
Abstract
Background
Vector control is still a pivotal method for preventing malaria, and its potency is weakened by the increasing resistance of vectors to chemical insecticides. As the most abundant and vital malaria vector in Southeast Asia, the chemical insecticide resistance status in Anopheles sinensis remains elusive in Laos, which makes it imperative to evaluate the true nature of chemical insecticide resistance-associated genetic mutations in An. sinensis in Laos.
Methods
Adult An. sinensis were collected from three border regions in Laos. DNA was extracted from individual mosquitoes. PCR amplification and DNA sequencing of a fragment containing codon 1014 of the voltage-gated sodium channel (vgsc) gene were completed to study the kdr allele frequency distribution, kdr intron polymorphism, population genetic diversity, and the evolutionary status of the kdr codon. The mitochondrial cytochrome c oxidase subunit II gene (COII) was amplified and sequenced to examine population variations, genetic differentiation, spatial population structure, population expansion, and gene flow patterns.
Results
Nine wild kdr haplotypes of the vgsc gene were detected in this study, and eight of them, namely 1014L1, 1014L2, 1014L4, 1014L7, 1014L9, 1014L10, 1014L11, and 1014L21, were discovered in the China–Laos border (northern Laos), while 1014L3 was only detected in the Thailand–Laos border (northwestern Laos) and Cambodia–Laos border (southern Laos). The newly identified haplotype, 1014L21, was uniquely distributed in the China–Laos border and was not identified in other countries. Based on sequence analysis of the mitochondrial COII genes, significant genetic differentiation and limited gene flow were detected between the China–Laos and Cambodia–Laos An. sinensis populations, which suggested that those two regions were genetically isolated. The distinct distribution of the kdr haplotype frequencies is probably the result of geographical isolation in mosquito populations.
Conclusions
Lack of kdr mutations in the vgsc gene was probably due to genetic isolation and the absence of intense selection pressure in the three border regions of Laos. This study reveals that pyrethroid-based chemical insecticides are still appropriate for battling An. sinensis in parts of Laos, and routine monitoring of chemical insecticide resistance should be continuously implemented and focused on more restricted areas as part of chemical insecticide resistance management.
Graphical Abstract

Similar content being viewed by others
Background
Malaria is a potentially fatal vector-borne disease in tropical and subtropical regions, with 241 million case reports and 627,000 deaths documented worldwide in 2020 [1]. The malaria burden in the Greater Mekong Subregion (GMS) remains a major public health threat affecting the health and lives of a large proportion of people [2]. In the Lao People’s Democratic Republic (Lao PDR), malaria is endemic, whereas the transmission intensity is heterogeneous, with the more intense transmission in remote and forested regions, especially in the southern part of the country [3,4,5]. Even though Laos has decreased malaria prevalence by 50% since 2000, reoccurrence has been documented since 2011, with over 260,000 patients recorded in 2015 [2]. In recent years, there has been an increase in the number of Plasmodium vivax reports in Lao PDR (nearly 50% of Plasmodium species from indigenous cases in 2018), although a large proportion of the reported cases of infection have long been related to Plasmodium falciparum [4]. Chemical control of vectors has played an essential role in controlling and eliminating malaria [6]. In Laos, as in the majority of GMS nations, malaria vector control depends on the utilization of insecticide-treated materials (i.e., long-lasting insecticide-treated nets [LLIN]) and indoor residual spraying (IRS) [2]. Before using insecticide-treated bed nets (ITNs), residual spraying with DDT (organochlorine family) was employed to prevent malaria. The utilization of DDT was terminated in 1990 (formally abolished in 2010, [7]), and from then on, insecticides based on pyrethroids (e.g., permethrin, deltamethrin, alpha-cypermethrin, and lambda-cyhalothrin) have been used for IRS and/or ITNs [8].
The voltage-gated sodium channel (VGSC) protein is the major target for pyrethroids and DDT [9]. The spectrum of mutations at the vgsc gene is remarkably conservative across insect species, revealing convergent evolution [9]. Despite the controversy, many studies have demonstrated that mutations at codon 1014 of VGSC induce resistance to pyrethroids and DDT in substantial arthropod species [9,10,11]. The main target-site resistance mechanism in Anopheles mosquitoes involves three non-synonymous variants (L1014F, L1014C, and L1014S) at the kdr codon L1014 of the vgsc gene, causing resistance to pyrethroids [10]. In Asia, those variants have been recorded in certain Anopheles populations from India [12, 13], Sri Lanka [14], Indonesia [15], China [16, 17], Vietnam, and Cambodia [18]. In Anopheles sinensis, the most abundant and important malaria vector in Southeast Asia [19,20,21,22,23], a remarkable positive association between kdr allele frequency and bioassay-based resistance phenotype has been recorded [17, 24,25,26]. Therefore, kdr mutation has been utilized as a molecular marker for monitoring the resistance of pyrethroids in An. sinensis [17]. In An. sinensis, four non-synonymous variants at codon L1014 of the vgsc gene are identified, namely the L1014F [17, 24, 25, 27,28,29], L1014S [18, 25], L1014C [17, 24, 27,28,29], and L1014W [25] mutations.
Due to the use of various insecticides in vector control and continuous use in agricultural activities, the geographical distribution and density of malaria vectors may change, and insecticide resistance is also expected [30]. Various factors could influence the distribution of kdr allele variants in An. sinensis throughout Southeast Asia. The intricacy of landscapes can impede genetic flow between mosquito populations. Selection based upon the intensity and duration of pyrethroid use can determine the allele distribution pattern of kdr as well [31]. Additionally, the genetic variation based on neutral biomarkers can reflect the population structure resulting from demographic factors. In this case, the cytochrome c oxidase genes of the mitochondria genome are utilized as the neutral reference [31]. Based on its unique features of maternal inheritance, no recombination, high variability compared with nuclear DNA, and low population size required, mitochondrial DNA (mtDNA) has become a popular biomarker for studies on genetic diversity and population structure [32].
The status of chemical insecticide resistance in An. sinensis remains elusive in the Lao PDR. Given that vector control is still a pivotal method in preventing malaria and that its potency is attenuated by the increasing resistance of vectors to insecticides, it is imperative to evaluate the actual occurrence of chemical insecticide resistance-related genetic variants in An. sinensis in Laos. In this study, the kdr allele distribution was investigated in An. sinensis adult samples collected from multiple sites, including the China–Laos border, Cambodia–Laos border, and Thailand–Laos border. Additionally, the possible evolutionary origin of kdr haplotypes was studied, and the role of the demographic history of An. sinensis on the evolutionary process of kdr variants was examined using mtDNA sequencing information.
Methods
Mosquito collection and identification
Using overnight trapping with battery-operated Centers for Disease Control and Prevention (CDC) light traps (model 1012, John W. Hock Inc., USA) in cattle/pig pens or human rooms from 8:00 pm to 8:00 am, adult mosquitoes were collected in Pathoomphone County (Champasak Province) in 2017, as well as in Pak lay County (Xayabuli Province) and Yot Ou County (Phongsaly Province) in 2019 in accordance with our previous study [33]. The live adult mosquitoes were killed by freezing in a refrigerator. The subsequent morphological identification was carried out by sex, species, and subgroup, using a dissecting microscope with the keys of Das et al. [34]. Each morphologically identified specimen was kept individually in a 1.5-ml microcentrifuge tube with 75% ethanol and stored at 4 °C for molecular species confirmation and further processing.
After that, the molecular identification of An. sinensis based on cytochrome c oxidase subunit II (COII) was carried out to avoid any variations in further analysis. Ninety-eight percent sequence identity with the voucher specimens/sequences in the NCBI Nucleotide database is required for final species confirmation. To avoid the issue of inadequate results for the voucher sequence produced by COII alone, we performed amplification and sequencing on ITS2. Hence, ITS2 and COII database comparisons of each morphologically identified An. sinensis sample were paired to determine the species of An. sinensis in accordance with previous studies [35, 36] (Additional file 1: Table S1).
DNA extraction, COII/kdr amplification, and sequencing
A total of 134 morphologically identified An. sinensis specimens were further screened for DNA extraction, COII/kdr amplification, and sequencing (Additional file 1: Table S1), and 89 of them were eventually molecularly identified as An. sinensis based on both ITS2 and COII (Additional file 1: Table S1). The extraction of genomic DNA in individual mosquitoes was carried out following the manufacturer’s instructions (QIAamp® DNA Mini Kit, Germany). The amplification for approximately 650 bp of the COII gene was carried out using primers LEU-F (5′-TCTAATATGGCAGATTAGTGCA-3′) and LYS-R (5′-ACTTGCTTTCAGTCATCTAATG-3′) [33]; a fragment containing codon 1014 of the An. sinensis vgsc gene (325-bp polymerase chain reaction [PCR] product of the kdr gene) was amplified using primers KDR-F (5′-TGCCACTCCGTGTGTTTAGA-3′) and KDR-R (5′-GAGCGATGATGATCCGAAAT-3′) [24]. For amplification of COII, the cycling parameter included 95 °C, 5 min; 95 °C/1 min, 51 °C/1 min, 72 °C/2 min for 35 cycles; with a final extension of 72 °C for 10 min. For amplification of KDR, the cycling parameter included 95 °C, 5 min; 94 °C/30 s, 55 °C/30 s, 72 °C/30 s for 35 cycles; with a final extension of 72 °C for 10 min. The PCR mixture (25 μl) consisted of 12.5 μl 2 × Taq PCR Mix (Tiangen Biotech, Beijing, China), 5 μl of template DNA (< 1 μg), 1 μl of 10 μM each primer, and 5.5 μl water. The PCR products were analyzed by 1.5% agarose gel electrophoresis stained with GoldView (Solarbio, China) under ultraviolet (UV) transillumination. PCR products were purified by an agarose gel DNA recovery kit (DP219, Tiangen Biotech, Beijing, China) before sequencing. The sequencing reaction was performed in both directions using an ABI Big Dye Terminator Kit v.3.1 (Applied Biosystems, Warrington, UK) and was analyzed using the ABI Prism 3500xL Genetic Analysis Tool (Applied Biosystems, CA, USA) in Shanghai (Sangon Biotech).
Sequence alignment and phylogenetic analysis based on COII/kdr sequences
The researchers used the keywords “(species name) & COII/kdr” for searching COII or kdr sequences of An. sinensis deposited in GenBank. Additional file 2: Table S2 shows the mentioned sequences of COII, while kdr sequences are listed in Table 1. The COII and kdr sequence data set was combined with our original data and records retrieved from GenBank. A multiple sequence alignment was conducted in MEGA-X [37], and the manual adjustment was conducted using BioEdit V7.0.9 if required [38]. Gaps were excluded from the analysis, and characters were unweighted. A phylogenetic tree was generated using a neighbor-joining algorithm bootstrapped with 1000 replicates [39] based on MEGA-X [37]. The visualization of this phylogram was performed using FigTree v1.4.2 [40].
Genetic diversity analysis and neutrality test based on COII/kdr sequences
DnaSP v.5.0 [41] was used to calculate the average of nucleotide differences per site (K), nucleotide diversity (π), haplotype diversity (Hd), and haplotypes (H) based on the COII and kdr sequences. To compare the COII and/or kdr haplotypes between Laos and other geographical regions, existing data in GenBank from other countries were also analyzed. The parsimony framework was applied using Network 4.0 [42] on both genes, and Tajima’s D [43] and Fu’s Fs [44] were calculated for haplotype data using DnaSP v.5.0 [41] to test the hypothesis of strict neutrality in the An. sinensis population.
Statistical analysis based on COII sequences
Based on COII sequences, Arlequin v.3.5 [45] was used to calculate pairwise FST for estimating population differentiation by complying with a difference in haplotype frequency, Nei’s Nm estimated gene flow conformed to GST [46], and analysis of molecular variance (AMOVA) for determining the distribution of genetic variation in the population using 1000 permutations. Isolation by distance (IBD) was examined using a nonparametric Mantel test with the web-based computer program IBDWS v.3.16 [47]. For distinguishing smooth unimodal distribution from multimodal or ragged distribution, the mismatch distribution (simulated in Arlequin v.3.5) was used [48,49,50].
Results
Sequence polymorphisms of the An. sinensis vgsc gene
The 267-bp DNA fragments individually amplified from a total of 89 An. sinensis mosquitoes were used for sequence polymorphism analysis. This sequence covered partial exon 19 (contains the codon 1014), intron 19, and partial exon 20 of the An. sinensis vgsc gene (KE525266.1). In general, 17 nucleotide polymorphic sites (PSs) were identified. The first to fifth PSs were located on exon 19, the sixth to 15th PSs on intron 19, and the 16th and 17th PSs on exon 20. The polymorphisms in the fourth and fifth PSs resulted in amino acid substitutions (L/F/S) at codon 1014, and the nucleotide variations in the first, second, third, 16th, and 17th PSs represented synonymous mutations (Fig. 1).

The nucleotide region of the An. sinensis vgsc gene addressed in this study. Dots indicate the polymorphic sites (PSs) in the obtained sequences. The red dots represent sites leading to nonsynonymous mutations. The positions of PSs in the 267-bp sequence are numbered below the dots. The nucleotides for each PS are given
Diversity and frequency of kdr haplotypes
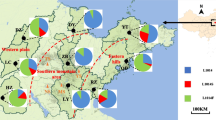
Point mutations at position 1014 of the domain II of the VGSC protein have been documented to confer kdr [9, 10]. To better understand the diversity and frequency of kdr haplotypes in this area, we downloaded 70 kdr sequences of An. sinensis from NCBI, combined with our original data from 89 samples collected in Laos. Thirty-nine haplotypes were identified from 159 An. sinensis individuals (Table 1). Seven haplotypes (i.e., 1014L19, 1014L20, 1014L21, 1014F7, 1014F8, 1014F9, 1014W1) were newly identified in other research except for one haplotype, 1014L21, which was newly identified in this study. In the present study, only a wild haplotype of kdr was detected, TTG (1014L); other mutations such as TTT (1014F), TCG (1014S), and TGT (1014C) were not found. Accordingly, nine wild haplotypes, namely 1014L1, 1014L2, 1014L3, 1014L4, 1014L7, 1014L9, 1014L10, 1014L11, and 1014L21, respectively, were identified (Table 1), in the frequencies ranging from 1.12% (1/89) to 43.82% (39/89) in the three Laos populations (Fig. 2). In the LPY (Yot Ou County, Phongsaly Province) population, 1014L1 (46.99%, 39/83), 1014L4(9.64%, 8/83), 1014L7 (12.05%, 10/83), 1014L9 (7.23%, 6/83), and 1014L10 (19.28%, 16/83) had higher frequencies than other haplotypes. In the LCP (Pathoomphone County, Champasak Province) population, only 1014L3 was identified and accounted for 100% (4/4). In the LXP (Pak lay County, Xayabuli Province) population, 1014L3 and 1014L9 were both 50% (1/2). In addition, the newly identified susceptible haplotype, 1014L21, was uniquely distributed at LPY at a low frequency (1.20%, 1/83). Due to the time limit and different sample sizes between each sampling site, further evaluation is needed.

Distribution and frequency of kdr alleles in An. sinensis populations along the China–Laos border, Thailand–Laos border, and Cambodia–Laos border. Pak lay County (Xayabuli Province); Yot Ou County (Phongsaly Province); Pathoomphone County (Champasak Province). Red line = Mekong River. The shapefile map of Laos was downloaded and prepared by using Pixel Map Generator-Beta online (amCharts, Vilnius, Lithuania) (https://pixelmap.amcharts.com/), which is copyright-free
To analyze the genetic diversity indices and neutrality tests (Fu’s Fs and Tajima’s D) based on the kdr intron of An. sinensis, 89 kdr intron sequences in this study and 65 kdr intron sequences retrieved from the GenBank database (Additional file 3: Table S3a) were used in the subsequent analysis. A total of 14 haplotypes were found in nine populations. Genetic diversity varied significantly among geographical regions. The overall haplotype diversity (Hd) and nucleotide diversity (Pi) were 0.788 and 0.02190, respectively. Compared to the three An. sinensis populations in Laos, high haplotype diversity and nucleotide diversity were found in six populations from China. Moreover, significant departures from neutrality were detected by Fu’s Fs test in all the populations (−5.17700, P < 0.05), CN-GX (Guangxi Province, Southwest China) population (−4.51700, P < 0.02), and the CN-HaN (Hainan Province, Southwest China) population (−3.66700, P < 0.02), whereas they were not detected in all the Laos populations by Fu’s Fs or Tajima’s D test. Additionally, slightly insignificant departures from neutrality were detected by Fu’s Fs test in the CN-AH (Anhui Province, Central China) population (−1.50700, P < 0.1) and CN-YN (Yunnan Province, Southwest China) population (−1.19500, P < 0.1) (Additional file 3: Table S3a). Since the Fu’s Fs statistic is particularly sensitive to demographic effects, it is difficult to conclude whether positive selection or demographic history (e.g., population expansion) accounts for the observed pattern.
Genealogical analysis of kdr mutations
Network analysis showed that haplotypes H6-1014L7 and H8-1014L11 were derived from single mutational steps through T231C and G202T, respectively, in the intron 19 from ancestor H1-1014L1, while H1-1014L21 reported in this study derived from H1-1014L1 with a single mutation at C245T in the exon 20. Haplotype H3-1014L3, which was only detected in LXP and LCP populations, and H6-1014L10, were derived from single mutational steps through G202T and T215C, respectively, in the intron 19 from ancestor H2-1014L2. Additionally, H4-1014L4 was derived from H3-1014L3 with a single mutation at T215C in intron 19, while H4-1014L9 exhibited one more single mutation at C245T in exon 20 from the ancestor H3-1014L3 (Fig. 3a).

The network of kdr haplotypes identified in An. sinensis populations. a Networks showing the genealogical relationship in Laos. Yellow, green, and blue circles represent 1014L haplotypes from LPY (Phongsaly Province: Yot Ou County), LCP (Champasak Province: Pathoomphone County), and LXP (Xayabuli Province: Pak lay County) populations, respectively. The size of each circle is proportional to its corresponding frequencies. H represents the type of intron haplotypes (numbers in brackets). The straight line indicates the possible mutational step. The note above the line referred to the mutation position and base. b Networks showing the genealogical relationship among different kdr haplotypes from the present study and NCBI data. Yellow, blue, green, red, and pink circles represent 1014L, 1014S, 1014C, 1014F, and 1014 W haplotypes, respectively. The size of each circle is proportional to its corresponding frequencies
To estimate the evolutionary relationship of kdr mutations, nine haplotypes identified in this study and 30 haplotypes retrieved from the GenBank database (Table 1) were used to construct a network using Network 4.0. The network analysis revealed complex reticulate patterns and multiple independent mutation events leading to kdr haplotypes. The genealogical analysis revealed that a single mutation might result in the resistant phenotype from the susceptible one. For example, 1014S2 shared an identical intron sequence with 1014L2, L5, and S1, and a single mutation (T165C) might change the susceptible 1014L2 to the resistant 1014S2. Haplotypes 1014F1, F2, F3, F5, F7, and F9 were possibly derived from 1014L1, 1014L3, 1014L3, 1014L7, 1014L2, and 1014L11, respectively, while 1014F6 and F8 derived from the ancestor 1014L1 with two mutational steps, i.e., the additional mutations in exon 19 (T140A) or intron 19 (T229A) of 1014F1. Likewise, 1014F4 was derived from the ancestor 1014L11 with two mutational steps, i.e., the additional mutations in exon 19 (T140A) of 1014F9. Haplotypes 1014S1, S2, S3, S4, S5, and S6 were perhaps the results of an independent mutational step from six different wild haplotypes 1014L5, 1014L2, 1014L3, 1014L4, 1014L1, and 1014L6, respectively. Additionally, 1014W1 was derived from the ancestor 1014L1, while 1014C1 and 1014C2 were derived from 1014F1 with two or more mutational steps (Fig. 3b, Table 1).
Mitochondrial DNA sequence variation
Eighty-nine sequences for COII were generated for the three populations (Additional file 3: Table S3b). The COII sequence alignment revealed 26 variable sites. The overall haplotype diversity (Hd), number of haplotypes, and nucleotide diversity (Pi) were 0.799, 22, and 0.00351, respectively. Significant departures from neutrality were both detected by Fu’s Fs test (−11.48900, P < 0.001) and Tajima’s D test (−1.79501, P < 0.02) in all the populations. In contrary to the LXP and LCP populations, high haplotype diversity (0.783), number of haplotypes (20), and nucleotide diversity (0.00282) were found in the LPY population. Additionally, significant departures from neutrality were both detected by Fu’s Fs test (−11.80300, P < 0.001) and Tajima’s D test (−1.61923, P < 0.05) in the LPY population, whereas not detected in the LXP and LCP population.
Population structure and genetic differentiation
The median-joining network based on 89 COII sequences denoted the distribution pattern exhibited by 22 haplotypes in An. sinensis populations. The An. sinensis populations fell into two main clusters. Cluster 1 consisted of all the haplotypes except for one haplotype (H22) from LCP; cluster 2 consisted of only one haplotype (H22). Within cluster 1, two sub-clusters were also found; sub-cluster 1 consisted of all the haplotypes in LPY and LXP, while sub-cluster 2 consisted of only one haplotype (H21) in LCP (Fig. 4a). The most common haplotypes referred to H1 (n = 13), H4 (n = 37), and H6 (n = 7), as only identified in 67.47% (56/83) of LPY and 50% (1/2) of LXP. H21 (n = 3) and H22 (n = 1) were only identified in LCP (Fig. 4a). The unweighted pair group method with arithmetic mean (UPGMA) dendrogram based on Nei’s unbiased genetic distances between haplotypes indicated that H22 constituted one cluster, while the other haplotypes constituted the second (Fig. 4b). Additionally, two sub-clusters were also found in the dendrogram, which was consistent with the results of the median-joining network.

Phylogenetic analysis based on the COII sequences in An. sinensis populations in Laos. a Phylogenetic network of 22 mitochondrial haplotypes of the COII gene in An. sinensis. Localities are indicated by different colors (bottom right). The size of each circle is proportional to its corresponding frequencies. b UPGMA dendrogram based on Nei’s unbiased genetic distance between the 22 haplotypes of An. sinensis. Yellow, green, and blue circles/rectangles represent haplotypes from LPY (Phongsaly Province: Yot Ou County), LCP (Champasak Province: Pathoomphone County), and LXP (Xayabuli Province: Pak lay County) populations, respectively
To draw a broader comparison in haplotype from Laos and other geographical regions, we downloaded and analyzed available data in GenBank from neighboring nations (Additional file 2: Table S2). In general, a total of 148 An. sinensis COII sequences were generated for seven populations, including LPY (n = 83), LCP (n = 4), LXP (n = 2), JP (Japan, n = 3), KR (South Korea, n = 19), TH (Thailand, n = 3), and CN (China, n = 34), and 46 haplotypes were found in the median-joining network. The An. sinensis populations fell into two main clusters. Cluster 1 consisted of all the haplotypes except for one haplotype (H27) from LCP; cluster 2 consisted of only one haplotype (H27). Within cluster 1, three sub-clusters were also found: sub-cluster 1 consisted of haplotype (H26) from LCP; sub-cluster 2 consisted of five haplotypes (H29, H35, H37, H42, and H43) from China; sub-cluster 3 consisted of other haplotypes (Additional file 4: Fig. S1a). The UPGMA dendrogram based on Nei’s unbiased genetic distances between haplotypes indicated that H27 constituted one cluster, while the other haplotypes constituted the second (Additional file 4: Fig. S1b). Additionally, three sub-clusters were also found in the dendrogram, which was consistent with the results of the median-joining network.
AMOVA analysis based on COII sequences demonstrated that most of the variances were found among group variation (58.43%) rather than within populations (38.93%) and among populations within groups (2.64%), suggesting that these populations could fall into several groups. However, no statistical significance was found in evaluating the fixation index among groups (FCT), among populations within groups (FSC). In contrast, the fixation index within populations (FST) showed statistical significance (P < 0.05) (Additional file 5: Table S4). Due to the time limit and different sample sizes between each sampling site, further evaluation is needed.
The maximal level of genetic differentiation by the fixation index FST based on sequences analysis of COII was between LCP and LPY (FST = 0.61657, P ˂ 0.05). while no significant genetic differentiation was found between LPY and LXP populations (0.08915, P > 0.05) or between LXP and LCP populations (0.09565, P > 0.05). Furthermore, the minimal estimate of gene flow (Nm) was between LCP and LPY (Nm = 0.31093). In contrast, high gene flows were found between LPY and LXP populations (Nm = 5.10835), as well as between LXP and LCP populations (Nm = 4.72727) (Additional file 6: Table S5).
Spatial genetic structure analysis, demographic history, and neutrality test
A Mantel test revealed a significant correlation between geographical and genetic distances in all populations (Z = 655.5437, r = 0.7995, P ≤ 0.0010), suggesting the genetic structure observed in An. sinensis populations could be partially explained by distance isolation based on COII sequence analysis (LCP and LPY populations) (Additional file 7: Fig. S2a). As indicated from Tajima’s D and Fu’s Fs tests based on COII, the LPY population exhibited significant negativity (P < 0.05, P < 0.001), suggesting a recent population expansion or selection (Table S3b). Furthermore, the observed smooth and unimodal mismatch distribution in the LPY population suggested a sudden population expansion, conforming to the mismatch distribution derived under the model of sudden expansion (Additional file 7: Fig. S2 b–d).
Discussion
The geographical isolation of An. sinensis populations from Lao PDR
Climate, society, and environmental factors can influence the spatial distribution of malaria vectors and disease transmission [51]. Geologically, 80% of the land in Laos is covered by mountains and plateaus, most of which are forested. The whole country is divided into Upper Laos, Central Laos, and Lower Laos from north to south. The terrain of Laos is high in the north and low in the south. In the north, Phongsaly Province borders China’s Yunnan-Guizhou Plateau, whereas Champasak Province borders Cambodia’s Stung Treng Province in the south, and the terrain is relatively flat. There was a strong association between Anopheles presence estimation and malaria prevalence. The malaria incidence is highest in southern Laos and lowest in the north, where malaria transmission is sporadic and local [3, 4].
A number of genetic markers were used to analyze population genomic data in Anopheles. mtDNA has been frequently used to resolve the issues of molecular taxonomy, phylogenetic relationships, and population structure in malaria vectors due to high copy numbers and the availability of conserved primers and PCR techniques [32]. In the case of recently diverged taxa or cryptic species of mosquitoes, however, ribosomal DNA (rDNA) has been shown to be more reliable than mtDNA in addressing evolutionary problems [52]. Microsatellites have been developed and used primarily to assess patterns and rates of gene flow among Anopheles populations due to their high mutation rate, while mtDNA polymorphisms are superior for detecting large-scale geographical differences [32]. In addition, for genotyping single biallelic single-nucleotide polymorphisms (SNPs) in large numbers of parasite or vector samples, the malaria genomics field has employed techniques such as Sequenom/Agena, TaqMan, and high-resolution melting curves. Nonetheless, SNP genotyping is inexpensive at scale but requires specialized instrumentation [53]. In the present study, significant genetic differentiation was identified between populations from the China–Laos border (LPY) and Cambodia–Laos border (LCP) by using mtDNA-COII sequencing (Fig. 4). The evident differences in the geographical distribution of COII allele between populations in the northern and the southern Laos are probably a result of the geographical barriers restricting gene flow, which was induced by the intricate landscape of Laos (Additional file 6: Table S5) (Additional file 7: Fig. S2a). In other words, spatial distance and heterogeneous landscape can be factors suppressing gene flow between the China–Laos border and Cambodia–Laos populations. The findings of distinct An. sinensis populations identified in Laos imply that local governments should undertake more targeted and effective malaria prevention and elimination measures.
Effects of insecticide-associated selection
The intense utilization of insecticides for agricultural pest control and insecticide utilized in vector control might cause strong selective pressure for the resistance development in malaria vectors [54, 55]. Insecticide resistance occurs when the frequency of resistance genes in the mosquito population increases after exposure to insecticides [51]. Theoretically, selective pressure might exert an effect on shaping the frequency of insecticide resistance-related mutation in a population [30]. According to Souris’ study, the possibility of insecticide resistance was speculated by spatio-temporal models for Anopheles species’ presence, and was more remarkable in the southwest parts of the nation, particularly in the Champasak Province, owing to insecticide utilization [51]. However, in the present study, kdr mutations in the vgsc gene were not found in the Cambodia–Laos border (Champasak Province) (Fig. 2, Table 1). Nevertheless, it was improbable to evaluate the effect of a regional selective process of insecticides on the existing kdr distribution pattern, as the history of insecticide utilization was unknown for the specimens discussed herein.
Previous studies revealed that in the widespread species An. sinensis, the L1014F/L1014S/L1014C/L1014W/N1013S kdr mutations were identified in China [24, 25, 29, 56], while L1014F/L1014C were identified in Korea [28]. However, few studies focus on kdr mutations of An. sinensis in Laos. According to Verhaeghen’s study, kdr of An. sinensis (L1014S) was only found in southern Vietnam and in Cambodia near the Vietnamese border, but not in Laos [18]. Likewise, the present study demonstrates that only a wild haplotype was detected, TTG (1014L), which suggested that a further susceptibility bioassay or research on other common polymorphisms were needed to investigate the actual status of pyrethroid-based chemical insecticide resistance in Laos, especially the southwest part of the country. Intriguingly, there was geographical heterogeneity of wild kdr haplotypes and the presence of region-specific haplotypes (Fig. 3a, Table 1). For instance, 1014L3 was only identified along the Thailand–Laos border and Cambodia–Laos border. The newly identified haplotype, 1014L21, was only discovered on the China–Laos border and has not been identified in other countries (Fig. 3a, Table 1) [30, 57]. The region-specific distributions of kdr haplotypes highlight the necessity to continue the monitoring of chemical insecticide susceptibility to promptly detect potential occurrence and/or migration of chemical insecticide resistance in malaria vectors in Lao PDR.
Furthermore, previous studies revealed that the geographical distribution pattern and the genealogical analysis of kdr haplotypes strongly suggest that kdr variants have multiple origins [30, 31, 57]. Likewise, in the present study, we found the same pattern of the evolutionary relationship of 1014S and 1014F haplotypes when conducting network analysis combined with haplotypes retrieved from the GenBank (Fig. 3b). Interestingly, four newly identified kdr haplotypes were observed, namely 1014F7, 1014F8, 1014F9, and 1014W1. Haplotypes 1014F8 and 1014F9 were only identified in Zhejiang Province (Southeast China) and Anhui Province (Central China), while 1014W1 was only identified in Henan Province (Central China). The distinct distribution patterns of the 1014F/W allele are probably a result of independent mutation events in diverse geographical regions (Fig. 3b, Table1).
In addition, application of genetic hitchhiking by using neutrality tests revealed that the significant negative Fu’s Fs value in two southwest China populations (Guangxi and Hainan Provinces) could reflect the impact of intense selective pressure by the increased utilization of chemical insecticide on the kdr locus and the flanking regions of kdr, such as the kdr intron. In contrast, neutrality tests failed to provide evidence of selection at the kdr intron in Laos. The lack of strong selection pressure might partially elucidate the absence of chemical insecticide-resistant alleles in Laos (Additional file 3: Table S3a).
History of insecticide-associated selection in Laos
Knowledge of insecticide resistance in Anopheles species is imperative for guiding malaria vector control strategies [58]. Exposing malaria vectors to insecticides is possibly predominantly driven by agricultural insecticides because of the intense agricultural insecticide input in contrast to public health insecticides [51]. However, the risk of insecticide resistance in malaria vectors in Southeast Asia is an underlying challenge for preventing malaria and the achievements seen during recent years [51]. Insecticide resistance in major malaria vectors has been identified in neighboring countries like Cambodia, Vietnam, and Thailand [55, 59], and the National Malaria Control Program has to determine potential regions of insecticide resistance emergence.
In Lao PDR, after the ban of DDT and the termination of the mass drug administration (MDA) program, ITNs and LLINs were introduced in 2000 [51]. Since then, vector control has relied on IRS and/or ITNs with the use of pyrethroids in this country [58]. According to the World Health Organization (WHO), approximately 50,403 LLINs were distributed to societies in 2018, accounting for a net coverage of 20% [4]. The greatest risk for insecticide resistance development is in the southwest part of Lao PDR; to be specific, rice and other agricultural systems along the Mekong River and the rest of the agricultural regions in Champasak [51]. However, previous limited studies revealed that no resistance to pyrethroids was found in several primary vectors including An. maculatus, An. minimus, and An. sinensis in Laos [18, 57], indicating that those insecticides remained suitable for malaria vector control. Likewise, in the present study, the lack of kdr mutations in An. sinensis samples from Laos is probably a result of the lack of intense local selection in combination with the geographical isolation in the mosquito populations. Since no regularly conducted vector control programs have been executed in the remote villages studied in Laos, the absence of pyrethroid selection pressure on the mosquito populations might explain why kdr is rare (Table 1).
Based on the findings in this study, we suggest that pyrethroids are still appropriate for controlling An. sinensis in Laos, and LLINs or IRS using pyrethroids remain valid for protecting people from indoor Anopheles bites and ought to be used extensively, particularly in the southern part of the country where malaria is endemic. A potent management program of chemical insecticide resistance should be executed to maintain the susceptibility of vgsc alleles. Furthermore, the application of pyrethroids should not be the only measure for malaria vector control and should be used in combination with insecticides with alternative modes of action or in a rotational manner [57]. As the precise quantity regarding insecticide usage in Lao PDR is unclear, a national registration system for insecticides should be established to guarantee the smooth monitoring of insecticides [51]. On the other hand, our findings can aid local governments in implementing targeted and effective vector control strategies for malaria prevention and elimination among the most vulnerable populations. Local chemical insecticide resistance surveillance systems in Anopheles mosquito populations should be implemented focusing on more restricted areas.
Conclusions
The present study compares the genetic patterns revealed by a functional gene, kdr, with a neutral marker, COII, and demonstrates the combined impact of demographic and selection factors on the An. sinensis population structure. The strong genetic differentiation and limited gene flow between the China–Laos (northern Laos) and Cambodia–Laos (southern Laos) An. sinensis populations based on COII analysis suggest that these two regions are genetically isolated. Only nine wild haplotypes of kdr, TTG (1014L) were observed in An. sinensis populations, as well as distinct distribution of kdr wild haplotypes among different geological regions in Laos.
Lack of kdr mutations in the vgsc gene in the three border regions of Laos is likely a consequence of genetic isolation and the absence of intense selection pressure. The present study suggests that pyrethroids remain suitable for use against An. sinensis in Laos, but routine monitoring of chemical insecticide resistance should be continued and focused on more restricted areas in this country as part of chemical insecticide resistance management.
Availability of data and materials
Data supporting the conclusions of this article are included within the article and its additional files. The current study’s datasets generated and/or analyzed are available in GenBank (http://www.ncbi.nlm.nih.gov/). The nucleotide sequence data reported in this study have been deposited in the GenBank DNA database (https://www.ncbi.nlm.nih.gov/genbank/) with accession number ON400573-ON400661.
Abbreviations
- GMS:
-
Greater Mekong Subregion
- Lao PDR:
-
Lao People’s Democratic Republic
- LLIN:
-
Long-lasting insecticide-treated nets
- IRS:
-
Indoor residual spraying
- VGSC:
-
Voltage-gated sodium channel protein
- mtDNA:
-
Mitochondrial DNA
- COII :
-
Cytochrome c oxidase subunit II
References
WHO. World malaria report 2020. Geneva: World Health Organization; 2020.
WHO. World malaria report 2016. Geneva: World Health Organization; 2016.
Jorgensen P, Nambanya S, Gopinath D, Hongvanthong B, Luangphengsouk K, Bell D, et al. High heterogeneity in Plasmodium falciparum risk illustrates the need for detailed mapping to guide resource allocation: a new malaria risk map of the Lao People’s Democratic Republic. Malar J. 2010;9:59.
WHO. World malaria report 2019. Geneva: World Health Organization; 2019.
Kounnavong S, Gopinath D, Hongvanthong B, Khamkong C, Sichanthongthip O. Malaria elimination in Lao PDR: the challenges associated with population mobility. Infect Dis Poverty. 2017;6:81.
Wilson AL, Courtenay O, Kelly-Hope LA, Scott TW, Takken W, Torr SJ, et al. The importance of vector control for the control and elimination of vector-borne diseases. PLoS Negl Trop Dis. 2020;14:e0007831.
MNREPCD. Lao people’s democratic republic national implementation plan under Stockholm convention on persistant organic pollutants. New Delhi: Ministry of Natural Resources and Environment Pollution Control Department; 2016. p. 147.
Ministry of Health LP. National strategy for malaria control and pre-elimination, 2011–2015, Lao PDR. New Delhi: Ministry of Health; 2010.
Dong K, Du Y, Rinkevich F, Nomura Y, Xu P, Wang L, et al. Molecular biology of insect sodium channels and pyrethroid resistance. Insect Biochem Mol Biol. 2014;50:1–17.
Silva APB, Santos JMM, Martins AJ. Mutations in the voltage-gated sodium channel gene of anophelines and their association with resistance to pyrethroids-a review. Parasit Vectors. 2014;7:450.
Gomes B, Purkait B, Deb RM, Rama A, Singh RP, Foster GM, et al. Knockdown resistance mutations predict DDT resistance and pyrethroid tolerance in the visceral leishmaniasis vector Phlebotomus argentipes. PLoS Negl Trop Dis. 2017;11:e0005504.
Dykes C, Kushwah R, Das M, Sharma S, Bhatt R, Veer V, et al. Knockdown resistance (kdr) mutations in Indian Anopheles culicifacies populations. Parasit Vectors. 2015;8:333.
Dykes CL, Das MK, Eapen A, Batra CP, Ghosh SK, Vijayan VA, et al. Knockdown Resistance (kdr) Mutations in Indian Anopheles stephensi (Diptera: Culicidae) Populations. J Med Entomol. 2016;53:315–20.
Surendran SN, Jayadas TTP, Tharsan A, Thiruchenthooran V, Santhirasegaram S, Sivabalakrishnan K, et al. Anopheline bionomics, insecticide resistance and transnational dispersion in the context of controlling a possible recurrence of malaria transmission in Jaffna city in northern Sri Lanka. Parasit Vectors. 2020;13:156.
Syafruddin D, Hidayati AP, Asih PB, Hawley WA, Sukowati S, Lobo NF. Detection of 1014F kdr mutation in four major Anopheline malaria vectors in Indonesia. Malar J. 2010;9:315.
Tan WL, Li CX, Lv RC, Dong YD, Guo XX, Xing D, et al. The polymorphism and geographical distribution of knockdown resistance of adult Anopheles sinensis populations in eastern China. Malar J. 2019;18:164.
Wang Y, Yu W, Shi H, Yang Z, Xu J, Ma Y. Historical survey of the kdr mutations in the populations of Anopheles sinensis in China in 1996–2014. Malaria J. 2015;14:120.
Verhaeghen K, Van Bortel W, Trung HD, Sochantha T, Keokenchanh K, Coosemans M. Knockdown resistance in Anopheles vagus, An. sinensis, An. paraliae and An. peditaeniatus populations of the Mekong region. Parasit Vectors. 2010;3:59.
Chai JY. History and current status of malaria in Korea. Infect Chemother. 2020;52:441–52.
Feng X, Zhang S, Huang F, Zhang L, Feng J, Xia Z, et al. Biology, bionomics and molecular biology of Anopheles sinensis Wiedemann 1828 (Diptera: Culicidae), main malaria vector in China. Front Microbiol. 2017;8:1473.
Sinka M, Bangs M, Manguin S, Chareonviriyaphap T, Patil A, Temperley W, et al. The dominant Anopheles vectors of human malaria in the Asia-Pacific region: occurrence data, distribution maps and bionomic precis. Parasit Vectors. 2011;4:89.
Zhang C, Sorchampa S, Zhou H, Jiang J, Yang R, Zhang Y. Survey of asymptomatic malaria and mosquito vectors in Muang Khua District of Phongsaly Province, China-Laos Border. Int J Infect Dis. 2020;96:141–7.
Hii J, Rueda LM. Malaria vectors in the Greater Mekong Subregion: overview of malaria vectors and remaining challenges. Southeast Asian J Trop Med Public Health. 2013;44:73–165.
Zhong D, Chang X, Zhou G, He Z, Fu F, Yan Z, et al. Relationship between knockdown resistance, metabolic detoxification and organismal resistance to pyrethroids in Anopheles sinensis. PLoS ONE. 2013;8:e55475.
Tan WL, Li CX, Wang ZM, Liu MD, Dong YD, Feng XY, et al. First detection of multiple knockdown resistance (kdr)-like mutations in voltage-gated sodium channel using three new genotyping methods in Anopheles sinensis from Guangxi Province. China J Med Entomol. 2012;49:1012–20.
Qin Q, Li YJ, Zhong DB, Zhou N, Chang XL, Li C, et al. Insecticide resistance of Anopheles sinensis in Hainan Island, a malaria-endemic area of China. Parasit Vectors. 2014;7:92.
Chang X, Zhong D, Fang Q, Hartsel J, Zhou G, Shi L, et al. Multiple resistances and complex mechanisms of Anopheles sinensis mosquito: a major obstacle to mosquito-borne diseases control and elimination in China. PLoS Negl Trop Dis. 2014;8:e2889.
Kang S, Jung J, Lee S, Hwang H, Kim W. The polymorphism and the geographical distribution of the knockdown resistance (kdr) of Anopheles sinensis in the Republic of Korea. Malaria J. 2012;11:151.
Tan WL, Wang ZM, Li CX, Chu HL, Xu Y, Dong YD, et al. First report on co-occurrence knockdown resistance mutations and susceptibility to beta-cypermethrin in Anopheles sinensis from Jiangsu province, China. PLoS ONE. 2012;7:e29242.
Yang C, Feng X, Huang Z, Li M, Qiu X. Diversity and frequency of kdr mutations within Anopheles sinensis populations from Guangxi, China. Malar J. 2016;15:411.
Chang X, Zhong D, Lo E, Fang Q, Bonizzoni M, Wang X, et al. Landscape genetic structure and evolutionary genetics of insecticide resistance gene mutations in Anopheles sinensis. Parasit Vectors. 2016;9:228.
Loaiza JR, Bermingham E, Sanjur OI, Scott ME, Bickersmith SA, Conn JE. Review of genetic diversity in malaria vectors (Culicidae: Anophelinae). Infect Genet Evol. 2012;12:1–12.
Zhang C, Yang R, Wu L, Luo C, Guo X, Deng Y, et al. Molecular phylogeny of the Anopheles hyrcanus group (Diptera: Culicidae) based on rDNA-ITS2 and mtDNA-COII. Parasit Vectors. 2021;14:454.
Das BP, Rajagopal R, Akiyama J. Pictorial key to the species of Indian Anopheline mosquitoes. J Pure Appl Zool. 1990;2:131–62.
Zhang C, Luo C, Yang R, Yang Y, Guo X, Deng Y, et al. Morphological and molecular identification reveals a high diversity of Anopheles species in the forest region of the Cambodia-Laos border. Parasit Vectors. 2022;15:94.
Davidson JR, Wahid I, Sudirman R, Small ST, Hendershot AL, Baskin RN, et al. Molecular analysis reveals a high diversity of Anopheles species in Karama West Sulawesi Indonesia. Parasit Vectors. 2020;13:379.
Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35:1547–9.
Hall T. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser. 1999;41:95–8.
Tamura K, Nei M, Kumar S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Natl Acad Sci USA. 2004;101:11030–5.
Rambaut A. FigTree. 2012. https://github.com/rambaut/figtree/releases. Accessed 20 Aug 2021.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2.
Bandelt HJ, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16:37–48.
Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–95.
Fu YX. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147:915–25.
Excofer L, Laval G, Schneider S. Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinform Online. 2005;1:47–50.
Nei M. Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci USA. 1973;70:3321–3.
Jensen JL, Bohonak AJ, Kelley ST. Isolation by distance, web service. BMC Genet. 2005;6:13.
Slatkin M, Hudson RR. Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genetics. 1991;129:555–62.
Rogers AR, Harpending H. Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol. 1992;9:552–69.
Rogers AR. Genetic evidence for a Pleistocene population explosion. Evolution. 1995;49:608–15.
Souris M, Marcombe S, Laforet J, Brey PT, Corbel V, Overgaard HJ. Modeling spatial variation in risk of presence and insecticide resistance for malaria vectors in Laos. PLoS ONE. 2017;12:e0177274.
Fang Y, Shi WQ, Zhang Y. Molecular phylogeny of Anopheles hyrcanus group members based on ITS2 rDNA. Parasit Vectors. 2017;10:417.
Neafsey DE, Taylor AR, MacInnis BL. Advances and opportunities in malaria population genomics. Nat Rev Genet. 2021;22:502–17.
Nkya TE, Akhouayri I, Kisinza W, David JP. Impact of environment on mosquito response to pyrethroid insecticides: facts, evidences and prospects. Insect Biochem Mol Biol. 2013;43:407–16.
Susanna D, Pratiwi D. Current status of insecticide resistance in malaria vectors in the Asian countries: a systematic review. F1000Res. 2022;10:200.
Chang XL, Xue YQ, Zhang AD, Zhu GD, Fang Q. Deltamethrin resistance, metabolic detoxification enzyme and kdr mutation in Anopheles sinensis in region along Huaihe River in Anhui Province. Zhongguo Xue Xi Chong Bing Fang Zhi Za Zhi. 2013;25:263–7.
Yang C, Feng X, Liu N, Li M, Qiu X. Target-site mutations (AChE-G119S and kdr) in Guangxi Anopheles sinensis populations along the China-Vietnam border. Parasit Vectors. 2019;12:77.
Marcombe S, Bobichon J, Somphong B, Phommavan N, Maithaviphet S, Nambanya S, et al. Insecticide resistance status of malaria vectors in Lao PDR. PLoS ONE. 2017;12:e0175984.
Quiñones ML, Norris DE, Conn JE, Moreno M, Burkot TR, Bugoro H, et al. Insecticide resistance in areas under investigation by the international centers of excellence for malaria research: a challenge for malaria control and elimination. Am J Trop Med Hyg. 2015;93:69–78.
Acknowledgements
We are grateful to the National Natural Science Foundation of China. We also thank the Yunnan Provincial Collaborative Innovation Center for Public Health and Disease Prevention and Control (Grant Number 2014YNPHXT03) and the China-ASEAN Public Health Cooperation Fund and Joint Control of Malaria and Dengue in Lancang-Mekong River Area for the sample collection, data collection, and performing the experiments. We thank Dr. Phoutnalong Vilay at the National Malaria Centre of Lao People's Democratic Republic who participated in the sampling and data collection. We are also grateful to Dr. Phanviengxay Khammavong, Hao Phommasa, and Chunfu Li for participating the fieldwork.
Funding
This study was supported by the National Natural Science Foundation of China (Grant Number 31601002), Yunnan Provincial Collaborative Innovation Center for Public Health and Disease Prevention and Control (Grant Number 2014YNPHXT03), and the China-ASEAN Public Health Cooperation Fund and Joint Control of Malaria and Dengue in Lancang-Mekong River Area.
Author information
Authors and Affiliations
Contributions
Data and statistical analysis, and manuscript preparation: YLZ and CLZ. Data collection: YLZ, CLZ, and RY. Mosquito collection and identification: CLZ, LBW, CHL, and XFG. Performed the experiments: CLZ. Procurement of funding: RY and YLZ. Study conception/design and critical review of the manuscript: CLZ, RY, and YLZ. All authors reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Full list of 89 An. sinensis specimens collected in China–Laos, Thailand–Laos, and Cambodia–Laos borders. N/A, not identified. LXP, Pak lay County of Xayabuli Province (Thailand–Laos border); LPY, Yot Ou County of Phongsaly Province (China–Laos border); LCP, Pathoomphone County of Champasak Province (Cambodia–Laos border).
Additional file 2: Table S2.
COII sequences of An. sinensis were downloaded from the NCBI. KR-YC, Yeoncheon (South Korea); KR-IC, Incheon (South Korea); KR-GR, Guryongpo (South Korea); CN-YN, Yunnan (China); Yunnan; CN-CQ, Chongqing (China); CN-AH, Anhui (China); TH-CM, Chiang Mai (Thailand); JP-NS, Nagasaki (Japan). N/A, no data.
Additional file 3: Table S3. a
Genetic diversity indices and neutrality tests (Fu’s Fs and Tajima’s D) based on the kdr intron of An. sinensis. b Genetic diversity indices and neutrality tests (Fu’s Fs and Tajima’s D) based on the COII gene of An. sinensis. nd not determined; #P < 0.10; *P < 0.05; **P < 0.02; ***P < 0.001. n number of sequences; s number of polymorphic sites; pi nucleotide diversity; h number of haplotypes; Hd haplotype diversity; LPY Yot Ou County (Phongsaly Province); LXP Pak lay County (Xayabuli Province); LCP Pathoomphone County (Champasak Province); CN-AH Anhui Province (China); CN-GX Guangxi Province (China); CN-YN Yunnan Province (China); CN-HaN Hainan Province (China); CN-HeN Henan Province (China); CN-ZJ Zhejiang Province (China).
Additional file 4: Figure S1.
Phylogenetic analysis based on the COII sequences in An. sinensis populations from Laos and other countries. a Phylogenetic network of 46 mitochondrial haplotypes of the COII gene in An. sinensis. Localities are indicated by different colors (bottom right). The size of each circle is proportional to its corresponding frequencies. b UPGMA dendrogram based on Nei’s unbiased genetic distance between the 46 haplotypes of An. sinensis. Yellow, green, and blue circles/rectangles represent haplotypes found in this study from LPY (Phongsaly Province: Yot Ou County), LCP (Champasak Province: Pathoomphone County), and LXP (Xayabuli Province: Pak lay County) populations, respectively. Black, white, red, and sky blue circles/rectangles represent haplotypes found in NCBI data from Korea, Japan, China, and Thailand (Tables S2), respectively.
Additional file 5: Table S4.
Analysis of molecular variance (AMOVA) of 10 An. sinensis populations based on COII. FCT, fixation index among groups; FSC, among populations within groups; FST, within populations.
Additional file 6: Table S5.
Genetic differentiation and gene flow among the geographical groups of An. sinensis based on COII. The pairwise FST values and Nm values based on the COII are shown below and above the diagonal, respectively. Characters in bold indicated the significance (P < 0.05). Inf, infinite. LPY, Yot Ou County, Phongsaly Province; LXP, Pak lay County, Xayabuli Province; LCP, Pathoomphone County, Champasak Province.
Additional file 7: Figure S2. a
Isolation by distance; the relationship between geographical and genetic distances based on COII sequences in An. sinensis populations. Isolation by distance (IBD) was examined using a nonparametric Mantel test with the web-based computer program IBDWS v.3.16. b–d Mismatch distribution graphs for An. sinensis population based on COII sequences. The x- and y-axis show the number of pairwise differences and the frequency of the pairwise comparisons, respectively. The observed frequencies are represented by a dotted line. The frequency expected under the hypothesis of the constant population model is depicted by a solid line. LPY, Yot Ou County, Phongsaly Province; LXP, Pak lay County, Xayabuli Province; LCP, Pathoomphone County, Champasak Province.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, Y., Zhang, C., Wu, L. et al. Population genetic structure and evolutionary genetics of Anopheles sinensis based on knockdown resistance (kdr) mutations and mtDNA-COII gene in China–Laos, Thailand–Laos, and Cambodia–Laos borders. Parasites Vectors 15, 229 (2022). https://doi.org/10.1186/s13071-022-05366-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-022-05366-9