
Abstract
Background
Aedes albopictus (Skuse) is an important vector of chikungunya, dengue, yellow fever and Zika viruses. In the absence of anti-viral medication and with limited availability of a commercial vaccine for public health use, vector control remains an effective means for reducing Aedes-borne disease morbidity. Knowledge about genetic mutations associated with insecticide resistance (IR) is a prerequisite for developing rapid resistance diagnosis, and the distribution and frequency of IR conferring mutations is important information for making smart vector control decisions.
Methods
Partial DNA sequences of domain II and domain III of Ae. albopictus voltage gated sodium channel (VGSC) gene were amplified from a total of 426 individuals, collected from 17 sites in the Beijing municipality. These DNA fragments were sequenced to discover the possible genetic mutations mediating knockdown resistance (kdr) to pyrethroids. The frequency and distribution of kdr mutations were assessed in the 17 Ae. albopictus populations. The origin of kdr mutations was investigated by haplotype clarification and phylogenetic analysis.
Results
Sequence alignments revealed the existence of multiple mutations (V1016G, I1532T, F1534S and F1534L) in VGSC. The highest frequency of the mutant 1016G allele (0.647) was found in Haidian, while 1016G was not detected in Huai Rou, Yan Qing, Ping Gu and Shun Yi. The frequency of 1532T was highest (0.537) in the population from the Olympic Forest Park (OFP, Chao Yang District), but not detectable in Huai Rou and Mi Yun. Two mutations were observed at codon 1534 with different distribution patterns: 1534L was only found in Tong Zhou (TZ) with a frequency of 0.017, while 1534S was distributed in TZ, OFP, Fang Shan, Da Xing and Shi Jing Shan with frequencies ranging from 0.019 (OFP) to 0.276 (TZ). One 1016G, one 1532T, one 1534L and two 1534S haplotypes were identified.
Conclusions
Multiple mutations (V1016G, I1532T, F1534L/S) in VGSC were found in Ae. albopictus in Beijing. This represents the first report of V1016G in Ae. albopictus. Sequence alignment and phylogenetic analysis revealed multiple origins of 1534S. The spatial heterogeneity in distribution and frequency of kdr mutations calls for a site-specific strategy for the monitoring of insecticide resistance. The relatively high frequencies of V1016G warn of a risk of pyrethroid resistance in mosquitoes in the urban zones.
Similar content being viewed by others


Background
The Asian tiger mosquito Aedes albopictus is a major vector of four important arboviruses, chikungunya virus, dengue virus, yellow fever virus and Zika virus [1]. Although Ae. albopictus is native to Southeast Asia, the Western Pacific islands and islands of the Indian Ocean, it has gradually spread in recent decades [2]. The current worldwide distribution of Ae. albopictus greatly increases the risk of vector-borne disease outbreaks and poses a global threat to public health [3,4,5]. In China, Ae. albopictus is the primary vector of dengue fever [6,7,8] and is susceptible to Zika virus [9].
Vector control remains an important means for the prevention and control of vector-borne epidemics by reducing the density of vector insects [10, 11]. Control of Aedes vectors currently relies on application of insecticides and habitat management [12]. However, continuous and intensive use of insecticides in the fields or in domestic setting has artificially created a direct or indirect selection pressure on vector insects, leading to the development of insecticide resistance (IR) [1, 12].
There are two major mechanisms conferring insecticide resistance in Aedes mosquitoes. One is metabolic resistance, mainly mediated by P450 monooxygenases [13, 14], glutathione transferases (GST) [15] and esterases (Est) [16]. The other is called target resistance, which is caused by point mutations in target proteins of insecticides, such as voltage gated sodium channels (VGSC), acetylcholinesterases and gamma aminobutyric acid receptors [17].
Pyrethroids are the frequently used insecticides for the control of adult mosquitoes because of its low mammalian toxicity and high and rapid activity in insects. This class of insecticide has been widely used as indoor/outdoor residual or space sprays for mosquito control in China since the 1980s [18]. VGSC are the primary target of pyrethroids in insects. VGSC mutation mediated knockdown resistance (kdr) is the common and main cause of resistance to pyrethroids in insects. Previous studies on Ae. aegypti have documented several point mutations (V410L, S989P, I1011M/V, V1016G/I, I1532T, F1534S/L/C, D1763Y; Musca domestica numbering) of VGSC [19,20,21,22,23,24,25,26,27,28], most of which locate in the S6 segment of domain II (IIS6) and IIIS6. These mutations are usually confined to specific geographical areas, and vary in frequency and effect on resistance [12]. However, only a few including V410L, S989P, I1011M, V1016G and F1534C, alone or in combination, have been confirmed to be associated with resistant phenotypes or functionally corresponded well to reduce VGSC sensitivity to pyrethroids [19,20,21,22,23,24,25,26]. Co-occurrence of certain mutations is commonly associated with higher levels of resistance [28,29,30]. For example, the combination of S989P, V1016G and F1534C alters the sensitivity to permethrin and deltamethrin by 1100-fold and 90-fold, respectively [30].
By contrast, fewer molecular and functional studies have been conducted on Ae. albopictus. The first kdr mutation (F1534C) was discovered in a Singapore population [31], and two different mutations at codon 1534 (F1534S/L) were later described [32,33,34,35]. In addition, a mutation at codon 1532 (I1532T) was detected in Italy [33] and China [18, 34]. So far, although amino acid replacements at residues 1532 and 1534 of VGSC have been documented in Chinese Ae. albopictus, mutations at other IR-related loci have not been well examined.
Beijing is the capital of China with a large human population. To reduce the risk of vector-borne diseases, mosquito control programs have been implemented for 65 years (since the patriotic health campaign was initiated in 1952 [36]). In particular, pest (including mosquito, fly, rat and cockroach) control campaigns have been conducted in order to build “healthy district” in all 16 districts of Beijing Municipality in the last 10 years. It is very likely that insecticide-resistant alleles have been selected in the field due to the regular and continual application of pyrethroids. However, no comprehensive study on IR status and IR associated genetic mutation has been reported on field Ae. albopictus populations in Beijing. In this study, we sequenced partial DNA sequences of the VGSC gene from field-caught Ae. albopictus samples, and evaluated the frequencies of kdr alleles within seventeen Ae. albopictus populations. In addition, we attempted to examine the origin of kdr mutations by haplotype clarification and phylogenetic analysis.
Methods
Samples
Aedes albopictus samples were obtained from 17 sites located in 16 administrative districts of Beijing. Adult mosquitoes were captured by CDC light trap baited with CO2 from 16:00 to 19:00 h during July-September in 2016 or 2017. Basic information about the sample collection is described in Table 1. The Aedes samples provided by the Beijing Center for Disease Control and Prevention (CDC) were further identified by a PCR method described by Higa et al. [37] using species-specific primers based on the rDNA-ITS sequence. The molecularly re-confirmed Ae. albopictus specimens were then used for kdr genotyping.
Amplification and sequence analysis of VGSC fragments
Primers V2F (5′-GAC AAT GTG GAT CGC TTC CC-3′) and V2R (5′-GCA ATC TGG CTT GTT AAC TTG-3′) [31] were used to amplify the partial sequence of domain II covering codons encoding amino acid residues 989, 1011 and 1016. The reaction was done in a final volume of 30 μl, comprising 0.3 μl of Ex Taq DNA polymerase (Takara Bio, Dalian, China), 3 μl of 10× buffer, 2.4 μl of dNTP, 0.6 μl of 10 μM V2F, 0.6 μl of 10 μM V2R, 200 ng of DNA template, and ddH2O to make up to 30 μl. The reaction procedure was as follows: 94 °C for 5 min, followed by 38 cycles of 94 °C for 30 s, 60 °C for 30 s and 72 °C for 40 s, with a final extension at 72 °C for 10 min. The PCR products were gel-purified and directly sequenced from both directions using the forward (V2F) and reverse (V2R) primers, respectively.
Primers V3F (5′-GAG AAC TCG CCG ATG AAC TT-3′) and V3R (5′-TAG CTT TCA GCG GCT TCT TC-3′) [31] were used to amplify the fragment of domain III containing codons 1532 and 1534. The reaction system comprised 0.3 μl of Ex Taq DNA polymerase (Takara Bio), 3 μl of 10× buffer, 2.4 μl of dNTP, 1.2 μl of 10 μM V3F, 1.2 μl of 10 μM V3R, 200 ng of DNA template, and ddH2O, in a total volume of 30 μl. The reaction procedure was as follows: 94 °C for 5 min, followed by 38 cycles of 94 °C for 30 s, 58 °C for 30 s and 72 °C for 40 s, with a final extension at 72 °C for 10 min. The PCR products were sequenced using the reverse primer (V3R).
The sequences of the fragments of domain II (D2) and the fragments of domain III (D3) were aligned by MUSCLE v.3.8 [38], and nucleotide polymorphisms (SNPs) were documented.
Haplotype identification
The haplotypes of VGSC alleles were identified by directly reading from homozygotes, or by splitting one from the other from heterozygotes carrying one-site variations, or by clone sequencing of heterozygotes with multiple-site variations. For clone sequencing, purified PCR products were ligated with the pEasy-T1 vector and transformed into competent cells of the Escherichia coli Trans 5α strain (Transgen, Beijing, China). Then, three to ten clones were sequenced. The sequences obtained from clone sequencing and from direct PCR product sequencing were cross-checked to clarify the two haplotypes in each heterozygote.
Phylogenetic analysis
The intron + exon sequences of confirmed haplotypes were used for phylogenetic analysis by the maximum likelihood method based on the Tamura-Nei model [39] using MEGA v.7 [40].
Results
Genetic mutations in Ae. albopictus VGSC
The domain II fragment (D2) contained partial exon 20, the full intron 20 and partial exon 21 of the Ae. albopictus VGSC gene (Fig. 1). Two non-synonymous mutations, S1000Y (TCC to TAC) and V1016G (GTA to GGA), were detected (Fig. 2). 1000Y was found in seven individuals from TZ, and existed in the heterozygous form. 1016G was found in both heterozygotes and homozygotes.

Schematic representation of the two regions of Ae. albopictus VGSC gene analyzed in this study. The intron-exon structure between the predicted initiation codon and stop codon is identified based on the genomic DNA sequence (MNAF02001058.1) and cDNA sequence (XM_019696540.1)

Example chromatograms showing the non-synonymous mutations identified in this study
The domain III fragment (D3) contained partial exon 28, the full intron 28 and partial exon 29 (Fig. 1). Three non-synonymous mutations I1532T (ATC or ATA to ACC), F1534S (TTC to TCC) and F1534L (TTC to TTG) were identified in exon 29 (Fig. 2). 1532T and 1534S existed in both heterozygous and homozygous forms. 1534L was only found in one heterozygote from TZ. The 1534C allele was not detected in our samples.
Frequency and distribution of kdr mutations
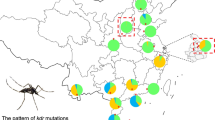
In the examined 17 populations, IR-related (kdr) mutations at three loci (1016, 1532 and 1534) were detected (Table 2, Fig. 2). The difference in distribution of each kdr mutation is obvious in Fig. 3. For example, diverse mutations at multiple sites were detected in TZ, while kdr mutation was not observed in HR. 1016G was detected in 13 of the 17 populations with a frequency ranging from 0.019 (MY) to 0.647 (HD). Specifically, the frequency of the 1016G allele was relatively high in the urban zones (e.g. DC, HD, SJS, FT), but low in rural areas. In particular, 1016G was not detected in four rural districts HR, YQ, PG and SY.

Distribution and frequency of kdr mutations in Ae. albopictus populations in Beijing. a Geographical position of the sampling sites. The shape file was downloaded from the GADM database of Global Administrative Areas (http://gadm.org/) on December 24, 2018. b 1016G mutation. c 1532T mutation. d 1534L/S mutations
1532T was found in 15 populations, with the highest frequency of 0.537 being in OFP. Two substitutions at codon 1534 (i.e. 1534L and 1534S) were detected in TZ, with the frequency of 1534S (0.276) being higher than that of 1534L (0.017). 1534S was also detected in OFP, FS, DX and SJS at low frequencies (< 0.050).
Frequency and distribution of triple-locus genotype combinations in the Ae. albopictus VGSC gene
From our samples in a total of 426 individuals, we found 11 types of triple-locus genotype combinations in Ae. albopictus (Table 3). The number of triple-locus genotype combinations observed in each sampling site ranged from 1 (HR) to 10 (TZ). Notably, similar genotype distribution patterns were observed between DC and XC, and between DX and SJS. In addition, two combinations (Type 4 and Type 8) were uniquely present in TZ.
Triple-locus wild homozygotes (Type 1) were observed in 16 populations. This type occurred at high frequencies in HR (1.000), MY (0.963), YQ (0.917), FS (0.885), SY (0.833), MTG (0.800) and CP (0.733), but at low frequencies in TZ and OFP. The presence of single mutation was observed at loci 1016, 1532 and 1534 respectively (Types 2, 3, 4, 5, 6, 7 and 8). The combinations (Type 5 and Type 6) heterozygous at one IR-related locus (either 1016 or 1532) were widely distributed. Combinations (Types 9, 10 and 11) heterozygous at two of the three kdr loci (i.e. 1016 + 1532, 1016 + 1534, or 1532 + 1534) were detected. Single mutant homozygotes 1016GG (Type 2), 1532TT (Type 3) and 1534SS (Type 4) were present, while there were no double-loci mutant homozygotes or triple-locus mutant individuals.
Nucleotide polymorphism and haplotype diversity of the Ae. albopictus VGSC gene
VGSC haplotypes were experimentally clarified from our samples. Seventeen haplotypes were identified based on 200 sequences of D2 from 151 individuals in this study (Table 4). Sequence analysis showed that there were length polymorphisms in intron-20 among these haplotypes; the shortest intron was 71 bp, while the longest one was 91 bp. In the exon region of D2 (230 bp in length), 11 sites were polymorphic. The nucleotide variations resulted in two amino acid substitutions (S1000Y and V1016G) (Fig. 2).
Similarly, 19 haplotypes were identified based on 165 sequences of D3 from 124 individuals (Table 5). Length polymorphisms were also observed in intron-28 among these haplotypes. The shortest intron was 67 bp, while the longest one was 83 bp. In the coding region (246 bp in length), there were 10 polymorphic sites, leading to non-synonymous mutations at codons 1532 and 1534 (Fig. 2).
Haplotypes related to insecticide resistance
By investigating 32 out of the 66 individuals harboring the 1016G substitution, only one identical 1016G haplotype (D2H07) was identified. Similarly, the 1532T mutation was detected in 161 mosquitoes, and only one identical 1532T haplotype (D3H01) was found after analyzing 35 homozygotes and 30 heterozygotes sampled across populations.
The 1534S mutation was found in 20 individuals, of which 16 (12 from TZ, one each from DX, FS, OFP and SJS) were used for haplotype identification. Two different 1534S haplotypes (D3H03 and D3H13) were found. Sequence alignment showed that the two haplotypes differed in both exon and intron regions (Fig. 4). Interestingly, their distribution was different: D3H03 was found only in TZ, while D3H13 was exclusively detected in the other four sites (DX, FS, OFP and SJS). A BLAST search showed that the sequence of D3H03 was identical to that of MH384960.1 (Ae. albopictus voucher HK48 voltage-gated sodium channel gene), and D3H13 was identical to MH384956.1 (Ae. albopictus voucher YP01 voltage-gated sodium channel gene) [18] and MH384954.1 (Ae. albopictus voucher HZ45 voltage-gated sodium channel gene) [18].

Alignment of nucleotide sequences of two 1534S haplotypes. The intron is highlighted in yellow. The codon encoding the 1534 residue is underlined. Asterisks indicate identical nucleotides
There was only one individual that harbored the 1534L in our samples, leading to the identification of one 1534L haplotype (D3H12). D3H12 was highly similar to MH384952.1 (Ae. albopictus voucher JN08 voltage-gated sodium channel gene) [18]; they encoded the same amino acid sequence with only one variation (G/A) in the third position of codon 1534 (Fig. 5). No haplotype harboring both 1532 and 1534 mutations was observed after examining all 12 specimens that carried both 1532T and 1534S.

Alignment of nucleotide sequences of mutant VGSC haplotypes. The 5' and 3' regions that are identical are not presented in the figure. The intron is highlighted in yellow. The codons encoding 1532 and 1534 residues are underlined. Asterisks indicate identical nucleotides
Evolutionary analysis of kdr haplotypes
To investigate the phylogenetic relationship of kdr haplotypes with other wild counterparts, two maximum likelihood trees were constructed based on terminal-truncated D2 and D3 sequences, and some corresponding sequences retrieved from GenBank (Figs. 6, 7). Figure 6 shows that the D2H07 harboring 1016G substitution is closely clustered with D2H05 and D2H03 in an independent branch. Inferred from the tree in Fig. 7, haplotypes carrying either 1532T or 1534L were clustered in one of the two major clades, respectively, while those with a 1534S mutation were divided into two different major clades. Although belonging to the same major clade, the two 1534L haplotypes were closely clustered, while 1534S2 and 1534S3 were located in different sub-clades.

Molecular phylogenetic analysis by the maximum likelihood method based on D2 sequences. The GenBank accessions for the sequences identified in this study (named as D2Hx) are provided in Table 4. The tree with the highest log likelihood (−917.40) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach, and then selecting the topology with the superior log-likelihood value. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. The analysis involved 27 nucleotide sequences. There were a total of 317 positions in the final dataset

Molecular phylogenetic analysis by the Maximum Likelihood method based on D3 sequences. The GenBank accessions for the sequences identified in this study (named as D3Hx) are provided in Table 5. The evolutionary history was inferred by using the maximum likelihood method based on the Tamura-Nei model [1]. The tree with the highest log-likelihood (−744.91) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach, and then selecting the topology with the superior log-likelihood value. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. The analysis involved 16 nucleotide sequences. There were a total of 306 positions in the final dataset
To track the evolutionary origin of the kdr haplotypes, sequence alignments of the closely clustered haplotypes were conducted. The results, shown in Fig. 8, indicate that the resistant haplotypes D2H07 (1016G) might be derived from the wild one (D2H05) through one mutational step. Similarly, one nucleotide alternation in D3H02 could create three mutants (1534L1, 1534L2 and 1534S3), while one nucleotide replacement in D3H04 could lead to the formation of 1534S1 (Fig. 8). The double-mutation (1532T + 1534S) haplotype of Ae. albopictus voucher YP37 voltage-gated sodium channel gene (MH384958.1) [18] differed from either D3H01 (1532T) or D3H13 (1534S2) by only one nucleotide (Fig. 8), suggesting that these kdr haplotypes share a common wild ancestor (D3H08).

Alignment of nucleotide sequences of kdr haplotypes. The sequences (c.300 bp in length) in each alignment are highly similar, differing by 1 or 2 nucleotides. Only the region with variations is showed in the figure. The intron is highlighted in yellow. The codons encoding the insecticide resistance related amino acid residues are underlined. Asterisks indicate identical nucleotides
Discussion
This study represents a comprehensive investigation of kdr-related mutations in Ae. albopictus mosquitoes across Beijing. Besides the known mutations (I1532T, F1534S/L), we identified a novel V1016G substitution in Ae. albopictus. The information about the frequency and distribution of kdr mutations could help us to understand the genetic mechanism of insecticide resistance in Ae. albopictus. The defined VGSC haplotypes are useful for tracing the evolutionary origin of kdr mutations.
Sequence alignments of D2 revealed a mutation at loci 1016 (V1016G) in our Ae. albopictus samples. Although a conserved V1016G kdr mutation has been documented in Aedes aegypti [41], to our knowledge, this is the first identification of the V1016G mutation in Ae. albopictus. Notably, 1016G was detected in 13 of the 17 populations with the highest frequency of 0.65 (65%), and 1016G/G homozygotes were found in five populations. These results indicate that 1016G resistant allele frequently occurs at least in Beijing. A literature review showed that most previous studies focused on genetic mutations in domain III [18, 34], the occurrence of 1016G in Ae. albopictus is probably underestimated. Given that V1016G substitution in Aedes aegypti can lead to insensitivity to both Type I and Type II pyrethroids, a global survey of the distribution of the V1016G mutation in Ae. albopictus is strongly suggested.
I1532T mutation has been previously reported in Ae. albopictus from Italy [33] and different areas of China [18, 34, 42]. In this study, I1532T mutation was observed in 161 individuals from 15 of 17 populations with a frequency ranging from 0.038 to 0.537. These findings demonstrate that 1532T has a widespread geographical distribution in Chinese Ae. albopictus. Thus far, the association of phenotype with I1532T substitution remains unknown. Given the prevalence of this allele in China, it is important to pursue further studies on the biological and pharmacological effects of I1532T.
Different from 1016G and 1532T, mutations at codon 1534 are multiple. Two of the three previously documented amino acid substitutions at the 1534 locus (1534L and 1534S) were detected in Beijing. Notably, only one of 426 individuals was found to carry 1534L, suggesting that this mutation has just emerged in or was recently introduced into Beijing. Interestingly, the nucleotide alterations leading to F1534L varied among populations: a transversion at the third position occurred in our specimen (TTC to TTG), and another different transversion at the same position (i.e. TTC to TTA) was observed in mosquitoes from the JN population in a study covering five field populations from China [18], while the F1534L mutation identified in a survey in the USA resulted from a nucleotide transition at the first position (TTC to CTC) [32]. The 1534C detected in Singapore [31], Greece [33], Brazil [43], India [44] and even south China [18, 42], was not found in our samples. In contrast, 1534S mutation is widely distributed in China: it has been found in samples from southern, eastern, central and northern China in addition to this study [18, 42].
This study reveals obvious spatial heterogeneity of kdr mutations within Beijing Ae. albopictus populations. The number of mutations and frequency shows considerable geographical variations (Tables 2, 3; Fig. 3). Such an irregular pattern reflects that factors affecting the evolution and distribution of kdr mutations in Ae. albopictus are complex. Overall, 1016G and 1532T alleles were commonly present in Beijing, with 1534S occurring mainly in TZ (Fig. 4). Compared to the rural districts, higher frequencies of 1016G were detected in the urban districts (DC, XC, CY, HD, FT, SJS), which is in-line with more intensive insecticide use and higher residential density.
Given that a significant association of V1016G and F1534S/C mutations in Aedes VGSC with resistance phenotypes has been established by a large amount of susceptibility bioassay or/and electrophysiological evidence [18,19,20,21,22, 24,25,26, 29, 33,34,35], the distribution and frequency of conserved kdr mutations (Fig. 3) could serve to predict the status of insecticide resistance in Beijing Ae. albopictus. The relatively high frequency of 1016G in the urban districts (especially in HD), and the prevalent co-occurrence of 1016G and 1534S in TZ (Fig. 4), strongly indicates a risk of pyrethroid resistance in these areas. In addition, it is worth noting that 1016G homozygotes were present in CP, DX, FT and SJS, strongly suggesting a pyrethroid resistance risk. Unfortunately, since no parallel susceptibility bioassays were performed on the samples used for genotyping, we could not establish a clear association of IR levels with specific kdr alleles. However, a loss of susceptibility to beta cypermethrin was observed in HD, OFP and TZ (personal communications).
The results from phylogenetic analysis and alignments of DNA sequences of wild and mutant haplotypes may help trace the origin of kdr mutations. Considering that only one 1016G and one 1532T haplotype was discovered, we are unable to determine if 1016G or 1532T is singly or multiply originated. To determine the number of origins of 1016G or 1532T alleles, examination of a sufficient number of samples from a broad geographical range is required. However, the sequence alignments (Fig. 8) suggest that haplotypes D2H07 (1016G) and D3H01 (1532T) may have evolved from wild haplotypes D2H05 and D3H08, respectively.
Regarding codon 1534, multiple mutations have been documented. Moreover, it is already known that nucleotide mutations may happen at all the three positions of codon 1534 (i.e. T to C at the first position, T to G or C at the second position, and C to G or A at the third position), which leads to three different amino acid substitutions (F1534C/L/S). These observations indicate that codon 1534 is sensitive to mutation. The cross-continent distribution of conserved F1534C/L/S mutation may reflect the importance of this amino acid residue in adaptating to pyrethroid selection pressure. The large difference in DNA sequence between 1534S1 and 1534S2 (Fig. 4) makes us conclude that 1534S has multiple independent origins. By contrast, the extremely high similarity among 1534L1, 1534L2 and 1534S3 (Fig. 8) strongly suggests that these kdr haplotypes share a common origin. Similarly, we hypothesize that the three kdr haplotypes 1534S2 and 1532T (D3H01), and 1532T and 1534S (MH384958) may evolve from a common susceptible progenitor.
This work focused on kdr mutation. Other mechanisms such as enhanced detoxification of pyrethroids may possibly contribute to IR in field populations; therefore, further work is required to gain a full understanding about the status of pyrethroid resistance and underlying mechanisms in Beijing Ae. albopictus populations. Such studies are in our project list.
Conclusions
This study delineates the distribution of kdr mutations in the dengue vector Ae. albopictus across Beijing, China. Amino acid substitutions at multiple sites (1000, 1016, 1532, 1534) and two variations at codon 1534 have been documented. Phylogenetic analysis and sequence alignments have demonstrated multiple origins of 1534S. The presence of 1016G and 1534S/L alleles indicates a strong risk of pyrethroid resistance in Beijing Ae. albopictus. These findings highlight the importance to monitor and quantify the level of pyrethroid resistance in field mosquitoes in the urban districts. Taking action to limit the spread of kdr alleles into the rural areas would be helpful to prevent the development of insecticide resistance.
Abbreviations
- IR:
-
insecticide resistance
- kdr :
-
knockdown resistance
- PCR:
-
polymerase chain reaction
- VGSC:
-
voltage-gated sodium channel(s)
References
Smith LB, Kasai S, Scott JG. Pyrethroid resistance in Aedes aegypti and Aedes albopictus: important mosquito vectors of human diseases. Pestic Biochem Physiol. 2016;133:1–12.
Enserink M. A mosquito goes global. Science. 2008;320:864–6.
Gratz NG. Critical review of the vector status of Ae albopictus. Med Vet Entomol. 2004;18:215–27.
Benedict MQ, Levine RS, Hawley WA, Lounibos LP. Spread of the tiger: global risk of invasion by the mosquito Ae. albopictus. Vector Borne Zoonotic Dis. 2007;7:76–85.
van den Hurk AF, Nicholson J, Beebe NW, Davis J, Muzari OM, Russell RC, et al. Ten years of the Tiger: Ae. albopictus presence in Australia since its discovery in the Torres Strait in 2005. One Health. 2016;2:19–24.
Peng HJ, Lai HB, Zhang QL, Xu BY, Zhang H, Liu WH, et al. A local outbreak of dengue caused by an imported case in Dongguan China. BMC Public Health. 2012;12:83–90.
Li Y, Kamara F, Zhou G, Puthiyakunnon S, Li C, Liu Y, et al. Urbanization increases Aedes albopictus larval habitats and accelerates mosquito development and survivorship. PLoS Negl Trop Dis. 2014;8:e3301.
Zhao H, Zhang FC, Zhu Q, Wang J, Hong WX, Zhao LZ, et al. Epidemiological and virological characterizations of the 2014 dengue outbreak in Guangzhou, China. PLoS One. 2016;11:e0156548.
Liu Z, Zhou T, Lai Z, Zhang Z, Jia Z, Zhou G, et al. Competence of Aedes aegypti, Ae. albopictus, and Culex quinquefasciatus mosquitoes as Zika virus vectors, China. Emerg Infect Dis. 2017;23:1085–91.
Zhang QL, He JF, Wu D, Wang ZJ, Zhong XG, Zhong HJ, et al. Maiden outbreak of chikungunya in Dongguan city, Guangdong Province, China: epidemiological characteristics. PLoS One. 2012;7:e42830.
Wu D, Wu J, Zhang Q, Zhong H, Ke C, Deng X, et al. Chikungunya outbreak in Guangdong Province, China, 2010. Emerg Infect Dis. 2012;18:493–5.
Moyes CL, Vontas J, Martins AJ, Ng LC, Koou SY, Dusfour I, et al. Contemporary status of insecticide resistance in the major Aedes vectors of arboviruses infecting humans. PLoS Negl Trop Dis. 2017;11:e0005625.
Stevenson BJ, Pignatelli P, Nikou D, Paine MJI. Pinpointing P450s associated with pyrethroid metabolism in the dengue vector, Aedes aegypti: developing new tools to combat insecticide resistance. PLoS Negl Trop Dis. 2012;6:e1595.
Ishak IH, Riveron JM, Ibrahim SS, Stott R, Longbottom J, Irving H, et al. The cytochrome P450 gene CYP6P12 confers pyrethroid resistance in kdr-free Malaysian populations of the dengue vector Aedes albopictus. Sci Rep. 2016;6:24707.
Lumjuan N, Rajatileka S, Changsom D, Wicheer J, Leelapat P, Prapanthadara LA, et al. The role of the Aedes aegypti epsilon glutathione transferases in conferring resistance to DDT and pyrethroid insecticides. Insect Biochem Mol Biol. 2011;41:203–9.
Poupardin R, Srisukontarat W, Yunta C, Ranson H. Identification of carboxylesterase genes implicated in temephos resistance in the dengue vector Aedes aegypti. PLoS Negl Trop Dis. 2014;8:e2743.
Ffrench-Constant RH, Daborn PJ, Le Goff G. The genetics and genomics of insecticide resistance. Trends Genet. 2004;2:163–70.
Gao J, Chen H, Shi H, Peng H, Ma Y. Correlation between adult pyrethroid resistance and knockdown resistance (kdr) mutations in Aedes albopictus (Diptera: Culicidae) field populations in China. Infect Dis Poverty. 2018;7:86.
Srisawat R, Komalamisra N, Eshita Y, Zheng M, Ono K, Itoh TQ, et al. Position mutations in domain II of the voltage-gated sodium channel gene in deltamethrin-resistant Ae. aegypti (Diptera: Culicidae). Appl Entomol Zool. 2010;45:275–82.
Sayono S, Hidayati AP, Fahri S, Sumanto D, Dharmana E, Hadisaputro S, et al. Distribution of voltage-gated sodium channel (Nav) alleles among the Ae. aegypti populations in Central Java Province and its association with resistance to pyrethroid insecticides. PLoS One. 2016;11:e0150577.
Kawada K, Higa Y, Komagata O, Kasai S, Tomita T, Yenet NT, et al. Widespread distribution of a newly found position mutation in voltage-gated sodium channel in pyrethroid-resistant Ae aegypti populations in Vietnam. PLoS Negl Trop Dis. 2009;3:e527.
Harris AF, Rajatileka S, Ranson H. Pyrethroid resistance in Aedes aegypti from Grand Cayman. Am J Trop Med Hyg. 2010;83:277–84.
Kawada H, Oo SZM, Thaung S, Kawashima E, Maung YNM, Thu HM, et al. Co-occurrence of position mutations in the voltage-gated sodium channel of pyrethroid-resistant Ae. aegypti populations in Myanmar. PLoS Negl Trop Dis. 2014;8:e3032.
Stenhouse SA, Plernsub S, Yanola J, Lumjuan N, Dantrakool A, Choochote W, et al. Detection of the V1016G mutation in the voltage-gated sodium channel gene of Aedes aegypti (Diptera: Culicidae) by allele-specific PCR assay, and its distribution and effect on deltamethrin resistance in Thailand. Parasit Vectors. 2013;6:253.
Li CX, Kaufman PE, Xue RD, Zhao MH, Wang G, Yan T, et al. Relationship between insecticide resistance and kdr mutations in the dengue vector Aedes aegypti in southern China. Parasit Vectors. 2015;8:325.
Muthusamy R, Shivakumar MS. Involvement of metabolic resistance and F1534C kdr mutation in the pyrethroid resistance mechanisms of Ae. aegypti in India. Acta Trop. 2015;148:137–41.
Brengues C, Hawkes NJ, Chandre F, McCarroll L, Duchon S, Guillet P, et al. Pyrethroid and DDT cross-resistance in Ae. aegypti is correlated with novel mutations in the voltage-gated sodium channel gene. Med Vet Entomol. 2003;17:87–94.
Plernsub S, Saingamsook J, Yanola J, Lumjuan N, Tippawangkosol P, Walton C, et al. Temporal frequency of knockdown resistance mutations, F1534C and V1016G, in Ae. aegypti in Chiang Mai city, Thailand and the impact of the mutations on the efficiency of thermal fogging spray with pyrethroids. Acta Trop. 2016;162:125–32.
Du Y, Nomura Y, Satar G, Hu Z, Nauen R, He SY, et al. Molecular evidence for dual pyrethroid-receptor sites on a mosquito sodium channel. Proc Natl Acad Sci USA. 2013;110:11785–90.
Hirata K, Komagata O, Itokawa K, Yamamoto A, Tomita T, Kasai S. A single crossing-over event in voltage-sensitive Na+ channel genes may cause critical failure of dengue mosquito control by insecticides. PLoS Negl Trop Dis. 2014;8:e3085.
Kasai S, Ng LC, Lam-Phua SG, Tang CS, Itokawa K, Komagata O, et al. First detection of a putative knockdown resistance gene in major mosquito vector, Aedes albopictus. Jpn J Infect Dis. 2011;64:217–21.
Marcombe S, Farajollahi A, Healy SP, Clark GG, Fonseca DM. Insecticide resistance status of United States populations of Aedes albopictus and mechanisms involved. PLoS One. 2014;9:e101992.
Xu J, Bonizzoni M, Zhong D, Zhou G, Cai S, Li Y, et al. Multi-country survey revealed prevalent and novel f1534s mutation in voltage-gated sodium channel (VGSC) gene in Aedes albopictus. PLoS Negl Trop Dis. 2016;10:e0004696.
Chen H, Li K, Wang X, Yang X, Lin Y, Cai F, et al. First identification of kdr allele F1534S in VGSC gene and its association with resistance to pyrethroid insecticides in Aedes albopictus populations from Haikou City, Hainan Island, China. Infect Dis Poverty. 2016;5:31.
Li Y, Xu J, Zhong D, Zhang H, Yang W, Zhou G, et al. Evidence for multiple-insecticide resistance in urban Aedes albopictus populations in southern China. Parasit Vectors. 2018;11:4.
Zhang SN, Liu ZB, Gu ZW. Disease control and prevention in China in the 20th century and prospects for the new millennium. Environ Health Prev Med. 2002;7:132–7.
Higa Y, Toma T, Tsuda Y, Miyagi I. A multiplex PCR-based molecular identification of five morphologically related, medically important subgenus Stegomyia mosquitoes from the genus Aedes (Diptera: Culicidae) found in the Ryukyu Archipelago, Japan. Jpn J Infect Dis. 2010;63:312–6.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7.
Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 1993;10:512–26.
Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Kawada H, Higa Y, Futami K, Muranami Y, Kawashima E, Osei JH, et al. Discovery of point mutations in the voltage-gated sodium channel from African Aedes aegypti populations: potential phylogenetic reasons for gene introgression. PLoS Negl Trop Dis. 2016;10:e0004780.
Chen H, Gao J, Jiang J, Peng H, Ma Y. Detection of the I1532 and F1534 kdr mutations and a novel mutant allele I1532T in VGSC gene in the field populations of Aedes albopictus from China. Chin J Vector Biol Control 2018;29:120–5.
Aguirre-Obando OA, Martins AJ, Navarro-Silva MA. First report of the Phe1534Cys kdr mutation in natural populations of Aedes albopictus from Brazil. Parasit Vectors. 2017;10:160.
Rath A, Mohanty I, Hazra RK. Insecticide susceptibility status of invasive Aedes albopictus across dengue endemic districts of Odisha, India. Pest Manag Sci. 2018;74:1431–40.
Authors’ contributions
Conceived and designed the experiments: QXH, ZXP, DY and ZXJ. Performed the experiments: YC, LN, LM and ZXJ. Analyzed the data: QXH, YC, LN, ZXJ and ZXP. Contributed reagents/materials/analysis tools: LM, ZXJ and DY. Wrote the paper: QXH, YC, ZXJ and ZXP. All authors read and approved the final manuscript.
Acknowledgements
We are grateful to the 16 district CDCs of Beijing for assistance with mosquito sample collection. We would thank the anonymous reviewers and editors for their comments and editing.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Data supporting the conclusions of this article are provided within the article. The newly generated sequences were submitted to the GenBank database under the accession numbers MK201602-MK201637.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
This work was supported by Science and Technology Development Project of Beijing Municipal Medical Research Institutes (PXM2017_026278_000006) and the State Key Laboratory of Integrated Management of Pest Insects and Rodents (IPM1812). The funders had no role in the study design, data collection, analysis, decision to publish or preparation of the manuscript.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhou, X., Yang, C., Liu, N. et al. Knockdown resistance (kdr) mutations within seventeen field populations of Aedes albopictus from Beijing China: first report of a novel V1016G mutation and evolutionary origins of kdr haplotypes. Parasites Vectors 12, 180 (2019). https://doi.org/10.1186/s13071-019-3423-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-019-3423-x