
Abstract
Background
Mosquitoes are important vectors for a range of diseases, contributing to high rates of morbidity and mortality in the human population. Culex pipiens pallens is dominant species of Culex mosquito in northern China and a major vector for both West Nile virus and Bancroftian filariasis. Insecticide application were largely applied to control the mosquito-mediated spread of these diseases, contributing to increasing rates of resistance in the mosquito population. The voltage-gated sodium channel (Vgsc) gene is the target site of pyrethroids, and mutations in this gene cause knockdown resistance (kdr). While these kdr mutations are known to be critical to pyrethroid resistance, their evolutionary origins remain poorly understood. Clarifying the origins of these mutations is potential to guide further vector control and disease prevention efforts. Accordingly, the present study was designed to study the evolutionary genetics of kdr mutations and their association with the population structure of Cx. p. pallens in Shandong province, China.
Methods
Adult Culex females were collected from Shandong province and subjected to morphological identification under a dissection microscope. Genomic DNA were extracted from the collected mosquitoes, the Vgsc gene were amplified via PCR and sequenced to assess kdr allele frequencies, intron polymorphisms, and kdr codon evolution. In addition, population genetic diversity and related population characteristics were assessed by amplifying and sequencing the mitochondrial cytochrome C oxidase I (COI) gene.
Results
Totally, 263 Cx. p. pallens specimens were used for DNA barcoding and sequencing analyses to assess kdr allele frequencies in nine Culex populations. The kdr codon L1014 in the Vgsc gene identified two non-synonymous mutations (L1014F and L1014S) in the analyzed population. These mutations were present in the eastern hilly area and west plain region of Shandong Province. However, only L1014F mutation was detected in the southern mountainous area and Dongying city of Shandong Province, where the mutation frequency was low. Compared to other cities, population in Qingdao revealed significant genetic differentiation. Spatial kdr mutation patterns are likely attributable to some combination of prolonged insecticide-mediated selection coupled with the genetic isolation of these mosquito populations.
Conclusions
These data suggest that multiple kdr alleles associated with insecticide resistance are present within the Cx. p. pallens populations of Shandong Province, China. The geographical distributions of kdr mutations in this province are likely that the result of prolonged and extensive insecticide application in agricultural contexts together with frequent mosquito population migrations. In contrast, the low-frequency kdr mutation detected in central Shandong Province populations may originate from the limited selection pressure in this area and the relative genetic isolation. Overall, the study compares the genetic patterns revealed by a functional gene with a neutral marker and demonstrates the combined impact of demographic and selection factors on population structure.
Similar content being viewed by others


Background
Mosquitoes are important vectors for a wide range of pathogenic organisms [1]. Culex species are the most common mosquitos in the world, contributing to pathogen emergence and spread through their ability to consume blood meals from humans and animals [2]. Rates of Culex-mediated disease outbreaks are becoming increasingly frequent, as in the case of the 2019 outbreak of Eastern equine encephalitis virus (EEEV) in the United States, which caused 34 infections and 11 deaths [3]. Culex mosquitos facilitate these spillover events in which EEEV and other deadly diseases are transmitted into humans, horses, and other so-called ‘dead end’ host species [4]. Predicting these outbreaks, however, remains very challenging owing to differences associated with monitoring and predicting mosquito distributions, environmental conditions, human activity, and reservoir-host interactions [5]. In northern China, the most prevalent domestic species of mosquito is Cx. p. pallens, which serves as the primary vector for bancroftian filariasis in Shandong Province [6], in addition to potentially facilitating West Nile Virus (WNV) transmission in some areas of China [7]. Studies of Cx. p. pallens population genetic structure and evolutionary genetics is potential to inform efforts to prevent vector-borne disease spread and to better evaluate the disease risk in different areas.
Insecticide application is the predominant mode of disease control at present [8, 9]. However, excessive and widespread use has provoked the emergence of high levels of insecticide resistance which are defined by the Insecticide Resistance Action Committee (IRAC) as “the selection of a heritable characteristic in an insect population resulting in the repeated failure of an insecticide product to provide the intended level of control when used as recommended ”[10, 11]. Such resistance continues to hamper efforts to limit the risk of mosquito-borne disease owing to their high levels of rapid toxicity in target insect populations together with relatively limited mammalian toxicity, pyrethroid insecticides have been frequently employed for both indoor and outdoor spraying aimed at controlling mosquito populations in China since the 1980s [12, 13]. Vgsc mutation-mediated knockdown resistance is among the leading causes of pyrethroid resistance [14]. Liu et al. explored the relationship between insecticide resistance and kdr mutations among mosquito populations in Shandong province, revealing a positive correlation between kdr mutations and mosquito survival rate, suggesting that analyses of kdr mutations can offer value as a biomarker for studies of pyrethroid resistance in Cx. p. pallens populations [15].
It is worth nFoting that some seemingly contradictory results have been reported in prior studies. For one, there is some level of disagreement between studies screening mosquitoes for resistance and studying the role of knockdown resistance in the development of resistance. For example, Su et al. applied deltamethrin to a sensitive population of Aedes albopictus to screen for resistance and found that the F1534S mutation was significantly associated with early resistance in this species, and that when resistance rates were high, a novel F1534L mutation was closely associated with resistance [16]. When screening a natural Cx. p. pallens population using pyrethroids, Shi et al. found kdr to be a major contributor to the early stages of resistance development, with a positive correlation between L1014F mutation frequency and resistance levels together with reductions in kdr gene polymorphisms such that the final population only contained a single haplotype [17]. At present, evolutionists believe that the above results are not truly contradictory, as the former is the result of de novo mutations at the target site under the selective pressure of insecticides, while the latter is the result of standing variation screening. Second, diametrically opposing results can also occur when monitoring wild populations. For example, Gao et al. suggested that the I1532T mutation was negatively correlated only with deltamethrin resistance in Ae. albopictus [18], while Liu et al. provided the first demonstration that the I1532T mutation was positively correlated with the deltamethrin-resistant phenotype [19]. This may be caused by the different resistance levels observed in wild populations. Researchers believe that mutation frequencies associated with drug resistance phenotypes caused by single-gene mutations increase exponentially. During the initial stage, the incidence of these mutations remains very low such that they are almost undetectable. However, when the resistance reaches a certain level, this mutation frequency will increase rapidly. In addition, knockdown resistance genes can also be affected by multi-site mutations. For example, the V1016G mutation can induce resistance alone, while the presence of both V1016G and S989P can induce stronger resistance. The V1016I does not directly cause resistance but can enhance resistance phenotypes associated with the F1534C mutation. The insecticide resistance phenotypes of mosquitoes are the result of multiple genes or multiple loci mutations in the same gene, together with joint action and long-term inheritance. Therefore, kdr mutation polymorphisms, frequencies, and haplotypes are used to trace the origins and development of resistance [20]. Knowledge of mosquito pyrethroid resistance characteristics would offer important guidance for vector control efforts. Detailed genetic analyses of the loci associated with insecticide resistance in particular local populations would also provide insight into the different mechanisms that contribute to such insecticide resistance in that region [21]. While kdr mutations are known to be key drivers of resistance to pyrethroid, the precise evolutionary origins of these mutations and their development in Cx. p. pallens populations from Shandong province remain to be characterized.
The present study was conducted to assess the patterns of kdr allele distribution among Cx. p. pallens in Shandong province, to determine whether these mutations were the result of one or more evolutionary origins, and to evaluate the impact of geographical isolation and demographic history on kdr mutation evolution in this mosquito population. Analyses of whether resistance-related mutations in a population are derived from a single emergence event and subsequent spreading or evolved independently on multiple occasions can highlight the relative importance that migration and mutation rates play in the dynamics of insecticide resistance. By analyzing neutral biomarker genes and assessing the genetic variation therein, it is possible to gain additional insight into demographic-related population structure. Mitochondrial DNA (mtDNA) markers have emerged as popular targets for analysis when studying genetic diversity and population structure given that they are maternally inherited, do not undergo recombination, are highly variable, and thus necessitate a relatively small target population to facilitate robust analyses [22, 23]. The present study was developed to assess neighboring kdr introns in the Vgsc gene while utilizing COI gene as a marker in order to simultaneously study the evolutionary history of kdr mutations and the population structure of Cx. p. pallens communities in Shandong Province, China.
Methods
Sample collection
Shandong Province is located in Eastern China along the lower reaches of the Yellow River, serving as a primary coastal province. The region exhibits a temperate monsoon climate that is often warm and rainy, making it well-suited to mosquito breeding. Cx. p. pallens breeding primarily takes place in moderately polluted standing bodies of water located near to human settlements. For this study, adult female Cx. p. pallens specimens were collected from Zibo and Linyi in the southern mountainous area of Shandong; Rizhao, Yantai, and Qingdao in the eastern hilly area of Shandong; and Heze, Dongying, Dezhou, and Liaocheng in the northwest plain region of Shandong from 2021 to 2022 (Table 1). Collected specimens were stored in 100% ethanol at − 80 °C. DNA was extracted from individual mosquitos with the Cador® Pathogen 96 QIAcube® HT Kit based on provided directions and stored at − 80 °C.
COI gene sequencing
The 651 bp COI gene was amplified with the LCOI490 (5′-GGT CAA CAA ATC ATA AAG ATA TTG G-3′) and HCO2198 (5′-TAA ACT TCA GGG TGA CCA AAA AAT CA-3′) primers via PCR. Each PCR reaction contained 12.5 μl of GoTaq Green Master Mix, 1 μl of each primer (10 μmol/l), 2 μl of template DNA, and nuclease-free water to a final volume of 25 μl. Thermocycler settings were: 94 °C for 1 min; 5 cycles of 94 °C for 40 s, 45 °C for 40 s, and 72 °C for 1 min; 30 cycles of 94 °C for 40 s, 53 °C for 40 s, and 72 °C for 1 min; and 72 °C for 5 min. Amplicons were electrophoretically separated and directly sequenced with the Big-Dye kit (Sangon Biotech Co., Ltd., Shanghai, China).
Vgsc gene sequencing
The amplification of 521 bp of the Vgsc gene including the kdr codon at position 1014 was conducted with the kdr-F (5′-C CT GCC ACG GTG GAA CTT C-3′)and kdr-R (5′-GGA CAA AAG CAA GGC TAA GAA-3′) and primers each PCR reaction contained 12.5 μl of GoTaq Green Master Mix, 1 μl of each primer (10 μmol/l), 2 μl of template DNA, and nuclease-free water to a final volume of 25 μl. Thermocycler settings were: 94 °C for 1 min; 30 cycles of 98 °C for 10 s (denaturation), 55 °C for 15 s, and 68 °C for 30 s; and 68 °C for 1 min. Amplicons were electrophoretically separated and directly sequenced with the Big-Dye kit (Sangon Biotech Co., Ltd., Shanghai, China).
Data analysis
Compared to Cx. p. pallens gene sequences in the GenBank repository using NCBI’s (National Center for Biotechnology Information) online BLAST (Basic Local Alignment Search Tool) algorithmic program. The COI gene exhibited a > 99% match to the expected Cx. p. pallens gene sequence, and Bioedit v 7.0 was used to align COI and kdr sequences. DnaSPv6 [24] was used to assess numbers of haplotypes and variable sites, haplotype diversity, and nucleotide diversity. Arlequin 3.5 was used to assess the coefficient of fixation (Fst) and gene flow (Nm) among groups. Departures from the expected neutral DNA sequence variability were detected through statistical neutrality testing [25]. Tajima’s D [26] is based on differences between estimated numbers of segregating sites and average numbers of pairwise differences. Intraspecific molecular polymorphism data are necessary to conduct the D and F tests developed by Fu & Li, with the latter being based on haplotype frequency distributions [27]. All calculations were performed using DnaSPv6 [24], as were mismatch distribution analyses. PopART was used to construct a network evolution tree with the Neighbor-Joining Network model passed on kdr haplotypes. MEGA v.7.0 was used to construct a Neighbor-Joining phylogenetic tree model based upon genetic distance values [28].
Results
PCR amplification of the Vgsc gene in these Cx. p. pallens specimens generated a 521 bp PCR fragment. The sequence data for this gene were analyzed to assess kdr allele frequency distributions, kdr codon evolution, polymorphisms in the neighboring intron downstream of the kdr codon, and population genetic diversity. PCR amplification of the COI gene generated a 651 bp PCR fragment. As the mitochondrial genome is maternally inherited and haploid, drift towards different haplotype frequencies will occur more rapidly within isolated populations, resulting in differentiation roughly twice that observed for nuclear markers. Accordingly, these mtDNA sequences were analyzed to assess population variations, population expansion, spatial population structure, genetic differentiation, and patterns of gene flow.
Kdr allele frequency distributions
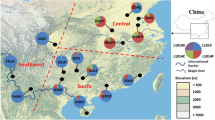
The wild-type allele (TTA/L) and the mutant alleles (TTT/F, TTC/F, TCA/S) were detected at codon 1014. A total of eight genotypes, including the wild-type genotype TTA/TTA (L/L), the wild-type/mutant heterozygotes TTA/TTT (L/F), TTA/TCA (L/S), and the mutant genotypes TCA/TCA (S/S), TTT/TTT (F/F), TTC/TTC (F/F), TTC/TTT (F/F) and TTT/TCA (F/S), were detected at codon 1014 (Table 2). The kdr mutant allele frequencies were found to vary with geographic location. L1014F and L1014S were observed in the northwest plain region (Dezhou, Liaocheng, Heze) and eastern hilly area (Qingdao, Rizhao, Yantai) of Shandong Province. In the northwest plain region, L1014F was the predominant mutation and L1014S was also common. However, only the L1014F was detected in the southern mountainous area and Dongying city at a low mutational frequency (Fig. 1).

Distribution and frequency of kdr mutations in Cx. p. pallens in Shandong province. The blue color in the pie charts represents the wild type kdr L1014 haplotype, the green color represents the mutant kdr L1014F haplotype and the red color represents the mutant kdr L1014S haplotype. DZ(Dezhou); LC(Liaocheng); HZ(Heze); DY(Dongying); ZB (Zibo); LY(Linyi); YT(Yantai); QD(Qingdao); RZ(Rizhao).TS(Taishan); YS(Yishan); MS(Mengshan); NS(Nishan)
Kdr haplotype diversity
In total, 28 polymorphic sites were identified in the study. Genetic diversity indices and neutrality tests (Fu’s Fs and Tajima’s D) were performed using the neighboring intron downstream of the kdr allele in Cx. p. pallens, with 520 kdr intron sequences being included in these analyses. As these polymorphic sites exhibited heterozygous phenotypes, a phase analysis approach was used for haplotype predictions, leading to the detection of 31 haplotypes from 9 populations (Table 3). Overall respective haplotype diversity (Hd) and nucleotide diversity (Pi) values were 0.664 and 0.0521. The Heze populations exhibited low Hd (0.351) and Pi (0.01261), whereas these values were high in other populations. No significant departures from neutrality were detected in any of these populations using Tajima’s D test, whereas Fu’s F test detected significant departures in the Qingdao and Zibo populations, with positive F test results indicating low levels of high-frequency populations consistent with a population size reduction and/or balancing selection (Table 3).
Genealogical analyses of kdr mutations
To estimate the evolutionary relationship of kdr mutations, 31 haplotypes were identified in this study. Two haplotypes were predominant in the populations: H01 (52.5%) and H02 (23.8%) (Fig. 2). The other detected haplotypes exhibited only limited geographic distributions or were unique to individual populations. Overall, there were 13 unique haplotypes and 18 shared haplotypes, with the former accounting for 58.1% of all haplotypes. The H01 and H02 haplotypes were shared among all study sites, while substantial haplotype sharing was also observed between the Liaocheng and Dezhou populations (including H06, H11, H13, and H15). Many shared haplotypes were also observed among the Qingdao, Dongying, Yantai, and Rizhao populations. In addition to these eastern coastal cities, the Qingdao population exhibited only limited sharing of haplotypes with other regions suggesting that this population is relatively differentiated from other populations, in line with the genetic differentiation coefficient Fst. Other than the H01 and H02 haplotypes, only one shared haplotype was present in the Zibo population. The geographically non-adjacent Yantai and Liaocheng populations shared seven haplotypes, with a negative genetic differentiation coefficient Fst is − 0.00859, suggesting that no appreciable genetic differentiation and high levels of communication between these populations. Given the limited sampling at each study site and associated time constraints, further analyses will be needed to confirm and expand upon these findings.

Geographic distribution of Intron haplotypes of kdr allele in the 9 Cx. p. pallens populations. The area of the circle represents the haplotype frequency, different colors indicate different haplotypes. DZ(Dezhou); LC(Liaocheng); HZ(Heze); DY(Dongying); ZB (Zibo); LY(Linyi); YT(Yantai); QD(Qingdao); RZ(Rizhao)
Network analysis results revealed a complex reticulate pattern and multiple independent mutational events resulting in the observed kdr haplotypes (Fig. 3). The H11 haplotype is largely in the center of this network as it is most closely related to the other haplotypes such that it may be a more evolutionarily ancient haplotype that can undergo mutation through one or more steps to yield the other haplotypes. Haplotypes H01-F and H01-S comprise the majority of samples in all populations and may be derived from ancestral H01-L, H08-L, H03-L, and H12-L haplotypes through one or two mutational steps. Likewise, the H02-F and H02-S haplotypes may be derived from the ancestral H02-L, H08-L, H010-L, and H18-L haplotypes through one or two mutational steps. The H05-F haplotype found in Heze stems from H01-L through one mutational step, while the H26-F haplotype from Yantai and Liaocheng is likely to have arisen from H11-L through three mutational steps. The H16-F and H29-F haplotypes are derived from a shared ancestor lacking the kdr haplotype. Overall, these genealogical analyses suggest that as few as one mutational step may differentiate between resistant and susceptible phenotypes among Cx. p. pallens.

kdr haplotype networks showing the genealogical relationship for Cx. p. pallens. Each haplotype is represented by a circle with size proportional to its frequency in the sample. Different colors indicate different populations. Lines represent variable asynchronous numbers; black solid circles represent missing haplotypes. The note above the circle referred to the mutation position and base. H: haplotype. H01LFS represent H01-L, H01-F and H01-S; H02LFS represent H02-L, H02-F and H02-S
Analyses of mtDNA sequence variation
A total of 520 mtDNA- COI sequences were next conducted polymorphism analyses, leading to the identification of 8 variable sites and 9 haplotypes (Table 4). The overall haplotype diversity (Hd) and nucleotide diversity (Pi) values were 0.306 and 0.00052, respectively. The low haplotype diversity and low nucleotide diversity indicated that different geographic populations may have experienced a “bottleneck effect”. Dezhou population exhibited the highest levels of diversity, whereas the Dongying and Zibo populations were the least diverse. The haplotype network graph constructed based on Median-joining method shows a radiative evolutionary center with a star-shaped distribution (Fig. 4). In total, 2 and 7 shared and unique haplotypes were identified, respectively. Of these, the H01 haplotype was the most widely distributed primitive haplotype, radiating out to one shared haplotype and multiple unique haplotypes. This star-shaped network phylogeny of this network also supports the notion that these Cx. p. pallens populations expanded following population bottleneck events, but that these populations are still undergoing expansion. The H04 haplotype was shared among the Rizhao, Qingdao, Dezhou, and Yantai populations. Other haplotypes were only present at low frequencies. H02, H03, and H09 were respectively unique to the Heze, Linyi, and Liaocheng populations, while H05-H08 were unique to the Dezhou population. The observation that these haplotypes were unique to these respective regions may be a consequence of limited sample selection or may be a result of genetic variation among these geographic environments. To better assess the Cx. p. pallens population structure and phylogenetic relationships among these populations, a Neighbor-joining (NJ) tree incorporating these 9 COI sequence-based haplotypes was constructed (Fig. 5), revealing that all haplotypes clustered in a single branch.

Phylogenetic network of 9 mitochondrial haplotypes of the COI gene in Cx. p. pallens in Shandong province. The haplotype network graph constructed based on Median-joining method. The size of each circle is proportional to its corresponding frequencies. Different colors indicate different populations. The number above the line referred to variable asynchronous numbers

Phylogenetic analysis based on COI haplotype variation. Marked with a solid red circle are haplotypes in the current study; others were retrieved from GenBank. Haplotypes marked with a solid blue square are associated with Cx. p. pallens. Neighbor-joining trees were constructed via the maximum composite likelihood substitution model using MEGA (version 7.0). Numbers at branches represent bootstrap values of 1000 replicates (values > 50 are shown). The scale-bar shows the number of nucleotide substitutions per site
Genetic differentiation among populations
An analysis of molecular variance (AMOVA) revealed that genetic differences of COI gene among and within populations respectively accounted for 18.3 and 81.7%, with a fixation index Fst value is 0.18293. As such, within-populations variance is the primary source of genetic variation among the Cx. p. pallens populations of Shandong Province. The pairwise Fst and Nm value estimates further highlight genetic differentiation among these populations (Table 5). A range of factors can contribute to the genetic differentiation of populations, including transport or other human activities, landscape barriers, and geographical distances among these populations. Fst values for the genetic differentiation index among these different mosquito populations ranged from − 0.00802 to 0.46939. The only negative Fst values were those between the Dezhou and Yantai populations. No significant differentiation was observed among 14 pairs of populations (Fst < 0.05; 38.9%), while moderate differentiation (0.05 < Fst < 0.15), high differentiation (0.15 < Fst < 0.25), and very high differentiation (Fst > 0.25) were respectively observed among 7 (19.4%), 8 (22.2%), and 7 (19.4%) pairs of populations. The highest Fst value was observed between the Qingdao-Heze, Qingdao-Zibo, Qingdao-Dongying populations (Table 5). Nm values corresponding to the gene flow among these populations were between − 31.43 and 134.26. Together, these findings were thus consistent with roughly half of the populations exhibiting high or very high levels of genetic differentiation and low levels of gene flow among these populations. Fixed coefficient Fst values between pairs of populations ranged between − 0.00802 and 0.46939 (Table 5), and the highest levels of genetic differentiation were observed when comparing the mosquito populations from Qingdao and either Dongying/Zibo (Fst = 0.46939; Nm = 0.28), owing to the presence of the H4 haplotype in Qingdao and its absence in Dongying/Zibo.
Population demography history
In an effort to infer the demographic history of Cx. p. pallens populations in Shandong Province, Tajima’s D and Fu’s Fs tests were next conducted, with the results and simulated P-values being shown in Table 4. Negative Tajima’s D and Fu’s Fs values were obtained for the Liaocheng, Dezhou, Linyi and Heze populations, suggesting high numbers of low-frequency mutations in these populations and that they are actively undergoing expansion. The frequency distributions for pairwise differences between sequences were also assessed to evaluate historical demographic expansions. Mismatch distributions can be used to visually assess historical population dynamics. Two models correspond to expected DNAsp values, with one assuming that the population size remains constant and the other assuming that the population grows or declines. When observations conform to one of these models, it indicates that the population is in line with the expected model-derived values and the population is constant or shrinking, whereas if the values deviate from expected values this indicates the population is expanding with a single-peak curve. The present results indicated that actual observations were consistent with hypothetical model values, thus indicating that the Cx. p. pallens populations in Shandong were in a state of genetic equilibrium.
Discussion
In the study, patterns of kdr mutations associated with insecticide resistance in Cx. p. pallens populations were studied by the Vgsc gene from mosquito populations in Shandong Province. The origins of these kdr mutations were explored through haplotype networks and comparisons of regional variability in mutation frequencies among these populations. The COI gene was further used to analyze the population structure in these study sites to clarify the differences among regions, supporting a model in which both geographical isolation and varying levels of selection pressure have shaped these communities.
Geographical isolation
Based on mtDNA sequencing, significant genetic differentiation was detected between populations from eastern hilly area of Shandong and southern mountainous area of Shandong. When comparing these two regions the Fst value was 0.28 consistent with substantial genetic differentiation. Notably, the Fst values when comparing the Qingdao population and other populations ranged from 0.059–0.469. Nm values were consistent with these findings, reaffirming the higher level of genetic differentiation for the Qingdao population as compared to the other studied populations. This may be a result of the fact that the sampling site was located in the Huangdao district of Qingdao. Historically, Huangdao was an isolated island surrounded by the sea, making travel to inland areas challenging. However, the Qingdao Jiao Zhou Bay Bridge and the Jiao Zhou Bay Subsea Tunnel were completed in 2010 and 2011 respectively. The development of transportation methods has facilitated transportation between Huangdao and other cities, and economic advances continue to contribute to rising transregional, transnational, and transcontinental vessel exchanges. Beginning in 2010, Liu et al. began investigating the association between kdr mutations and insecticide resistance in Cx. p. pallens of Shandong province, they found lower frequencies of mutations in Qingdao populations as compared to other study sites. The World Health Organization (WHO) standard resistance bioassay was applied to test insecticides resistance in Cx. p. pallens, Huangdao district populations exhibited a mortality rate of 83.8% consistent with preliminary resistance, whereas mortality rates in Jining populations were just 37.3% consistent with high levels of resistance [18]. Compared to studies from a decade ago, kdr mutations frequency in Qingdao populations significantly increased in this study, that may be because the Qingdao Jiao Zhou Bay Bridge and the Jiao Zhou Bay Subsea Tunnel were completed, promoting communication among different populations, but the main reason is related to the using of pesticides. The Huangdao port thus provides a potential site not only for the interactions among people and the dissemination of goods and services, but also for the spread of infectious diseases. The Huangdao port is currently an important node in over 150 international routes, engaging in trade with over 450 ports in over 130 countries. Over 60 international long-distance sea routes connect this port to far-flung areas including South Africa, the Black Sea, Europe, the Americas, the Mediterranean Sea, and Australia. Invasive mosquito vector species are being passively transported to new areas by humans for decades. However, current levels of mobility with a worldwide increased flow of people and commodities have raised the risk of the introduction of invasive mosquito species considerably. Mosquito surveillance in maritime ports of entry is crucial to allow the early detection of invasive mosquito species [29]. A total of 136 whole blood samples were collected from malaria suspect cases at Qingdao port from 2014 to 2019. Forty-six malaria cases were confirmed with the rate 33.82%. There were no local malaria cases in Qingdao City, the prevention and control of imported malaria is the focus of the future work [30]. Accordingly, there is a strong need to develop appropriate port quarantine protocols, to improve the health services provided in port cities, and to improve public health emergency response measures to ensure public safety.
The impact of insecticide-associated selection
Heze is situated in southwestern Shandong Province in the lower reaches of the Yellow River, and contains extensive areas of flat terrain with deep soil that is well-suited to agricultural use. It is a primary site of grain production in this province and bordering Anhui, Henan, and Jiangsu provinces to which good transportation is available. Vector-borne disease outbreaks have been historically common in Heze, and it was the last city in Shandong Province to eliminate new malaria cases [31]. Relative to other analyzed populations, the Cx. p. pallens population from Heze exhibited lower levels of genetic variation in the intronic regions associated with and a high frequency of kdr mutation (L1014F), indicating that the low levels of intronic variation may be attributable to a selective sweep, referring to a rapid increase in the frequency of a given allele as a consequence of directional selection, tied to high levels of intensive insecticide application and/or from the fixation of this resistance genotype. During a selective sweep, linked alleles in regions flanking the allele subject to selective pressure can also rise in frequency, contributing to a drop in overall genetic variation [32]. Comparisons of the DNA sequences flanking the kdr L1014 codon indicated a high degree of genetic differentiation between the Heze population and other analyzed populations consistent with strong local selection for this resistance allele in Heze. In contrast, these mutational frequencies were low in Dongying, Linyi, and Zibo, with only one mutant allele (L1014F) being present. Zibo is south of the Tai-Yi Mountain range, Linyi is adjacent to Zibo. From north to south, the three main mountains in Linyi are the Yi, Meng, and Ni mountains, which probably due to interrupted gene flow from eastern hilly or northwest plain region to southern mountainous area, caused by geographical isolations. While Dongying is in the northwest plain regions of Shandong province, it is near the end of the Yellow River and the coast of the Bohai sea where the flow of the Yellow River into the sea slows, forming a large delta that has arisen through the rapid deposition of high levels of silt sediment, resulting in shallow belowground water levels and extremely salinized soil that contributes to high levels of ecological fragility. On the one hand, currently, the saline-alkali land area in Dongying city is 350 km2, most crops cannot grow in saline-alkali soils, agricultural cultivation is relatively poor [33, 34]. Compared to other city, pesticides used less. On the other hand, saline-alkali stress reduces soil bacterial community diversity and soil enzyme activities and affects the metabolism of pesticides [35, 36]. Mosquito gut microbes may participate in the resistance of the host to insecticides to alleviate the effects of insecticides on the host [37]. Saline-alkali soil may affect the development of mosquito resistance, but the involved microorganism needs further research. When analyzing kdr codon L1014 sequences, significant departures from neutrality were observed in Linyi and Zibo, resulting from population bottleneck events. Rates of agricultural insecticide application in Dongying may be relatively low, potentially explaining the lack of insecticide resistance alleles in this population. Of the different neutrality tests, Fu’s Fs statistic exhibits the greatest power when detecting departures from neutrality based on a genetic hitchhiking model [32]. High Fu’s Fs measures for most of the study sites in Shandong province may be attributable to the high levels of insecticide use and strong selection placed on both the kdr locus and other regions of the genome. Over the past 50 years, insecticides have been widely used in agricultural settings and for vector control throughout central China [22]. The rapidly rising rates of resistance to these insecticides and the widespread kdr mutation distributions observed among these Cx. p. pallens populations underscore the need for the establishment of resistance surveillance plans and efforts to better manage insecticide efficacy.
The multiple origins of kdr mutations
The results of genealogical analyses of the intron downstream of the kdr codon together with the observed patterns of geographic distribution strongly indicate that these kdr mutations are derived from multiple mutational events rather than a single origin. Two non-synonymous codon L1014 mutations were detected among analyzed mosquito populations. While just one of these mutations was evident in central Shandong Province, both were found in the western and eastern coastal regions of the province. This is consistent with prior kdr genotyping results published for Cx. p. pallens, which revealed that the L1014F mutation was widely distributed throughout the Shandong region [38]. Two potential models can explain the emergence of kdr mutations. Specifically, these mutations may have arisen once and then spread over time, or they may have independently evolved in multiple geographically distinct populations. At least two independent mutational events were identified as having given rise to the L1014F or L1014S haplotypes through a single mutational step (Fig. 3). This is consistent with observations in Anopheles gambiae, which is the primary vector for malaria in Africa, in which a minimum of 4–5 independent non-synonymous mutations have been documented at position L1014 of the Vgsc gene [39]. Some Vgsc channel gene mutational events have been shown to be derived from a single mutation from a common ancestor, whereas others result from two mutational steps [40]. Network analyses revealed that just one mutational step can change H01-F/H01-S to H05-F (Fig. 3). Given these findings, H05-F may correspond to a kdr allele that evolved through sequential progression from H01-F/H01-S. Other species of insects have similarly been shown to exhibit multiple point mutation-mediated origins of resistance alleles in the voltage-gated sodium channel gene [39, 41], and multiple kdr mutation origins have been reported in Musca domestica and in the aphid Myzus persicae [42]. Moreover, kdr mutations of multiple origins are common among Culicinae mosquitoes, with relevant reports in Anopheles gambiae, Anopheles sinensis, and Aedes aegypti having been published to date [43, 44].
Conclusions
The geographic distribution of kdr haplotypes should reflect the interplay between the evolutionary forces of mutation, gene flow and selection. Overall, these data suggest that the multiple origins of kdr insecticide resistance alleles observed in Cx. p. pallens from Shandong Province are attributable to the prolonged and intensive application of insecticides in agricultural contexts. In contrast, the populations lacking these kdr mutations are more likely to be genetically isolated and free of substantial selection pressure. By comparing genetic patterns for both a gene functionally associated with insecticide resistance and a neutral marker gene, these findings simultaneously offer insight into the combined effects of selection and demographic factors on the resultant Cx. p. pallens population structure. In addition, more ecological studies are needed, relating levels of insecticide resistance with the genotypic composition at the kdr locus and with the analysis of other resistance mechanisms (e.g., metabolic, behavioral). These analyses would provide both valuable insight into the molecular evolution of pyrethroid resistance and a foundation for subsequent vector control efforts aimed at controlling the spread of mosquito-borne illness.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its additional files.
Abbreviations
- Vgsc :
-
Voltage-gated sodium channel
- kdr :
-
Knockdown resistance
- WNV:
-
West Nile virus
- EEEV:
-
Eastern equine encephalitis virus
- IRAC:
-
Insecticide Resistance Action Committee
- Hd :
-
Haplotype diversity
- Pi :
-
Nucleotide diversity
- COI :
-
Mitochondrial cytochrome C oxidase I gene
References
Wu SL, Sanchez CH, Henry JM, Citron DT, Zhang Q, Compton K, et al. Vector bionomics and vectorial capacity as emergent properties of mosquito behaviors and ecology. PLoS Comput Biol. 2020;16 4:e1007446. https://doi.org/10.1371/journal.pcbi.1007446. https://www.ncbi.nlm.nih.gov/pubmed/32320389
Gonzalez MA, Delacour-Estrella S, Bengoa M, Barcelo C, Bueno-Mari R, Eritja R, et al. A Survey on Native and Invasive Mosquitoes and Other Biting Dipterans in Northern Spain. Acta Parasitol. 2022;67(2):867–77. https://doi.org/10.1007/s11686-022-00529-1. https://www.ncbi.nlm.nih.gov/pubmed/35298775
Lindsey NP, Martin SW, Staples JE, Fischer M. Notes from the Field: Multistate Outbreak of Eastern Equine Encephalitis Virus - United States, 2019. MMWR Morb Mortal Wkly Rep. 2020;69(2):50–1. https://doi.org/10.15585/mmwr.mm6902a4. https://www.ncbi.nlm.nih.gov/pubmed/31945032
Morens DM, Folkers GK, Fauci AS. Eastern Equine Encephalitis Virus - Another Emergent Arbovirus in the United States. N Engl J Med. 2019;381(21):1989–92. https://doi.org/10.1056/NEJMp1914328. https://www.ncbi.nlm.nih.gov/pubmed/31747726
Bartlow AW, Manore C, Xu C, Kaufeld KA, Del Valle S, Ziemann A, et al. Forecasting Zoonotic Infectious Disease Response to Climate Change: Mosquito Vectors and a Changing Environment. Vet Sci. 2019;6 2 https://doi.org/10.3390/vetsci6020040. https://www.ncbi.nlm.nih.gov/pubmed/31064099
Huang X, Deng X, Kou J, Liu X, Wang H, Cheng P, et al. Elimination of Lymphatic Filariasis in Shandong Province, China, 1957–2015. Vector Borne Zoonotic Dis. 2020;20(12):875–81. https://doi.org/10.1089/vbz.2020.2624. https://www.ncbi.nlm.nih.gov/pubmed/32795160
Wondji CS, Priyanka De Silva WA, Hemingway J, Ranson H, Parakrama Karunaratne SH. Characterization of knockdown resistance in DDT- and pyrethroid-resistant Culex quinquefasciatus populations from Sri Lanka. Trop Med Int Health. 2008;13(4):548–55. https://doi.org/10.1111/j.1365-3156.2008.02033.x. https://www.ncbi.nlm.nih.gov/pubmed/18312471
Shaw WR, Catteruccia F. Vector biology meets disease control: using basic research to fight vector-borne diseases. Nat Microbiol. 2019;4(1):20–34. https://doi.org/10.1038/s41564-018-0214-7. https://www.ncbi.nlm.nih.gov/pubmed/30150735
van den Berg H, Velayudhan R, Yadav RS. Management of insecticides for use in disease vector control: Lessons from six countries in Asia and the Middle East. PLoS Negl Trop Dis. 2021;15 4:e0009358. https://doi.org/10.1371/journal.pntd.0009358. https://www.ncbi.nlm.nih.gov/pubmed/33930033
Sparks TC, Storer N, Porter A, Slater R, Nauen R. Insecticide resistance management and industry: the origins and evolution of the Insecticide Resistance Action Committee (IRAC) and the mode of action classification scheme. Pest Manag Sci. 2021;77(6):2609–19. https://doi.org/10.1002/ps.6254. https://www.ncbi.nlm.nih.gov/pubmed/33421293
Sparks TC, Nauen R. IRAC: Mode of action classification and insecticide resistance management. Pestic Biochem Physiol. 2015;121:122–8. https://doi.org/10.1016/j.pestbp.2014.11.014. https://www.ncbi.nlm.nih.gov/pubmed/26047120
Martinez-Torres D, Chandre F, Williamson MS, Darriet F, Berge JB, Devonshire AL, et al. Molecular characterization of pyrethroid knockdown resistance (kdr) in the major malaria vector Anopheles gambiae s.s. Insect Mol Biol. 1998;7(2):179–84. https://doi.org/10.1046/j.1365-2583.1998.72062.x. https://www.ncbi.nlm.nih.gov/pubmed/9535162
Dong K, Du Y, Rinkevich F, Nomura Y, Xu P, Wang L, et al. Molecular biology of insect sodium channels and pyrethroid resistance. Insect Biochem Mol Biol. 2014;50:1–17. https://doi.org/10.1016/j.ibmb.2014.03.012. https://www.ncbi.nlm.nih.gov/pubmed/24704279
Hadiatullah H, Zhang Y, Samurkas A, Xie Y, Sundarraj R, Zuilhof H, et al. Recent progress in the structural study of ion channels as insecticide targets. Insect Sci. 2022; https://doi.org/10.1111/1744-7917.13032. https://www.ncbi.nlm.nih.gov/pubmed/35575601
Liu HM, Cheng P, Huang X, Dai YH, Wang HF, Liu LJ, et al. Identification of TCT, a novel knockdown resistance allele mutation and analysis of resistance detection methods in the voltage-gated Na(+) channel of Culex pipiens pallens from Shandong Province, China. Mol Med Rep. 2013;7(2):525–30. https://doi.org/10.3892/mmr.2012.1184. https://www.ncbi.nlm.nih.gov/pubmed/23151871
Su X, Guo Y, Deng J, Xu J, Zhou G, Zhou T, et al. Fast emerging insecticide resistance in Aedes albopictus in Guangzhou, China: Alarm to the dengue epidemic. PLoS Negl Trop Dis. 2019;13 9:e0007665. https://doi.org/10.1371/journal.pntd.0007665. https://www.ncbi.nlm.nih.gov/pubmed/31525199
Shi L, Hu H, Ma K, Zhou D, Yu J, Zhong D, et al. Development of Resistance to Pyrethroid in Culex pipiens pallens Population under Different Insecticide Selection Pressures. PLoS Negl Trop Dis. 2015;9 8:e0003928. https://doi.org/10.1371/journal.pntd.0003928. https://www.ncbi.nlm.nih.gov/pubmed/26275298
Gao JP, Chen HM, Shi H, Peng H, Ma YJ. Correlation between adult pyrethroid resistance and knockdown resistance (kdr) mutations in Aedes albopictus (Diptera: Culicidae) field populations in China. Infect Dis Poverty. 2018;7(1):86. https://doi.org/10.1186/s40249-018-0471-y. Published 2018 Sep 4. https://www.ncbi.nlm.nih.gov/pubmed/30176907
Liu H, Liu L, Cheng P, et al. Bionomics and insecticide resistance of Aedes albopictus in Shandong, a high latitude and high-risk dengue transmission area in China. Parasit Vectors. 2020;13(1):11. Published 2020 Jan 9. https://doi.org/10.1186/s13071-020-3880-2.
Chang X, Zhong D, Lo E, Fang Q, Bonizzoni M, Wang X, et al. Landscape genetic structure and evolutionary genetics of insecticide resistance gene mutations in Anopheles sinensis. Parasit Vectors. 2016;9:228. https://doi.org/10.1186/s13071-016-1513-6. https://www.ncbi.nlm.nih.gov/pubmed/27108406
Zhang Y, Zhang C, Wu L, Luo C, Guo X, Yang R, et al. Population genetic structure and evolutionary genetics of Anopheles sinensis based on knockdown resistance (kdr) mutations and mtDNA-COII gene in China-Laos, Thailand-Laos, and Cambodia-Laos borders. Parasit Vectors. 2022;15 1:229. https://doi.org/10.1186/s13071-022-05366-9. https://www.ncbi.nlm.nih.gov/pubmed/35754022
Loaiza JR, Bermingham E, Sanjur OI, Scott ME, Bickersmith SA, Conn JE. Review of genetic diversity in malaria vectors (Culicidae: Anophelinae). Infect Genet Evol. 2012;12(1):1–12. https://doi.org/10.1016/j.meegid.2011.08.004. https://www.ncbi.nlm.nih.gov/pubmed/21864721
Chang X, Zhong D, Fang Q, Hartsel J, Zhou G, Shi L, et al. Multiple resistances and complex mechanisms of Anopheles sinensis mosquito: a major obstacle to mosquito-borne diseases control and elimination in China. PLoS Negl Trop Dis. 2014;8 5:e2889. https://doi.org/10.1371/journal.pntd.0002889. https://www.ncbi.nlm.nih.gov/pubmed/24852174
Rozas J, Ferrer-Mata A, Sanchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, et al. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol Biol Evol. 2017;34(12):3299–302. https://doi.org/10.1093/molbev/msx248. https://www.ncbi.nlm.nih.gov/pubmed/29029172
Kimura M. Evolutionary rate at the molecular level. Nature. 1968;217(5129):624–6. https://doi.org/10.1038/217624a0. https://www.ncbi.nlm.nih.gov/pubmed/5637732
Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123(3):585–95. https://doi.org/10.1093/genetics/123.3.585. https://www.ncbi.nlm.nih.gov/pubmed/2513255
Fu YX. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147(2):915–25. https://doi.org/10.1093/genetics/147.2.915. https://www.ncbi.nlm.nih.gov/pubmed/9335623
Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol Biol Evol. 2016;33(7):1870–4. https://doi.org/10.1093/molbev/msw054. https://www.ncbi.nlm.nih.gov/pubmed/27004904
Wilke ABB, Vasquez C, Carvajal A, Moreno M, Petrie WD, Beier JC. Mosquito surveillance in maritime entry ports in Miami-Dade County, Florida to increase preparedness and allow the early detection of invasive mosquito species. PLoS One. 2022;17(4):e0267224. https://doi.org/10.1371/journal.pone.0267224. Published 2022 Apr 15. https://www.ncbi.nlm.nih.gov/pubmed/35427409
World Health Organization. World Malaria Report 2020[EB/OL]. (2020-11-30). https://apps.who.int/iris/handle/10665/337660.
Kong X, Liu X, Tu H, Xu Y, Niu J, Wang Y, et al. Malaria control and prevention towards elimination: data from an eleven-year surveillance in Shandong Province, China. Malar J. 2017;16 1:55. https://doi.org/10.1186/s12936-017-1708-0. https://www.ncbi.nlm.nih.gov/pubmed/28137270
Stephan W. Genetic hitchhiking versus background selection: the controversy and its implications. Philos Trans R Soc Lond B Biol Sci. 2010;365(1544):1245–53. https://doi.org/10.1098/rstb.2009.0278. https://www.ncbi.nlm.nih.gov/pubmed/20308100
Liu L, Wang B. Protection of Halophytes and Their Uses for Cultivation of Saline-Alkali Soil in China. Biology (Basel). 2021;10:5. https://doi.org/10.3390/biology10050353. https://www.ncbi.nlm.nih.gov/pubmed/33922035
Yang C, Sun J. Soil Salinity Drives the Distribution Patterns and Ecological Functions of Fungi in Saline-Alkali Land in the Yellow River Delta, China. Front Microbiol. 2020;11:594284. https://doi.org/10.3389/fmicb.2020.594284. https://www.ncbi.nlm.nih.gov/pubmed/33424797
Yang D, Tang L, Cui Y, Chen J, Liu L, Guo C. Saline-alkali stress reduces soil bacterial community diversity and soil enzyme activities. Ecotoxicology. 2022;31(9):1356–68. https://doi.org/10.1007/s10646-022-02595-7. https://www.ncbi.nlm.nih.gov/pubmed/36208367
Che J, Zhu YL, Li YH, Zhang R, Ruan ZY, Zhang W. Response of bacterial communities in saline-alkali soil to different pesticide stresses. Environ Sci Pollut Res Int. 2022;29(28):42709–19. https://doi.org/10.1007/s11356-021-16316-w. https://www.ncbi.nlm.nih.gov/pubmed/35088261
Soltani A, Vatandoost H, Oshaghi MA, Enayati AA, Chavshin AR. The role of midgut symbiotic bacteria in resistance of Anopheles stephensi (Diptera: Culicidae) to organophosphate insecticides. Pathog Glob Health. 2017;111(6):289–96. https://doi.org/10.1080/20477724.2017.1356052. https://www.ncbi.nlm.nih.gov/pubmed/28745553
Liu HM, Yang PP, Cheng P, Wang HF, Liu LJ, Huang X, et al. Resistance Level of Mosquito Species (Diptera: Culicidae) from Shandong Province, China. Int J Insect Sci. 2015;7:47–52. https://doi.org/10.4137/IJIS.S24232. https://www.ncbi.nlm.nih.gov/pubmed/26816489
Pinto J, Lynd A, Vicente JL, Santolamazza F, Randle NP, Gentile G, et al. Multiple origins of knockdown resistance mutations in the Afrotropical mosquito vector Anopheles gambiae. PLoS One. 2007;11:e1243. https://doi.org/10.1371/journal.pone.0001243. https://www.ncbi.nlm.nih.gov/pubmed/18043750
Gauthier N, Clouet C, Perrakis A, Kapantaidaki D, Peterschmitt M, Tsagkarakou A. Genetic structure of Bemisia tabaci Med populations from home-range countries, inferred by nuclear and cytoplasmic markers: impact on the distribution of the insecticide resistance genes. Pest Manag Sci. 2014;70(10):1477–91. https://doi.org/10.1002/ps.3733. https://www.ncbi.nlm.nih.gov/pubmed/24458589
Rinkevich FD, Hedtke SM, Leichter CA, Harris SA, Su C, Brady SG, et al. Multiple origins of kdr-type resistance in the house fly, Musca domestica. PLoS One. 2012;7 12:e52761. https://doi.org/10.1371/journal.pone.0052761. https://www.ncbi.nlm.nih.gov/pubmed/23285178
Alon M, Benting J, Lueke B, Ponge T, Alon F, Morin S. Multiple origins of pyrethroid resistance in sympatric biotypes of Bemisia tabaci (Hemiptera: Aleyrodidae). Insect Biochem Mol Biol. 2006;36(1):71–9. https://doi.org/10.1016/j.ibmb.2005.10.007. https://www.ncbi.nlm.nih.gov/pubmed/16360952
Zhou X, Yang C, Liu N, Li M, Tong Y, Zeng X, et al. Knockdown resistance (kdr) mutations within seventeen field populations of Aedes albopictus from Beijing China: first report of a novel V1016G mutation and evolutionary origins of kdr haplotypes. Parasit Vectors. 2019;12 1:180. https://doi.org/10.1186/s13071-019-3423-x. https://www.ncbi.nlm.nih.gov/pubmed/31014392
Cosme LV, Gloria-Soria A, Caccone A, Powell JR, Martins AJ. Evolution of kdr haplotypes in worldwide populations of Aedes aegypti: Independent origins of the F1534C kdr mutation. PLoS Negl Trop Dis. 2020;14 4:e0008219. https://doi.org/10.1371/journal.pntd.0008219. https://www.ncbi.nlm.nih.gov/pubmed/32298261
Acknowledgements
The authors would like to thank all the reviewers who participated in the review and MJEditor (www.mjeditor.com) for its linguistic assistance during the preparation of this manuscript.
Funding
This study was supported by grants from the National Natural Science Foundation of China [81871685 to MQG], Shandong Provincial Natural Science Foundation (ZR2020KH001 to HML), NHC Key Laboratory of Parasite and Vector Biology(National Institute of Parasitic Diseases, Chinese Center for Diseases Control and Prevention, NHCKFKT2021–02 to HML),Academic promotion programme of Shandong First Medical University (2019QL005)and The Innovation Project of Shandong Academy of Medical Sciences.
Author information
Authors and Affiliations
Contributions
CHZ, XJW, PC, LJL, XXG, HFW, ZWL, JJL, WQW and YTW provided the experimental data and wrote the manuscript. MQG and HML reviewed and edited the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zang, C., Wang, X., Cheng, P. et al. Evaluation of the evolutionary genetics and population structure of Culex pipiens pallens in Shandong province, China based on knockdown resistance (kdr) mutations and the mtDNA-COI gene. BMC Genomics 24, 145 (2023). https://doi.org/10.1186/s12864-023-09243-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-023-09243-2