determined by the complete clustering method
In addition, the hierarchical clustering of the endophytic bacterial communities (OTUs level at 97% similarity) in both brown and black seeds was conducted based on the unweighted uni-fraction method (Fig. 5b), which reflects that the bacterial communities appeared different between the two seed types. The result of the hierarchical clustering was similar to that of PCoA analysis.
Interestingly, these results also revealed high heterogeneity within the bacterial communities associated with brown seeds. In contrast, the bacterial community composition of black seeds was very similar in all treatments. Hence, it was concluded that black seeds offer a more stable and less easily disturbed environment compared to brown seeds.
Differences in the endophytic community compositions
Linear Discriminant Analysis Effect Size (LEfSe) analysis was used to determine the significant differences of the bacterial communities between brown and black seeds. Illumina MiSeq sequencing data demonstrated that the relative abundances of bacterial taxa have displayed significant differences between brown and black seeds at the phylum, class, order, family and genus level (Fig. 6).

LEfSe analyses of bacterial community in the brown seeds and black seeds. Br: brown seeds; Bl: black seeds. a Histogram of the microbiota of brown seeds and black seeds with a threshold value of 4; P < 0.05 considered significant. b Cladogram representing the abundance of the taxa in the brown seeds and black seeds
At the phylum level, the dominant phyla (relative abundance > 5.0% at least in one sample) were Proteobacteria (Br = 57.4 ± 19.9% and Bl = 58.6 ± 22.8%), Firmicutes (Br = 27.3 ± 30.9% and Bl = 40.8 ± 22.5%) and Actinobacteria (Br = 13.0 ± 13.2% and Bl = 0.2 ± 0.1%). Of them, Proteobacteria was the most dominant phylum in both brown and black seeds. The differences in the relative abundance of Actinobacteria (LDA = 4.790, p = 0.0495) exhibited a significant difference between brown and black seeds. Compared with black seeds, Actinobacteria was significantly enriched in brown seeds. On the contrary, the relative abundance of Firmicutes in brown seeds was less than that of black seeds, but there was no significant difference between the two groups. Detailed the relative abundance information of endophytic bacteria at phylum level in brown seeds and black seeds can be found in the Additional file 4: Table S2.
In the observed 102 identified genera, the dominant genera (relative abundance > 5.0% at least in one sample) were Kushneria (Br = 30.8 ± 27.2% and Bl = 24.0 ± 15.1%), Halomonas (Br = 1.0 ± 0.9% and Bl = 33.5 ± 28.1%), Bacillus (Br = 19.9 ± 34.2% and Bl = 13.1 ± 22.7%), Marinilactibacillus (Br = 0.06 ± 0.1% and Bl = 27.2 ± 15.2%), Rhodococcus (Br = 12.1 ± 12.3% and Bl = 0.2 ± 0.16%), Ralstonia (Br = 11.7 ± 13.8% and Bl = 0.14 ± 0.16%), Pelomonas (Br = 4.6 ± 5.0% and Bl = 0.09 ± 0.03%) and Bradyrhizobium (Br = 2.1 ± 2.7% and Bl = 0.03 ± 0.01%). Of them, Marinilactibacillus (LDA = 5.134, p = 0.0495), Rhodococcus (LDA = 4.785, p = 0.0495), Ralstonia (LDA = 4.734, p = 0.0495), Pelomonas (LDA = 4.4.373, p = 0.0495) and Bradyrhizobium (LDA = 4.033, p = 0.0495) exhibited significantly differences between the two groups (Fig. 6a). The relative abundances of Rhodococcus, Ralstonia, Pelomonas and Bradyrhizobium were significantly enriched in brown seeds, while Marinilactibacillus was enriched in black seeds. Although, Kushneria, Halomonas and Bacillus haven’t exhibited significant differences between the two groups, but they presented different distribution proportions between the two groups. Detailed the relative abundance information of endophytic bacteria at genus level in brown seeds and black seeds can be found inthe Additional file 3: Table S1.
The relationship between two sample groups and dominant endophytic bacteria at species level could be found in Additional file 2:Fig. S2. The results showed that Bacillus krulwichiae, Rhodococcus erythropolis, Ralstonia solanacearum, Pelomonas (unclassified), and Bradyrhizobium elkanii had higher relative abundance in brown seeds than in the black seeds, whereas the black seeds harbored a high relative abundance of unclassified Halomonas (unclassified), Marinilactibacillus (unclassified) and Bacillus gibsonii.
The results showed that the relative abundance of bacterial community distribution patterns between brown and black seeds collected from the same natural environment differed significantly.
Functional analysis of the microbiota
The presumptive functions of the endophytic microbiota of the dimorphic seeds collected from the same natural environment were evaluated using PICRUSt2. The predicted genes were classified by aligning them to the MetaCyc databases (https://metacyc.org/). A total of 370 metabolic pathways were identified and were further selected to analyze significant differences between the two groups. Within the top 20 relative abundance categories, the abundance of only pentose phosphate pathway (non-oxidative branch) exhibited significantly difference between the two groups (Fig. 7), which significantly higher in black seeds than in brown seeds.

Comparison of the relative abundance in top 20 MetaCyc metabolic pathways between brown seeds and black seeds. Br: brown seeds; Bl: black seeds. * stands for 0.01 ≤ p < 0.05, ** stands for 0.001 ≤ p < 0.01 and *** stands for p < 0.001 according to Student’s T-test
Discussion
In this study, high-throughput sequencing technology was used to reveal the diversity of endophytic bacterial communities in the dimorphic seeds of S. glauca obtained from the same natural environment. Our findings demonstrated that seed dimorphism had little impact on the diversity and richness of endophytic bacterial communities in brown and black seeds, but significantly different relative abundances of the endophytic bacterial taxa were detected in brown and black seeds of S. glauca.
Many studies have shown that the seed dimorphism of Suaeda spp. usually associated with differences in seed shape, seed size, seed coat color [2, 6,7,8, 11, 12, 14, 15], seed coat structure [16], seed germinability [7, 11, 12, 15] and seed phytochemical properties [16,17,18]. In this study, we observed that seed coat structure and seed phytochemical properties of brown and black seeds of S. glauca significantly differed. Our results revealed that black seeds of S. glauca had two layers of the seed coat, including a layer of hard, cuticle exotesta and a layer of soft, membranous endotesta compared to single-layer membranous testa in brown seeds. A similar result has also been reported in Borszczowia aralocaspica (S. aralocaspica) [16]. Seed coat acts asa modulator between seed and environment and can regulate gaseous exchange and water imbibition [40, 41]. A previous study has indicated that black seeds of S. glauca had an intermediate physical dormancy and exhibited a low germination percentage, but it was water-permeable [6]. Brits et al. [42, 43] demonstrated that the intact hard testa may partially reduce oxygen diffusion to the embryo, contribute to hypoxic constraints. Brits and Manning [44] found the seeds of Leucospermum cordifolium have also two layers of seed coat (exotesta and endotesta), and exhibit water-permeable and oxygen-impermeable, which named as “anoxia PY (physical dormancy)”. Besides, Wang et al. [10] reported that the seed coat of black seeds of S. salsa contains a high content of waxes compared to brown seeds. These results implied that the difference in both structure and chemical composition of seed coat leads to differences in oxygen exchange capacity between black and brown seeds of S. glauca. The black seeds may rather have a limited capacity for gas exchange compared to brown seeds. Interestingly, Tegtmeier et al. [45] found that oxygen availability can influence colonization patterns of microbes in the gut microbiota.
Our results revealed that the content of soluble protein, soluble starch and soluble sugar was significantly higher in extracts obtained from brown seeds than those of black seeds; in contrast, the fat content of brown seeds was lower than that of black seeds. The different abilities of nutrition accumulation in dimorphic seeds have also been reported in S. salsa [17, 36] and S. aralocaspica [16, 18]. For example, Song et al. [16] found that the content of soluble sugar, soluble protein, total nitrogen, total phosphorus and inorganic ions (K+, Na+, K+/Na+) in brown seeds were significantly higher than those of black seeds in S. aralocaspica. In addition, we also detected higher content of total phenols in brown seeds compared to that of black seeds. A similar result was also reported in S. salsa [17]. Overall, these results suggest that there were significant differences in seed phytochemical properties between the dimorphic seeds of S. glauca. Interestingly, numerous studies have determined that the compositions of seed endophytic microbiota have been influenced the seed phytochemical traits [34, 35].
In the present study, alpha-diversity indices were used to evaluate the seed endophytic bacterial community richness and diversity. The results showed no significant differences in alpha-diversity indices between brown and black seeds. It was quite surprising that the significant differences in the seed coat structure and seed phytochemical characteristics between brown and black seeds had little impact on the diversity and richness of endophytic bacterial communities in the dimorphic seeds. A similar result has also been reported by Zhang et al. [46], who found five rice genotypes have little impact on the diversity and richness of endophytic bacteria.
In the present study, 9 prokaryotic phyla were observed, of which Proteobacteria, Firmicutes and Actinobacteria were dominant. These above-mentioned phyla have also been reported as dominant endophytes of other plant seeds [46, 47]. Kushneria, Halomonas, Bacillus, Marinilactibacillus, Rhodococcus, Ralstonia, Pelomonas and Bradyrhizobium were the high relative abundant genera, of them, Kushneria, Halomonas and Bacillus were the core endophytic bacterial community. Interestingly, Kushneria, Halomonas and Bacillus have also been reported as dominant endophytes from roots of halophytes, such as Salicornia rubra, Sarcocornia utahensis and Allenrolfea occidentalis [48]. Previous studies have revealed that Kushneria strains were isolated mostly from saline environments [49], endosphere of halophyte Arthrocnemum macrostachyum [50] and Avicennia germinans [51], phyllosphere of halophyte Avicennia germinans [52] and rhizosphere of halophyte Saccharum spontaneum [53]. Some members of the genus Kushneria reported having plant growth-promoting activities, including siderophore production, indolacetic acid (IAA) production, nitrogen fixation and phosphate solubilization [50, 54]. Halomonas and Kushneria are closely related and were grouped in the same genus in the past [52]. Many Halomonas sp. exhibit salt tolerance and can improve plant growth under salt stress conditions [48, 55,56,57]. Bacillus is common genera among the endosphere niche of diverse plants, where they play an important role in plant protection and growth stimulation [58, 59]. The results suggested that these core taxa may play an important role in the seed endosphere of halophyte S. glauca, and these taxa can assist the plant to resist stress environments. Besides, the Venn diagram revealed that greater taxa presented in brown seeds, and also had high heterogeneity within the bacterial communities compared to black seeds (Fig. 5). One possible explanation was that brown seed with a single layer membraneous seed coat and abundant nutrients could contribute to colonize microorganisms present in the carposphere of utricles, and easily susceptible to the carposphere environment. Recent studies have shown that seed bacterial endophytes may also originate from the phyllosphere, anthosphere and carposphere [26, 60].
Based on alpha diversity analysis, PCoA analysis, and hierarchical clustering tree results, seed dimorphism had no significant impact on diversity indices as a whole, it influenced significantly the relative abundance of endophytic bacterial taxa between brown and black seeds. Our comprehensive comparison revealed that the relative abundances of endophytic bacterial communities of dimorphic seeds were significantly different from each other at phylum, class, order, family and genus level. At the phylum level, we observed 9 phyla. Interestingly, the relative abundance of Actinobacteria was higher in brown seeds than in black seeds, which means Actinobacteria may be enriched in brown seeds. This might attribute to brown seeds with single-layer membranous seed coat and fast germinability were easily susceptible to soil-borne pathogens compared to black seeds, while Actinobacteria may protect brown seeds against pathogens and promotes plant growth [61, 62]. Gripenberg et al. [63] found that there was a potential trade-off between seed chemical and mechanical defense, polyphenols are one of the most common seed defenses, which are most likely to be present in large seeds with short seed dormancy and low investment in mechanical seed defense. Compared to black seeds with high investment in mechanical seed defense (two layers seed coat, including hard exotesta and soft, membranous endotesta), brown seeds had a high level of phenolic content. Hence, we speculated that a high abundance of Actinobacteria, combined with high levels of total phenols, can protect brown seeds from pathogens in the soil.
At the genus level, 5 genera of the 8 dominant genera possessed significant differences between brown and black seeds. Rhodococcus, Ralstonia, Pelomonas, Bradyrhizobium and Marinilactibacillus exhibited a significant difference between the two groups. Notably, we found that Rhodococcus, Ralstonia, Pelomonas and Bradyrhizobium tend to be enriched in brown seeds and present in high proportion compared to black seeds. Our results revealed that Rhodococcus erythropolis, Ralstonia solanacearam, Pelomonas (species unclassified) and Bradyrhizobium elkanii, were the dominant species in brown seeds (Fig. S2). Rhodococcus have been found living in close association with various plant parts, such as the rhizosphere [64], phyllosphere [65, 66] and endosphere [67,68,69,70]. R. erythropolis can colonize plant roots [70], and also prevent plant disease by degrading N-acyl-homoserine lactone signaling molecules [71]. Moreover, several members of the genus Rhodococcus also show plant growth-promoting activities, including ACC deaminase, IAA production, siderophore production and phosphate solubilization [72,73,74,75]. Some strains of the genus Pelomonas were detected in the endosphere of Typha angustifolia [76] and reported to fix nitrogen [77]. Bradyrhizobium, a genus of Gram-positive that was initially proposed as a group of slow-growing, alkaline-producing root nodule nitrogen-fixing bacteria [78]. B. elkanii isolated from the root nodules of Acacia confusa, exhibit the nitrogen-fixing ability and can enhance the growth and root development of A. confuse [79]. Numerous studies revealed that endophytic bacteria can improve plant fitness by enhancing nutrient mobilization, nitrogen fixation, phosphate solubilization and conferring resistant against pathogens [27, 80]. Thus, we speculated that brown seeds harbor a large number of microorganisms with plant growth-promoting (PGP) traits, which contribute to the establishment and development of seedlings of brown seeds. Since brown seeds without dormancy behavior were the main source of early spring seedling of S. glauca [6]. In addition, we also detected strains of Ralstonia in brown seeds, such as R. solanacearam, which is an important soil-borne plant pathogen [81]. Taken together, it seemed that brown seeds served not only as a host for diverse plant-probiotic bacterial strains but also for putative opportunistic pathogenic bacteria.
In our study, compared to the endophytic microbiota of brown seeds, we found that Marinilactibacillus tends to be enriched in black seeds, and had higher proportions. Remarkably, Marinilactibacillus has also firstly reported as one of the most abundant genera in the endosphere of halophyte Halimione portulacoides [82]. A previous study revealed that Marinilacibacillus piezotolerans was a facultatively anaerobic lactobacillus [83, 84]. The results implied that Marinilactibacillus may adapt to the inner hypoxia environment of black seeds, since two layers of the seed coat of black seeds prevent gas-exchange. Truyens et al. [30] found that selection of seed endophytes partly relies on bacterial properties, and only bacteria with competitive and adaptive colonization characteristics can inhabit the seeds [85]. We also found that pentose phosphate pathway (non-oxidative branch) tends to be enriched in black seeds and had higher proportions compared to brown seeds. Stincone et al. [86] reported that the non-oxidation branch of the pentose phosphate pathway (PPP) is critical to maintain redox balance under stress situations. Fidalgo et al. [82] found that Marinilactibacillus spp. isolates tested positive for cellulolytic, proteolytic and xylanolytic enzymatic activities. Strain B. gibsonii (Fig. S2) was also an enriched species in black seeds, which was an efficient alkaline pectinase producer [87]. Together, the results suggested that oxygen availability may affect the competitive capacity of bacteria in endophytic microbiota of black seeds, and selectively enriched strains might reduce the mechanical resistance of hard exotesta of black seeds, which contributed to enhance the germinability of black seeds. Mayer and Poljakoo-Mayber [88] found that one of the possible reasons for the loss of impermeability of seeds was the action of microbes.
Conclusion
In summary, our work revealed that seeds characteristics might play an important role in the endophytic bacterial composition of the dimorphic seeds of S. glauca collected from the same natural environment. The present study suggests that there were significant differences in seed coat structure and seed phytochemical properties between brown and black seeds of S. glauca. Although seed dimorphism had little impact on the diversity and richness of endophytic bacteria communities in brown and black seeds, a significant difference in the relative abundance of endophytic bacteria was detected. This study showed that under the same natural environmental conditions, the endophytic bacterial communities of the dimorphic seeds might be influenced mainly by the seed coat structure and partly by seed phytochemical characteristics. Moreover, this study also showed that seed fitness was closely associated with the variations of endophytic bacterial communities between brown and black seeds. Brown seeds harbored a large number of bacteria with plant-growth-promoting traits, whereas black seeds presented bacteria with enzyme activities (i.e. pectinase, cellulolytic and xylanolytic activities). These findings might provide valuable information for a better understanding of the ecological adaptation strategy of dimorphic seeds.
Methods
Seed collection and surface sterilization
The wild, naturally growing halophyte S. glauca were obtained from their natural habitats in yingchengzi coastal saline beach (121.36° E, 38.99° N) in Dalian, Liaoning, China. Mature seeds from naturally grown plants that colonized in the same natural environment were harvested (at least 100 mother plants collected on October 25th,2018) and air-dried for 10 days at room temperature.
The dimorphic seedswere separated according to their phenotypic characteristics, and then two types of seeds were placed into 50 ml sterile conical tubes. Each seed sample type was replicated three times. To avoid environmental bacterial contamination, seed surface sterilization was done according to the following procedure: First, the seeds were rinsed with 30 ml sterilized distilled water at least 5 times or until no cloudiness was observed in the wash. Second, the washed seeds were immersed in 1.0% sodium hypochlorite for 2 min. Third, the bleached seeds were rinsed with 30 ml sterile distilled water for 1 min and then immersed in 30 ml 70% ethanol for 1 min. Fourth, the ethanol was removed and seeds were rinsed five times with sterilized distilled water. Finally, the surface-sterilized seeds were air-dried for 12 h in the sterilized 90 mm Petri-dish with double filter paper. To check the effect of surface sterilization, some seeds per treatment were randomly picked and placed on the TSA agar medium (TSA, Qingdao Hope Bio-Technology Co., Ltd., Qingdao, P.R. China). The plates were incubated for 3 days at 25 °C. The sterilized seed samples were put in 50 ml sterile conical tube, frozen in liquid nitrogen and then immediately stored at − 80 °C for later DNA extraction.
Seed morphological structure and phytochemical properties
The seed morphological structure was observed under the dissecting microscope. The crude fat, soluble sugar, soluble starch, total protein, total phenolic content was measured. To analyze the crude fat content, dry samples of dimorphic seeds (brown and black seeds, 1.0 g, respectively) were ground and petroleum ether (boiling range: 30 to 60 °C) was used as an extraction buffer. The crude fat extraction was performed using the Soxhlet apparatus. The crude fat content was determined following the AOAC method [89]. The procedure as described by Booij et al. [90] was followed for the extraction of soluble sugar with slight modification. 0.5 g dry seeds were crushed in a mortar using liquid nitrogen. Four milliliters of 80% ethanol was added and the mixture was incubated for 30 min at 100 °C.The extracts were centrifuged at 7000×g for 3 min at 4 °C. The supernatants were obtained and the extraction repeated twice. Soluble sugars were determined by the anthrone method at 625 nm using glucose as standard [91]. To analyze starch content, 0.5 g dry seeds were ground in a mortar using liquid nitrogen. The extraction was performed according to the method as described by Zhao et al. [92]. Starch content was determined with the anthrone method [93] at 640 nm using soluble starch as standard. For protein analysis, 0.5 g of dry seeds were ground and extracted according to the method as described by Piattoni et al. [94]. The total protein content was measured by using a spectrophotometer at 595 nm following the Bradford protocol [95]. Bovine Serum Albumin (BSA) standard curve was used to determine the total protein content. To analyze total phenolic content, 1.0 g dry seeds were ground and extracted according to the protocol described by Gallagher et al. [96]. Total phenolic content was determined using a spectrophotometer at 765 nm according to the Folin-Cocalteau reagent method [97]. Total phenolic concentrations were quantified by comparison with gallic acid as a standard curve.
DNA extraction, PCR amplification and sequencing
Total genomic DNA was extracted from six surface-sterilized seed samples (0.2 g/each sample) using the E.Z.N.A.® soil DNA Kit (Omega Bio-Tek, Norcross, GA, U.S.) according to the manufacturer’s protocols. The quality of the genomic DNA was checked using 1% agarose gel electrophoresis. DNA concentration and purity were determined by NanoDrop 2000 UV-visible spectrophotometer (Thermo Scientific, Wilmington, USA).
Two-step PCR amplification was performed to minimize the host rRNA gene contamination while analyzing microbial communities. The first PCR amplification of the 16S rRNA gene was carried out with the bacterial primer pairs 799F (5′-AACMGGATTAGATACCCKG-3′) and 1392R (5′-ACGGGCGGTGTGTRC-3′) [98] on the ABI GeneAmp®9700 PCR thermocycler (ABI, CA, USA). The 20 μl PCR reaction mixture contained 10 ng template DNA, 5 × TransStart FastPfu buffer 4 μL, 2.5 mM dNTPs 2 μL, forward primer (5 μM) 0.8 μL, reverse primer (5 μM) 0.8 μL, TransStart FastPfu DNA Polymerase 0.4 μL, BSA 0.2 μL and finally ddH2O up to 20 μL. The PCR amplification of 16S rRNA gene was performed as follows: initial denaturation at 95 °C for 3 min, followed by 27 cycles of denaturing at 95 °C for 30 s, annealing at 55 °C for 30 s and extension at 72 °C for 45 s, and single extension at 72 °C for 10 min, and end at 4 °C. The second PCR amplification was performed using 2 μL of the extraction product as a template, using the bacterial 16S rRNA gene primer pairs 799F (5′-AACMGGATTAGATACCCKG-3′) and 1193R (5′-ACGTCATCCCCACCTTCC-3′) [99]. All conditions for the second PCR step were identical except that thermocycling was done for 13 cycles instead of 27 cycles. PCR reactions were performed in triplicate. The PCR product was checked by using 2% agarose gel electrophoresis, and purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA) according to manufacturer’s instructions and quantified using Quantus™ Fluorometer (Promega, USA).
Purified PCR products were sequenced by paired-end sequencing performed on an Illumina MiSeq PE300 platform (Illumina, San Diego, USA) according to the standard protocols by Majorbio Bio-Pharm Technology Co., Ltd. (Shanghai, China).
16S rRNA gene sequence analysis
The raw 16S rRNA gene sequences reads were demultiplexed, quality-filtered by fastp (version 0.20.0) [100] and merged by FLASH (version 1.2.7) [101] with the following criteria: (i) the 300 bp reads were truncated at any site receiving an average quality score of < 20 over a 50 bp sliding window, and the truncated reads shorter than 50 bp were discarded, reads containing ambiguous characters were also discarded; (ii) only overlapping sequences longer than 10 bp were assembled. The maximum mismatch ratio of the overlap region was 0.2. Reads that could not be assembled were discarded; (iii) samples were distinguished according to the barcode and primers. Chimeric sequences were identified and removed using UCHIME [102]. The remaining high-quality sequences were clustered into operational taxonomic units (OTUs) with 97% similarity cut-off [103, 104] using UPARSE (version 7.0) [104]. The taxonomy of each OTU representative sequence was analyzed by RDP Classifier (version 2.2) [105] against the SILVA 16S rRNA database (Release 132) using a confidence threshold of 0.7.
Statistical analysis
To avoid biases introduced by primers, sequences belonging to chloroplasts (o_Chloroplast) or mitochondria (f_Mitochondria) were discarded and other OTUs of the libraries were used for microbial community analyses. Alpha-diversity was evaluated by calculating community richness parameters (Chao, ACE), community diversity parameters (Shannon, Simpson) and a sequencing depth index (Good’s coverage) using MOTHUR software (version v. 1.30.1) [106]. R package software (version 3.3.1) was used to generate the results of the Venn diagram, Bar diagram, Pie diagram and Circos diagram. Beta diversity analysis based on unweighted Unifrac was carried out to visualize the results of PCoA (Principal coordinates’ analysis) and hierarchical clustering analysis at the OUT level by using R package software. Besides, the prediction of the microbial gene functions was done using PICRUSt2 against the MetaCyc metabolic pathway database (https://metacyc.org/).
The student’s t-test (SPSS 19.0) was used to compare the difference of seed phytochemical properties, the Alpha-diversity index and the relative abundance of MetaCyc metabolic pathways between brown and black seeds. The differential bacterial taxa between brown and black seeds were analyzed using Linear discriminant analysis (LDA) effect size (LEfSe). Only taxa with an average relative abundance greater than 0.01% were considered. All reported values were the average of triplicate results (mean ± SD).
Availability of data and materials
The raw reads were deposited into the NCBI Sequence Read Archive (SRA) database (SRA, https://www.ncbi.nlm.nih.gov/sra) under accession number PRJNA664311 (https://dataview.ncbi.nlm.nih.gov/object/PRJNA664311?reviewer=nmr0cvtfvjbkm1lllc2tag9u5m). Other datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Abbreviations
- TSA:
-
Tryptone soy agar
- PCoA:
-
Principal co-ordinates analysis
- OTUs:
-
Operational taxonomic units
- LEfSe:
-
Linear discriminant analysis effect size
- LDA:
-
Linear discriminant analysis
- PICRUSt:
-
Phylogenetic Investigation of Communities by Reconstruction of Unobserved States
- BSA:
-
Bovine serum albumin
- PCR:
-
Polymerase chain reaction
- Br:
-
Brown seeds
- Bl:
-
Black seeds
References
Imbert E. Ecological consequences and ontogeny of seed heteromorphism. Perspect Plant Ecol Evol Syst. 2002;5(1):13–36. https://doi.org/10.1078/1433-8319-00021.
Redondo-Gómez S, Mateos-Naranjo E, Cambrollé J, Luque T, Figueroa ME, Davy AJ. Carry-over of differential salt tolerance in plants grown from dimorphic seeds of Suaeda splendens. Ann Bot. 2008;102(1):103–12. https://doi.org/10.1093/aob/mcn069.
Volis S, Bohrer G. Joint evolution of seed traits along an aridity gradient: seed size and dormancy are not two substitutable evolutionary traits in temporally heterogeneous environment. New Phytol. 2013;197(2):655–67. https://doi.org/10.1111/nph.12024.
El-Keblawy A, Bhatt A, Gairola S. Perianth colour affects germination behavior in wind-pollinated Salsola rubescens in the Arabian deserts. Botany. 2013;92(1):69–75. https://doi.org/10.1139/cjb-2013-0183.
Bhatt A, Santo A. Germination and recovery of heteromorphic seeds of Atriplex canescens (Amaranthaceae) under increasing salinity. Plant Ecol. 2016;217(9):1069–79. https://doi.org/10.1007/s11258-016-0633-6.
Wang HF, Kong L, Gao R, Abudureheman B, Li XY, Li QL. Germination biology of dimorphic seeds of the annual halophyte common seepweed (Suaeda glauca). Weed Sci. 2020;68(2):143–50. https://doi.org/10.1017/wsc.2019.74.
Li WQ, Liu XJ, Khan A, Yamaguchi S. The effect of plant growth regulators, nitric oxide, nitrite and light on the germination of dimorphic seeds of Suaeda salsa under saline conditions. J Plant Res. 2005a;118(3):207–14. https://doi.org/10.1007/s10265-005-0212-8.
Song J, Fan H, Zhao YY, Jia YH, Du XH, Wang BS. Effect of salinity on germination, seedling emergence, seedling growth and ion accumulation of a euhalophyte Suaeda salsa in an intertidal zone and on saline inland. Aqua Bot. 2008;88(4):331–7. https://doi.org/10.1016/j.aquabot.2007.11.004.
Song J, Shi WW, Liu RR, Xu YG, Sun N, Zhou JC, et al. The role of the seed coat in adaptation of dimorphic seeds of the euhalophyte Suaeda salsa to salinity. Plant Spec Biol. 2017;32(2):107–14. https://doi.org/10.1111/1442-1984.12132.
Wang FX, Xu YG, Wang S, Shi WW, Liu RR, Feng G, et al. Salinity affects production and salt tolerance of dimorphic seeds of Suaeda salsa. Plant Physiol Biochem. 2015;95:41–8. https://doi.org/10.1016/j.plaphy.2015.07.005.
Wang HL, Wang L, Tian CY, Huang ZY. Germination dimorphism in Suaeda acuminata: a new combination of dormancy types for heteromorphic seeds. S Afr J Bot. 2012a;78:270–5. https://doi.org/10.1016/j.sajb.2011.05.012.
Wang L, Huang ZY, Baskin CC, Baskin JM, Dong M. Germination of dimorphic seeds of the desert annual halophyte Suaeda aralocaspica (Chenopodiaceae), a C4 plant without Kranz anatomy. Ann Bot. 2008;102(5):757–69. https://doi.org/10.1093/aob/mcn158.
Wang L, Wang HL, Yin L, Tian CY. Transcriptome assembly in Suaeda aralocaspicato reveal the distinct temporal gene/miRNA alterations between the dimorphic seeds during germination. BMC Genomics. 2017;18(1):806. https://doi.org/10.1186/s12864-017-4209-1.
Yang F, Baskin JM, Baskin CC, Yang XJ, Cao DC, Huang ZY. Effects of germination time on seed morph ratio in a seed-dimorphic species and possible ecological significance. Ann Bot. 2015;115(1):137–45. https://doi.org/10.1093/aob/mcu210.
Khan MA, Gul B, Weber DJ. Germination of dimorphic seeds of Suaeda moquinii under high salinity stress. Aust J Bot. 2001;49:185–92. https://doi.org/10.1071/BT00020.
Song YG, Li L, Zhang XM, Pan XL, Zeng XH. Differences of seed coat structure and ions content between dimorphic seeds of Borszczowia aralocaspica. Bull Bot Res. 2012;32:290–5.
Zhao YQ, Yang Y, Song YP, Li Q, Song J. Analysis of storage compounds and inorganic ions in dimorphic seeds of euhalophyte Suaeda salsa. Plant Physiol Biochem. 2018a;130:511–6. https://doi.org/10.1016/j.plaphy.2018.08.003.
Wang L, Zhao ZY, Zhang K, Tian CY. Oil content and fatty acid composition of dimorphic seeds of desert halophyte Suaeda aralocaspica. Afr J Agr Res. 2012b;7(12):1910–4. https://doi.org/10.5897/AJAR11.1535.
Vandenkoornhuyse P, Quaiser A, Duhamel M, Le Van A, Dufresne A. The importance of the microbiome of the plant holobiont. New Phytol. 2015;206(4):1196–206. https://doi.org/10.1111/nph.13312.
Sanchez-Cañizares C, Jorrín B, Poole PS, Tkacz A. Understanding the holobiont: the interdependence of plants and their microbiome. Curr Opin Microbiol. 2017;38:188–96. https://doi.org/10.1016/j.mib.2017.07.001.
Bordenstein SR, Theis KR. Host biology in light of the microbiome: ten principles of holobionts and hologenomes. PLoS Biol. 2015;13(8):e1002226. https://doi.org/10.1371/journal.pbio.1002226.
Holland MA, Polacco JC. PPFMs and other covert contaminants: is there more to plant physiology than just plant? Annu Rev Plant Physiol Plant Mol Biol. 1994;45(1):197–209. https://doi.org/10.1146/annurev.pp.45.060194.001213.
Walitang DI, Kim K, Madhaiyan M, Kim Y, Kang YY, Sa TM. Characterizing endophytic competence and plant growth promotion of bacterial endophytes inhabiting the seed endosphere of rice. BMC Microbiol. 2017;17(1):209. https://doi.org/10.1186/s12866-017-1117-0.
Chee-Sanford JC, Williams MM, Davis AS, Sims GK. Do microorganisms influence seed-bank dynamics? Weed Sci. 2006;54(3):575–87. https://doi.org/10.1614/WS-05-055R.1.
Puente ME, Lib CY, Bashan Y. Endophytic bacteria in cacti seeds can improve the development of cactus seedlings. Environ Exp Bot. 2009;66(3):402–8. https://doi.org/10.1016/j.envexpbot.2009.04.007.
Compant S, Clement C, Sessitsch A. Plant growth-promoting bacteria in the rhizo- and endosphere of plants: their role, colonization, mechanisms involved and prospects for utilization. Soil Biol Biochem. 2010;42(5):669–78. https://doi.org/10.1016/j.soilbio.2009.11.024.
Hardoim PR, Hardoim CCP, van Overbeek LS, van Elsas JD. Dynamics of seed-borne rice endophytes on early plant growth stages. PLoS One. 2012;7(2):e30438. https://doi.org/10.1371/journal.pone.0030438.
Ferreira A, Quecine MC, Lacava PT, Oda S, Azevedo JL, Araujo WL. Diversity of endophytic bacteria from Eucalyptus species seeds and colonization of seedling by Pantoea afflomerans. FEMS Microbiol. 2008;287(1):8–14. https://doi.org/10.1111/j.1574-6968.2008.01258.x.
Yuan ZL, Druzhinina IS, Labbé J, Redman R, Qin Y, Rodriguez R, et al. Specialized microbiome of a halophyte and its role in helping non-host plants to with stand salinity. Sci Rep. 2016;6(1):32467. https://doi.org/10.1038/strep32467.
Truyens S, Weyens N, Cuypers A, Vangronsveld J. Changes in the population of seed bacteria of transgenerationally Cd-exposed Arabidopsis thaliana. Plant Biol. 2013;15(6):971–81. https://doi.org/10.1111/j.1438-8677.2012.00711.x.
Simon HM, Smith KP, Dodsworth JA, Guenthner B, Handelsman J, Goodman RM. Influence of tomato genotype on growth of inoculated and indigenous bacteria in the spermosphere. Appl Environ Microbiol. 2001;67(2):514–20. https://doi.org/10.1128/AEM.67.2.514-520.2001.
Liu Y, Zuo S, Xu LW, Zou YY, Song W. Study on diversity of endophytic bacterial communities in seeds of hybrid maize and their parental lines. Arch Microbiol. 2012;194(12):1001–12. https://doi.org/10.1007/s00203-012-0836-8.
Walitang DI, Kim CG, Kim KY, Kang YY, Kim YK, Sa TM. The influence of host genotype and salt stress on the seed endophytic community of salt-sensitive and salt-tolerant rice cultivars. BMC Plant Biol. 2018;18(1):51. https://doi.org/10.1186/s12870-018-1261-1.
Nakaew N, Sungthong R. Seed phytochemicals shape the community structures of cultivable actinobacteria-inhabiting plant interiors of Thai pigmented rice. Microbiol Open. 2018;7(4):e591. https://doi.org/10.1002/mbo3.591.
Mano H, Tanaka F, Watanabe A, Kaga H, Okunishi S, Morisaki H. Culturable surface and endophytic bacterial flora of the maturing seeds of rice plant (Oryza sativa) cultivated in a paddy field. Microbes Environ. 2006;21(2):86–100. https://doi.org/10.1264/jsme2.21.86.
Zhao YQ, Ma YC, Duan HM, Liu RR, Song J. Traits of fatty acid accumulation in dimorphic seeds of the euhalophyte Suaeda salsa in saline conditions. Plant Biosyst. 2019;153(4):514–20. https://doi.org/10.1080/11263504.2018.1508090.
Li WQ, Shinjiro Y, Ajmal KM, Ping A, Liu X, Tran LP. Roles of gibberellins and abscisic acid in regulating germination of Suaeda salsa dimorphic seeds under salt stress. Front Plant Sci. 2016;6:1–10. https://doi.org/10.3389/fpls.2015.01235.
Chen S, Yang Z, Wang MJ, Song J, Sui N, Fan H. Salinity improves chilling resistance in Suaeda salsa. Acta Physiol Plant. 2014;36(7):1823–30. https://doi.org/10.1007/s11738-014-1555-3.
Zhou JC, Fu TT, Sui N, Guo JR, Feng G, Fan JL, et al. The role of salinity in seed maturation of the euhalophyte Suaeda salsa. Plant Biosyst. 2016;150(1):83–90. https://doi.org/10.1080/11263504.2014.976294.
Dübbern De Souza FH, Marcos-Filho J. The seed coat as a modulator of seed-environment relationships in Fabaceae. Braz J Bot. 2001;24:365–75. https://doi.org/10.1590/S0100-84042001000400002.
Ma F, Cholewa EWA, Mohamed T, Peterson CA, Gijzen M. Cracks in the palisade cuticle of soybean seed coats correlate with their permeability to water. Ann Bot. 2004;94(2):213–28. https://doi.org/10.1093/aob/mch133.
Brits GJ, Calitz FJ, Brown NAC, Manning JC. Desiccation as the active principle in heat-stimulated seed germination of Leucospermum R. Br. (Proteaceae) in fynbos. New Phytol. 1993;125(2):397–403. https://doi.org/10.1111/j.1469-8137.1993.tb03892.x.
Brits GJ, Calitz FJ, Brown NAC. Heat desiccation as a seed scarifying agent in Leucospermum spp. (Proteaceae) and its effects on the testa, viability and germination. Seed Sci Technol. 1999;27:163–76.
Brits CJ, Manning JC. Seed structure and physiology in relation to recruitment ecology in Leucospermum (Proteaceae) in fynbos. Aust J Bot. 2019;67(4):290–308. https://doi.org/10.1071/BT18199.
Tegtmeier D, Tompson CL, Schauer C, Brune A. Oxygen affects gut bacterial colonization and metabolic activities in a gnotobiotic cockroach model. Appl Environ Microbiol. 2015;82(4):1080–9. https://doi.org/10.1128/AEM.03130-15.
Zhang J, Zhang CW, Yang J, Zhang RJ, Gao JS, Zhao X, et al. Insights into endophytic bacterial community structures of seeds among various Oryza sativa L. rice genotypes. J Plant Growth Regul. 2019;38(1):93–102. https://doi.org/10.1007/s00344-018-9812-0.
Truyens S, Weyens N, Cuypers A, Vangronsveld J. Bacterial seed endophytes: genera, vertical transmission and interaction with plants. Environ Microbiol Rep. 2015;7(1):40–50. https://doi.org/10.1111/1758-2229.12181.
Kearl J, McNary C, Lowman JS, Mei CS, Aanderud ZT, Smith ST, et al. Salt-tolerant halophyte rhizosphere bacteria stimulate growth of alfalfa in salty soil. Front Microbiol. 2019;10:1849. https://doi.org/10.3389/fmicb.2019.01849.
Yun JH, Sung H, Kim HS, Tak EJ, Kang W, Lee JY, et al. Complete genome sequence of the halophile bacterium Kushneria konosiri X49T, isolated from salt-fermented Konosirus punctatus. Stand Genomic Sci. 2018;13(1):19. https://doi.org/10.1186/s40793-018-0324-0.
Navarro-Torre S, Carro L, Rodríguez-Llorente ID, Pajuelo E, Caviedes MÁ, Igual JM, et al. Kushneria phyllosphaerae sp. nov. and Kushneria endophytica sp. nov., plant growth promoting endophytes isolated from the halophyte plant Arthrocnemum macrostachyum. Int J Syst Evol Microbiol. 2018;68(9):2800–6. https://doi.org/10.1099/ijsem.0.002897.
Soto-Ramírez N, Sánchez-Porro C, Rosas S, González W, Quiñones M, Ventosa A, et al. Halomonas avicenniae sp. nov., isolated from the salty leaves of the black mangrove Avicennia germinans in Puerto Rico. Int J Syst Evol Microbiol. 2007;57(5):900–5. https://doi.org/10.1099/ijs.0.64818-0.
Sánchez-Porro C, de la Haba RR, Soto-Ramírez NS, Márquez MC, Montalvo-Rodríguez R, Ventosa A. Description of Kushneria aurantia gen. nov., sp. nov., a novel member of the family Halomonadaceae, and a proposal for reclassification of Halomonas marisflavias Kushneria marisflavi comb. nov., of Halomonas indalinina as Kushneria indalinina comb. nov. and of Halomonas avicenniae as Kushneria avicenniae comb. nov. Int J Syst Evol Microbiol. 2009;59(2):397–405. https://doi.org/10.1099/ijs.0.001461-0.
Bangash A, Ahmed I, Abbas S, Kudo T, Shahzad A, Fujiwara T, et al. Kushneria pakistanensis sp. nov., a novel moderately halophilic bacterium isolated from rhizosphere of a plant (Saccharum spontaneum) growing in salt mines of the Karak area in Pakistan. Antonie Van Leeuwenhoek. 2015;107(4):991–1000. https://doi.org/10.1007/s10482-015-0391-9.
Navarro-Torre S, Mateos-Naranjo E, Caviedes MA, Pajuelo E, Rodríguez-Llorente ID. Isolation of plant-growth-promoting and metal-resistant cultivable bacteria from Arthrocnemum macrostachyumin the Odiel marshes with potential use in phytoremediation. Mar Pollut Bull. 2016;110(1):133–42. https://doi.org/10.1016/j.marpolbul.2016.06.070.
Lafi FF, Ramirez-Prado JS, Alam I, Bajic VB, Hirt H, Saad MM. Draft genome sequence of Halomonas elongata strain K4, an endophytic growth-promoting bacterium enhancing salinity tolerance in Planta. Genome Announc. 2016;4(6):e01214–6. https://doi.org/10.1128/genomeA.01214-16.
Zhang J, Wang PC, Tian HM, Tao Z, Guo TT. Transcriptome analysis of Ice plant growth-promoting endophytic bacterium Halomonas sp. strain MC1 to identify the genes involved in salt tolerance. Microorganisms. 2020;8:88. https://doi.org/10.3390/microorganisms8010088.
Mukherjee P, Mitra A, Roy M. Halomonas rhizobacteria of Avicennia marina of Indian sundarbans promote rice growth under saline and heavy metal stresses through exopolysaccharide production. Front Microbiol. 2019;10:1207. https://doi.org/10.3389/fmicb.2019.01207.
Shishido M, Breuil C, Chanway CP. Endophytic colonization of spruce by plant growth-promoting rhizobacteria. FEMS Microbiol Ecol. 1999;29(2):191–6. https://doi.org/10.1111/j.1574-6941.1999.tb00610.x.
Berg G, Zachow C, Lottmann J, Gotz M, Costa R, Smalla K. Impact of plant species and site on rhizosphere-associated fungi antagonistic to Verticillium dahlia Kleb. Appl Environ Microbiol. 2005;71(8):4203–13. https://doi.org/10.1128/AEM.71.8.4203-4213.2005.
Compant S, Mitter B, Coli-Mull JG, Gangl H, Sessitsch A. Endophytes of grapevine flowers, berries, and seeds: identification of cultivable bacteria, comparison with other plant parts, and visualization of niches of colonization. Microb Ecol. 2011;62(1):188–97. https://doi.org/10.1007/s00248-011-9883-y.
Schrey SD, Tarkka MT. Friends and foes: streptomycetes as modulators of plant disease and symbiosis. Antonie Van Leeuwenhoek. 2008;94(1):11–9. https://doi.org/10.1007/s10482-008-9241-3.
Joseph B, Sankarganesh P, Edwin BT, Raj SJ. Endophytic Streptomycetes from plants with novel green chemistry: review. Int J Biol Chem. 2012;6(2):42–5. https://doi.org/10.3923/ijbc.2012.42.52.
Gripenberg S, Rota J, Kim JM, Wright SJ, Garwood NC, Fricke EC, et al. Seed polyphenols in a diverse tropical plant community. J Ecol. 2018;106(1):87–100. https://doi.org/10.1111/1365-2745.12814.
Bafana A. Diversity and metabolic potential of culturable root-associated bacteria from Origanum vulgare in sub-Himalayan region. World J Microbiol Biotechnol. 2013;29(1):63–74. https://doi.org/10.1007/s11274-012-1158-3.
Bai Y, Müller DB, Srinivas G, Garrido-Oter R, Potthoff E, Rott M, et al. Functional overlap of the Arabidopsis leaf and root microbiota. Nature. 2015;528(7582):364–9. https://doi.org/10.1038/nature16192.
Ritpitakphong U, Falquet L, Vimoltust A, Berger A, Métraux JP, L’Haridon F. The microbiome of the leaf surface of Arabidopsis protects against a fungal pathogen. New Phytol. 2016;210(3):1033–43. https://doi.org/10.1111/nph.13808.
Li J, Zhao GZ, Chen HH, Qin S, Xu LH, Jiang CL, et al. Rhodococcus cercidiphylli sp. nov., a new endophytic actinobacterium isolated from a Cercidiphyllum japonicum leaf. Syst Appl Microbiol. 2008;31(2):108–13. https://doi.org/10.1016/j.syapm.2008.03.004.
Zhao GZ, Li J, Zhu WY, Tian SZ, Zhao LX, Yang LL, et al. Rhodococcus artemisiae sp. nov., an endophytic actinobacterium isolated from the pharmaceutical plant Artemisia annua L. Int J Syst Evol Microbiol. 2012;62(Pt_4):900–5. https://doi.org/10.1099/ijs.0.031930-0.
Maropola MKA, Ramond JB, Trindade M. Impact of metagenomic DNA extraction procedures on the identifiable endophytic bacterial diversity in Sorghum bicolor (L. Moench). J Microbiol Methods. 2015;112:104–17. https://doi.org/10.1016/j.mimet.2015.03.012.
Toussaint JP, Pham TTM, Barriault D, Sylvestre M. Plant exudates promote PCB degradation by a rhodococcal rhizobacteria. Appl Microbiol Biotechnol. 2012;95(6):1589–603. https://doi.org/10.1007/s00253-011-3824-z.
Latour X, Barbey C, Chane A, Groboillot A, Burini JF. Rhodococcus erythropolis and its Y-Lactone catabolic pathway: an unusual biocontrol system that disrupts pathogen quorum sensing communication. Agronomy. 2013;3(4):816–38. https://doi.org/10.3390/agronomy3040816.
Belimov AA, Safronova VI, Sergeyeva TA, Egorova TN, Matveyeva VA, Tsyganov VE, et al. Characterization of plant growth promoting rhizobacteria isolated from polluted soils and containing 1-aminocyclopropane-1-carboxylate deaminase. Can J Microbiol. 2001;47(7):642–52. https://doi.org/10.1139/w01-062.
Abbamondi GR, Tommonaro G, Weyens N, Thijs S, Sillen W, Gkorezis P, et al. Plant growth-promoting effects of rhizospheric and endophytic bacteria associated with different tomato cultivars and new tomato hybrids. Chem Biol Technol Agric. 2016;3(1):1–10. https://doi.org/10.1186/s40538-015-0051-3.
Hasuty A, Choliq A, Hidayat I. Production of indole acetic acid (IAA) by Serratia marcescens subsp. marcescens and Rhodococcus aff. qingshengii. Int J Agric Technol. 2018;14:299–312.
Murugappan RM, Benazir-Begun S, Usha C, Lok-Kirubahar S, Karthikeyan M. Growth promoting and probiotic potential of the endophytic bacterium Rhodococcus globerulus colonizing the medicinal plant Plectranthus amboinicus (Lour.) Spreng. Int J Curr Res Rev. 2017;9:7–13.
Li YH, Liu QF, Liu Y, Zhu JN, Zhang Q. Endophytic bacterial diversity in roots of Typha angustifolia L. in the constructed Beijing Cuihu Wetland (China). Res Microbiol. 2011;162(2):124–31. https://doi.org/10.1016/j.resmic.2010.09.021.
Xie CH, Yokota A. Reclassification of Alcaligenes latus strains IAM 12599T and IAM 12664 and Pseudomonas saccharophila as Azohydromonas lata gen. nov., comb. nov., Azohydromonas australica sp. nov. and Pelomonas saccharophila gen. nov., comb. nov., respectively. Int J Syst Evol Microbiol. 2005;55(6):2419–25. https://doi.org/10.1099/ijs.0.63733-0.
Somasegaran P, Hoben HJ. Handbook for rhizobia: methods in legume-rhizobium technology. New York: Springer-Verlag; 1994. https://doi.org/10.1007/978-1-4613-8375-8.
Lee JT, Tsai SM, Lin CH. The nitrogen-fixing Bradyrhizobium elkanii significantly stimulates root development and pullout resistance of Acacia confusa. Afr J Biotechnol. 2017;16(18):1067–77. https://doi.org/10.5897/AJB2017.15971.
Rosenblueth M, Martinez-Romero E. Bacterial endophytes and their interactions with hosts. Mol Plant-Microbe Interact. 2006;19(8):827–37. https://doi.org/10.1094/MPMI-19-0827.
Erill I, Puigvert M, Legrand L, Guarischi-Sousa R, Vandecasteele C, Setubal J, et al. Comparative analysis of Ralstonia solanacearum Methylomes. Front Plant Sci. 2017;8:504. https://doi.org/10.3389/fpls.2017.00504.
Fidalgo C, Henriques I, Rocha J, Tacão M, Alves A. Culturable endophytic bacteria from the salt marsh plant Halimione portulacoides: phylogenetic diversity, functional characterization, and influence of metal (loid) contamination. Environ Sci Pollut Res. 2016;23(10):10200–14. https://doi.org/10.1007/s11356-016-6208-1.
Toffin L, Zink K, Kato C, Pignet P, Bidault A, Bienvenu N, et al. Marinilactibacillus piezotolerans sp. nov., a novel marine lactic acid bacterium isolated from deep sub-seafloor sediment of the Nankai Trough. Int J Syst Evol Microbiol. 2005;55(1):345–51. https://doi.org/10.1099/ijs.0.63236-0.
Wei YL, Cao JW, Fang JS, Kato C, Cui WC. First complete genome sequence of Marinilactibacillus piezotolerans strain 15R, a marine lactobacillus isolated from Coal-bearing sediment 2.0 kilometers below the seafloor, determined by pacbio single-molecule real-time technology. Genome Announc. 2017;5(7):e01625–16. https://doi.org/10.1128/genomeA.01625-16.
Okunishi S, Sako K, Mano H, Imamura A, Morisaki H. Bacterial flora of endophytes in the maturing seed of cultivated rice (Oryza sativa). Microbes Environ. 2005;20(3):168–77. https://doi.org/10.1264/jsme2.20.168.
Stincone A, Prigione A, Cramer T, Wamelink MMC, Campbell K, Cheung E, et al. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol Rev. 2014;90(3):927–63. https://doi.org/10.1111/brv.12140.
Li ZM, Bai ZH, Zhang BG, Xie HJ, Hu Q, Hao CB, et al. Newly isolated Bacillus gibsonii S-2 capable of using sugar beet pulp for alkaline pectinase production. World J Microbiol Biotechnol. 2005b;21(8-9):1483–6. https://doi.org/10.1007/s11274-005-7025-8.
Mayer AM, Poljakoff-Mayber A. The germination of seeds. 4th ed. Oxford: Pergamon Press; 1989.
Helrich K. Official methods of analysis of the Association of Official Analytical Chemists. Volume 2, Food composition, additives, natural contaminants. Am J Public Health and Nations Health. 1990;41(4):465.
Booij I, Piombo G, Risterucci JM, Coupé M, Thomas D, Ferry M. Étude de la composition chimique de dates à différentsstades de maturité pour la caractérisationvariétales de divers cultivars de palmier dattier (Phoenix dactylifera L.). Fruits. 1992;47:667–77.
Morris DL. Quantitative determination of carbohydrates with Dreywood’santhrone reagent. Science. 1948;107(2775):254–5. https://doi.org/10.1126/science.107.2775.254.
Zhao M, Zhang HX, Yan H, Qiu L, Baskin CC. Mobilization and role of starch, protein, and fat reserves during seed germination of six wild grassland species. Front Plant Sci. 2018b;9:234. https://doi.org/10.3389/fpls.2018.00234.
Fernandes B, Dragone G, Abreu AP, Geada P, Teixeira J, Vicente A. Starch determination in Chlorella vulgaris-a comparison between acid and enzymatic methods. J Appl Phycol. 2012;24(5):1203–8. https://doi.org/10.1007/s10811-011-9761-5.
Piattoni CV, Ferrero DML, Dellaferrera I, Vegetti A, Iglesias AÁ. Cytosolic glyceraldehyde-3-phosphate dehydrogenase is phosphorylated during seed development. Front Plant Sci. 2017;8:522. https://doi.org/10.3389/fpls.2017.00522.
Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72(1-2):248–54. https://doi.org/10.1016/0003-2697(76)90527-3.
Gallagher RS, Ananth R, Granger K, Bradley B, Anderson JV, Fuerst EP. Phenolic and short-chained aliphatic organic acid constituents of wild oat (Avenafatua L.) seeds. J Agric Food Chem. 2010;58:218–25. https://doi.org/10.1021/jf9038106.
Serea CP, Barna O. Phenolic content and antioxidant activity in milling fractions of oat. J Agroaliment Process Technol. 2011;17:291–4.
Hanshew AS, Mason CJ, Raffa KF, Currie CR. Minimization of chloroplast contamination in 16S rRNA gene pyrosequencing of insect herbivore bacterial communities. J Microbiol Methods. 2013;95(2):149–55. https://doi.org/10.1016/j.mimet.2013.08.007.
Bulgarelli D, Rott M, Schlaeppi K, van Themaat EVL, Ahmadinejad N, Assenza F, et al. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature. 2012;488(7409):91–5. https://doi.org/10.1038/nature11336.
Chen S, Zhou Y, Chen Y, Gu J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–90. https://doi.org/10.1093/bioinformatics/bty560.
Magoč T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27(21):2957–63. https://doi.org/10.1093/bioinformatics/btr507.
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27(16):2194–200. https://doi.org/10.1093/bioinformatics/btr381.
Stackebrandt E, Goebel BM. Taxonomic note: a place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int J Syst Bacteriol. 1994;44(4):846–9. https://doi.org/10.1099/00207713-44-4-846.
Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10(10):996–8. https://doi.org/10.1038/nmeth.2604.
Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73(16):5261–7. https://doi.org/10.1128/AEM.00062-07.
Schloss PD, Gevers D, Westcott SL. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS One. 2011;6(12):e27310. https://doi.org/10.1371/journal.pone.0027310.
Acknowledgments
We acknowledge Shanghai Majorbio Bio-pharm Technology Co., Ltd., for the data were analyzed on the free online platform of Majorbio I-Sanger Cloud Platform (www.i-sanger.com).
Funding
This work was supported by the Natural Science Foundation of Liaoning Province (2019-ZD-0466), the Educational Department of Liaoning Province (603200052092) and National Natural Science Foundation of China (No. 32070352).
Author information
Authors and Affiliations
Contributions
HFW and QLL managed the project. HFW and WJL designed the experiments. HFW and RG prepared the samples. YLG and XYL performed the experiments. HFW, YLG and XYL completed the data analysis and preparation of Figure and Table. YGX submitted sequence data to GeneBank. HFW wrote the manuscript. MPNRrevised the manuscript and prepared its final version. All authors have read and agreed to the published version of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
Representative image of sterilized-surface dimorphic seeds on TSA agar medium incubated for 3 d at 25 °C. Br:brown seeds; Bl:black seeds.
Additional file 2: Fig. S2.
The relationship between two sample groups and dominant endophytic bacterium at the species level. Br:brown seeds; Bl:black seeds.
Additional file 3: Table S1.
The relative abundance of the genus in each sample (cutoff of 0.01).
Additional file 4: Table S2.
The relative abundance of the phylum in each sample (cutoff of 0.01).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, H., Narsing Rao, M., Gao, Y. et al. Insights into the endophytic bacterial community comparison and their potential role in the dimorphic seeds of halophyte Suaeda glauca. BMC Microbiol 21, 143 (2021). https://doi.org/10.1186/s12866-021-02206-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-021-02206-1