
Abstract
Background
Crossbreeding is an important way to improve production beef cattle performance. Pinan cattle is a new hybrid cattle obtained from crossing Piedmontese bulls with Nanyang cows. After more than 30 years of cross-breeding, Pinan cattle show a variety of excellent characteristics, including fast growth, early onset of puberty, and good meat quality. In this study, we analyzed the genetic diversity, population structure, and genomic region under the selection of Pinan cattle based on whole-genome sequencing data of 30 Pinan cattle and 169 published cattle genomic data worldwide.
Results
Estimating ancestry composition analysis showed that the composition proportions for our Pinan cattle were mainly Piedmontese and a small amount of Nanyang cattle. The analyses of nucleotide diversity and linkage disequilibrium decay indicated that the genomic diversity of Pinan cattle was higher than that of European cattle and lower than that of Chinese indigenous cattle. De-correlated composite of multiple selection signals, which combines four different statistics including θπ, CLR, FST, and XP-EHH, was computed to detect the signatures of selection in the Pinan cattle genome. A total of 83 genes were identified, affecting many economically important traits. Functional annotation revealed that these selected genes were related to immune (BOLA-DQA2, BOLA-DQB, LSM14A, SEC13, and NAALADL2), growth traits (CYP4A11, RPL26, and MYH10), embryo development (REV3L, NT5E, CDX2, KDM6B, and ADAMTS9), hornless traits (C1H21orf62), and climate adaptation (ANTXR2).
Conclusion
In this paper, we elucidated the genomic characteristics, ancestry composition, and selective signals related to important economic traits in Pinan cattle. These results will provide the basis for further genetic improvement of Pinan cattle and reference for other hybrid cattle related studies.
Similar content being viewed by others

Background
Cattle are one of the most important domestic animals for human beings. According to the origin of cattle, domestic cattle can be simply divided into two subspecies: Bos taurus and Bos indicus [1]. Or according to geographical region, it can also be divided into five groups: European taurine, Eurasian taurine, East Asian taurine, Chinese indicine, and Indian indicine [2]. The earliest domesticated cattle were mainly used for draft purposes. With the development of society, except for a few developing countries, cattle have been developed in the direction of specialization through continuous breeding and improvement. Today, there are hundreds of cattle breeds in the world, and China alone has more than 50 native breeds, including five excellent breeds (Yanbian, Luxi, Qinchuan, Jinnan, and Nanyang cattle). Because the cattle in China used to be mainly used for farming, the production performance is not as good as the specialized beef cattle breeds in European and American countries. To compensate for the perceived lower beef production potential of local cattle, these cattle are often crossed with exotic breeds to develop new breeds. Piedmontese cattle, which are native to Italy, are internationally recognized as terminal male due to their double muscle genes and have been introduced to more than 20 countries for crossbreeding. Grading up is often used to improve the performance of cattle. A new breed called Pinan cattle has been developed in Henan Province of China by introducing Piedmontese cattle as male parents and grading up with local Nanyang cattle. It is generally used for production after crossing three generations. Pinan cattle has many advantages, such as the early onset of puberty, fast-growth speed, and good meat quality, which greatly improved the breeding efficiency and meet the demands of people.
Whole-genome sequencing (WGS) has been used to find a large number of variations that can be used as molecular genetic markers. The application of WGS plays an important role in the study of human diseases, the breeding of animals and plants, as well as the origin and domestication of species. WGS studies have yielded many important results in the field of livestock, including domestication, adaptive mechanisms of indigenous breeds, candidate genes responsible for important economic traits, and historical population dynamics [3, 4]. In earlier years, it was mainly European commercial cattle that were studied using whole-genome sequencing [5]. Later, researchers also began to look at native varieties to find genomic variations associated with desired traits in native varieties [6,7,8,9,10], which could help improve breeding.
In the current study, we sequenced the genomes of 30 Pinan cattle to detect genetic variation, population structure, and selective sweep. Our result will lay a foundation for further studies on the genetic basis of important economic traits, provide ideas and basis for future improvement in Pinan cattle, and provide a reference for related studies in other hybrid breeds.
Results
Sequencing and SNP detection
A total of 5,402,336,411 clean reads were generated after genome sequencing in 30 Pinan cattle samples. The clean reads were aligned to the latest Bos taurus reference genome (ARS-UCD1.2) with an average depth of 9.67X using Sentieon [11] software. We identified 23,077,494 biallelic SNPs in 30 Pinan cattle using GATK. Among these SNPs, functional annotation showed that SNPs mainly existed in the intergenic region (59.6%) and intronic region (37.4%). Exons accounted for 0.77% of the total SNPs, including 71,193 non-synonymous SNPs and 99,811 synonymous SNPs (Table S3).
To place Pinan cattle into a global context, we also detected the single nucleotide polymorphisms in five "core" cattle populations [2], and two Chinese cattle breeds (Qinchuan and Jiaxian Red cattle). Among these breeds, Chinese indicine (37,087,338) has the highest number of SNPs, followed by crossbreed Jiaxian cattle (29,315,624), Qinchuan cattle (26,270,077), Nanyang cattle (25,468,742), Pinan cattle (23,077,494) and Indian indicine (23,005,288). The number of SNP in Bos taurus was lower than that in indicine cattle and hybrid cattle. The results of this SNP distribution are consistent with the observations of other studies [2, 8].
Population structure analysis
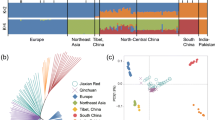
To explore the genetic relationships between Pinan cattle and other cattle breeds around the world, estimating ancestry, building neighbor-joining (NJ) trees and principal component analysis (PCA) were performed (Fig. 1). The first and second PCs explained 6.23% and 2.58% of the variation in the entire genomic data, respectively. The first PC separated Bos taurus and Bos indicus. The analysis revealed clear geographical patterns of cattle distribution. The proportions of individuals in Pinan cattle inferred by the ADMIXTURE are presented in Fig. 1 A. The case with a small CV error value (Table S4) was selected for drawing. When K = 2, these different cattle breeds can be divided into two groups: Bos taurus and Bos indicus; when K = 4, European taurine and Zebu cattle have a single genomic composition. We can see that the ancestry proportions inherited from Bos taurus are greater than that from Bos indicus in Pinan cattle. To be more precise, Pinan cattle composition is more similar to Eurasian taurine which indicates that the genetic influence of Piedmontese cattle was greater than that of Nanyang cattle. The results of the NJ tree (Fig. 1B and Figure S1) and PCA (Fig. 1C) analysis were similar. All "core" cattle groups form independent clusters. The individuals of the Pinan cattle are also mostly clustered together and close to the Piedmontese cattle.

Population structure of Pinan cattle and its relationship with several breeds in the world. (A) ADMIXTURE was used with K = 2 and K = 4 for model-based clustering among different cattle. Color them by geographical area and label them with the breed. Neighbor-joining trees (B) and principal component analysis (C) separated the cattle breeds (199 animals in total) into seven categories
Genetic diversity and linkage disequilibrium
To elucidate the genomic characteristics in Pinan cattle, nucleotide diversity was calculated (Fig. 2A). The results showed that Chinese indicine had the highest nucleotide diversity, followed by Nanyang, Indian, and Qinchuan cattle. The lowest nucleotide diversity was found in European cattle. The nucleotide diversity of our crossbreed Pinan cattle is lower than that of female parent Nanyang cattle but higher than that of male parent Piedmontese cattle. From the perspective of linkage disequilibrium (LD) (Fig. 2B), although the LD of each variety decreased rapidly, there were some differences among varieties. At distances between markers (> 50 kb), Indian indicine and Chinese indicine had the lowest LD level. The highest LD level in Jersey, followed by Pinan. East Asian taurine and European taurine had a medium LD level. Overall, the LD level of indicine breeds was lower than that of taurine breeds, consistent with previous studies [12]. It is worth noting that Pinan cattle had a higher LD level than either of its parents.

A The nucleotide diversity of 12 different cattle breeds. The black line in the boxplot is the median line and the outside points are outliers. B Genome-wide average LD decay is estimated from each breed. Different colored lines represent different breeds. The legend in the middle is shared by both figures
De-correlated composite of multiple signals
Within-population and cross-population statistics were combined separately in a single score using the De-correlated composite of multiple signals (DCMS) statistic [13]. After calculation of the DCMS statistics, the p-value was fitted to a normal distribution and corrected for multiple testing (FDR < 0.05). The analysis identified 92 candidate regions (Table S5) of the genome containing 83 genes. We identified several of the most significant genomic regions on BTA 9: 63.80–64.09 M and BTA 19:27.50 M-28.19 M (Fig. 3). All regions were annotated and some candidate genes associated with important traits were found (Table 1). Including hornless traits (C1H21orf62), growth traits (CYP4A11, RPL26, and MYH10), Disease-related traits (MACC1 and SNX14), climate adaptation (ANTXR2), lipid metabolism (ANGPTL4), embryo development (REV3L, NT5E, CDX2, KDM6B, and ADAMTS9), immune response (BOLA-DQA2, BOLA-DQB, LSM14A, SEC13, and NAALADL2), sperm flagella (DNAH2), milk yield (ODF4, ARHGEF15, and MYH10).

Manhattan plot of the selective signals detected by the DCMS method in the Pinan cattle. The dashed lines indicade the significant threshold level at a FDR of 5% (q-value < 0.05)
Discussion
The characterization of population structure and genetic diversity can help us to evaluate bovine genetic resources and play an important role in utilization and conservation. In this study, the population genetic structure of Pinan Cattle was studied in the background with different cattle breeds. As can be seen from the ADMIXTURE analysis (Fig. 1), the ancestral contributions of Pinan cattle were mainly from Piedmontese.
Although Pinan cattle are closer to the Piedmontese lineage, the number of genetic variations inherited from Nanyang cattle is much higher than that inherited from Piedmontese cattle. The nucleotide diversity of Pinan cattle is lower than that of Chinese indicine and other native Chinese cattle breeds, but about twice that of European cattle breeds such as Angus, Hereford, and Piedmontese. The higher genomic diversity of Pinan cattle may be due to the influence of female parent Nanyang cattle. Because Nanyang cattle is a hybrid between Bos taurus and Bos indicus, it has high genomic diversity. In addition, cattle breeds with higher genetic diversity tend to have lower LD decay, such as Qinchuan cattle. Interestingly, the LD decay level of Pinan cattle was higher than that of Nanyang cattle and Piedmontese, which may be due to the greater selection intensity experienced by the Pinan herd than Nanyang cattle and Piedmontese.
In addition, we found important selective scanning markers in Pinan cattle. Over the past 30 years, Pinan cattle have been used for intensive beef breeding, leading to genetic improvement of production traits, especially for their early onset of puberty. Several genes (REV3L, NT5E, CDX2, KDM6B, ADAMTS9) in the Pinan cattle genome show selectivity for embryo development. To better understand the selection pressure, we explored the biological function of these genes. REV3L is involved in the DNA repair process, and it was upregulated in cattle embryos derived from depleted cells [54]. Loss of REV3L resulted in chromosome instability and embryogenesis failure in mice [25]. Genome-wide association study has shown that REV3L is associated with embryonic development in Nellore cattle [55]. NT5E produces a protein that encodes an enzyme that converts adenosine monophosphate into adenosine [28]. The study has shown that the NT5E gene affects the cleavage rate of cattle [56]. In addition, two non-synonymous SNP in NT5E can be useful for improving Inosine 5’-monophosphate which contributes to the umami taste in beef in Japanese Black beef [57]. CDX2 is a transcription factor that is localized in the nucleus of the trophectoderm [58]. Studies have shown that CDX2 is essential for early development and gene expression involved in the differentiation of inner cell mass and trophectoderm lineages in cattle embryos [30, 59]. KDM6B, also known as JMJD3, is a histone demethylase expressed in bovine cleavage embryos [60]. KDM6B is transiently upregulated around embryo developmental stages when the embryo genome is activated [61]. Another study has shown that H3K27me3 demethylase activity mediated by KDM6B is required for normal bovine embryo development and reprogramming gene expression during the embryonic genome [41]. ADAMTS9 is widely expressed during mouse embryo development [49]. Other studies have shown that ADAMTS9 is necessary for ovarian development and may promote the rupture for follicular rupture in zebrafish [62, 63]. A study has shown that ADAMTS proteases play a role in remodeling the bovine ovulatory follicle in preparation for ovulation and the formation of the corpus luteum [64]. Interestingly, a GWAS study reported that ADAMTS9 was associated with chest size in goats [65]. This suggests it may also play an important role in cattle. These genes may be an important reason for the early maturity and faster growth of Pinan cattle than Nanyang cattle.
Native Chinese cattle tend to be more resistant and adaptable than foreign breeds. In a selective sweep of Pinan cattle populations, we identified several immune system-related genes (BOLA-DQA2, BOLA-DQB, LSM14A, and NAALADL2). BOLA-DQA2 and BOLA-DQB both belong to the Bovine Leukocyte Antigen (BOLA) class II genes which are involved in the immune response. For example, BOLA-DQA2 plays an important role in resistance to mastitis in dairy cows [52]. LSM14A plays a critical role in antiviral immune responses [33, 66]. NAALADL2 is reported to be responsible for immune homeostasis [67]. They may have a similar effect in cattle but there are currently few studies on that. In addition, we found a positive selection gene (ANTXR2) associated with climate adaptation [21]. These positive selection genes are likely to improve the survival of Pinan cattle.
In addition, we found several genes (CYP4A11, RPL26, and MYH10) related to growth traits. CYP4A11 is involved in fatty acid metabolism, blood pressure regulation, and kidney tubule absorption of ions [68]. The study has shown that CYP4A11 affects growth and fat deposition in cattle [17]. RPL26, a ribosomal protein gene, is reported to be related to residual feed intake in Holstein cows [69]. GWAS analysis has shown that one missense variant in RPL26 is associated with average daily gain. There is a report shows that MYH10 is strongly associated with milk production and body conformation traits of dairy cattle [70]. GWAS study also shows that MYH10 is a candidate gene for yearling weight in Chinese Simmental beef cattle. In addition, two other positive selection genes ODF4 and ARHGEF15 have been reported to be involved in the milk yield of dairy goats. This suggests they may have a similar role in cattle.
Conclusions
In this study, genomic variation of Pinan cattle was studied comprehensively using WGS data. The study on the population structure and genomic diversity of Pinan cattle will provide the basis for the genetic evaluation of this breed and point out the direction for the development of reasonable breeding strategies for future breeding and improvement. In addition, we have identified a series of candidate genes that may have important effects on survival, growth, early maturity, and meat quality traits of this variety. These results will provide a basis for further genomic studies of beef cattle and provide a reference for genomic studies of imported crossbred beef cattle in China or other parts of the world.
Methods
Samples and sequencing
We collected blood samples from 30 Pinan cattle in Xinye County, Henan Province using EDTA-K2 anticoagulant tubes. After dry ice storage, genomic DNA was extracted in the laboratory using the standard phenol–chloroform method [71]. The integrity of genomic DNA was verified by agarose gel electrophoresis. And we used Nanodrop to measure the purity of DNA. DNA samples containing more than 1.5 µg were used to build libraries. A paired-end library with an average insert length of 500 bp and an average read length of 150 bp was constructed for each individual. Sequencing was performed using Illumina Hiseq 2000 platform at the Novogene Bioinformatics, Beijing, China.
To better compare the genetic diversity of Pinan cattle with other cattle breeds in the world, we collected additional samples based on the five "core" groups proposed by Chen et al., [2] as well as the data of 30 Jiaxian cattle and 30 Qinchuan cattle of local breeds in China. The samples include European varieties (Angus, Hereford, Red Angus, Gelbvieh, Limousin, Simmental, Piedmontese, Korean breed (Hanwoo), Chinese breed (Tibetan, Qinchuan, Leiqiong, Guangfeng, Ji’an Wannan and India-Pakistan cattle breeds ((Tharparkar, Sahiwal, Hariana, Nelore, and Gir) (Table S2). A total of 199 samples were used in this study.
Reads mapping, SNP calling, and annotation
After obtaining the fastq file of the raw data, Trimmomatic software (v0.36) was used to trim sequence reads with the parameters: “LEADING:20, TRAILING:20, SLIDINGWINDWOE: 3:15, AVGQUAL:20, MINLEN:35, TOPHRED33”. The clean reads were aligned to the Bos taurus reference genome ARS-UCD1.2 by the “sentieon bwa mem” module with default parameters from sentieon software (versionV202010) [11]. After that “sentieon driver” module was used to filter duplicate reads and used to perform Base Quality Score Recalibration with default parameters. Single nucleotide polymorphisms (SNPs) were detected by the Genome Analysis Toolkit (GATK, version 4.1.8.1) [72]. The raw SNPs were called using the “SelectVariants” module of GATK. After SNPs were called, the “VariantFiltration” module was used to filter SNPs with the parameters “QualByDepth (QD) < 2.0, Quality (QUAL) < 30.0, StrandOddsRatio (SOR) > 3.0, FisherStrand (FS) > 60.0, RMSMappingQuality (MQ) < 40.0, MappingQualityRankSumTest (MQRankSum) < -12.5, and (ReadPosRankSumTest) ReadPosRankSum < -8.0”. Finally, a total of 53,174,296 biallelic SNPs were obtained using bcftools (version 1.8) with the parameters: F_MISSING < 0.1 & MAC > 2. After that, SNPs were filtered using VCFtools v0.1.16 with the parameters: –maf 0.05 and –max-missing 0.9. Based on the Bos taurus reference genome annotation file(https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/002/263/795/GCF_002263795.1_ARS-UCD1.2/GCF_002263795.1_ARS-UCD1.2_genomic.gff), Annovar [73] software was used to annotate the functions of each SNP.
Population genetic analysis
PLINK (version 1.9) [74] software was used to remove those sites with high LD with the parameter (–indep-pair-wise 50 5 0.2), and the remaining SNP sites were used for performing ADMIXTURE analysis. PCA was performed using the SmartPCA program in the Eigensoft V5.0 package [75]. ADMIXTURE v1.3 [76] was used for population structure analysis, the kinship set is from 2 to 8. The matrix of pairwise genetic distances supplied by plink was used for constructing an unrooted evolutionary tree. The visualization of evolutionary was done by MEGA7 v7.0.26 [77] and FigTree v1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/).
Selective sweep identification
In order to detect selective signals in the Pinan cattle, we used different strategies to scan the genomes of Pinan cattle. The nucleotide diversity (θπ) and the composite likelihood ratio (CLR) [78] method were used in the Pinan cattle population. First, the SNP loci with allele frequency less than 0.05 were removed. After that, we used VCFtools v0.1.16 [79] to estimate the nucleotide diversity by a sliding window approach in which the windows were 50 kb and the step size is 20 kb. The SweepFinder2 [80] was used for calculating the CLR with a grid size of 50 kb. In addition, fixation index (FST) and cross-population extended haplotype homozygosity (XP-EHH) were used for comparing the genomic difference between Pinan and Piedmontese cattle. We used VCFtools to analyze FST with a 50 kb window and a 20 kb step. Within each population, haplotype phasing was performed using BEAGLE 5.0 [81], and selscan V1.1 [82] was used to calculate the XP-EHH statistics for each population. For XP-EHH selection scans, our test statistics are the average of standardized XP-EHH scores for each 50 kb region.
DCMS estimation
In this study, we combined four statistics including θπ, CLR, FST, and XPEHH into a single DCMS framework as described in Ma et al. [13]. The analysis steps were carried out by referring to other studies [10, 83,84,85]. The DCMS statistic was calculated for each 50 kb window using the R MINOTAUR package [86]. The genome-wide P-value was calculated by the stat_to_pvalue function of the R MINOTAUR package, and appropriate unilateral tests were performed, i.e., a left tailed test for the θπ and CLR (two.tailed = FALSE, right.tailed = FALSE) and a right tailed test for the FST and XPEHH (two.tailed = FALSE, right.tailed = TRUE). Then, the n × n correlation matrix between the statistics is calculated using the covNAMcd function (alpha = 0.75, nsamp = 50,000) of the R rrcovNA package [87]. The matrix was used as the input of the DCMS function of the R MINOTAUR package to calculate the DCMS value of the whole genome. After calculating the DCMS value, these DCMS values were transformed to a normal distribution with the robust linear model using the rlm function (dcms ~ 1) of the R MASS package [88]. The output of the fitting model is the input of the pnorm R function to calculate the p-value of DCMS statistics. To control for multiple test false discovery rate (FDR) in the rejected null hypothesis, the p.adjust R function (p = dcms_pvalue, method = ”BH”, n = length(dcms_pvalue)) was adopted to convert the p-value of DCMS into the corresponding q-value according to Benjamini and Hochberg methods.
Candidate gene annotation
The region with DCMS q-value lower than 0.05 was regarded as the statistically significant region, and the genes contained in the significant region were annotated through the reference genome annotation file based on the ARS-UCD 1.2 reference genome. The function of these candidate genes was further determined through a literature review.
Availability of data and materials
Sequences are available from GenBank with the Bioproject accession number PRJNA698276 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA698276).
Abbreviations
- SNP:
-
Single nucleotide polymorphism
- WGS:
-
Whole-genome sequencing
- LD:
-
Linkage disequilibrium
- PCA:
-
Principal component analysis
- NJ:
-
Neighbor-joining
- θπ:
-
Nucleotide diversity
- CLR:
-
Composite likelihood ratio
- FST:
-
Fixation index
- XP-EHH:
-
Cross-population extended haplotype homozygosity
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
References
Decker JE, McKay SD, Rolf MM, Kim J, Molina Alcalá A, Sonstegard TS, Hanotte O, Götherström A, Seabury CM, Praharani L. Worldwide patterns of ancestry, divergence, and admixture in domesticated cattle. PLoS Genet. 2014;10(3):e1004254.
Chen N, Cai Y, Chen Q, Li R, Wang K, Huang Y, Hu S, Huang S, Zhang H, Zheng Z. Whole-genome resequencing reveals world-wide ancestry and adaptive introgression events of domesticated cattle in East Asia. Nat Commun. 2018;9(1):1–13.
Bai Y, Sartor M, Cavalcoli J. Current status and future perspectives for sequencing livestock genomes. J Anim Sci Biotechnol. 2012;3(1):1–6.
Georges M, Charlier C, Hayes B. Harnessing genomic information for livestock improvement. Nat Rev Genet. 2019;20(3):135–56.
Gutiérrez-Gil B, Arranz JJ, Wiener P. An interpretive review of selective sweep studies in Bos taurus cattle populations: identification of unique and shared selection signals across breeds. Front Genet. 2015;6:167.
Shen J, Hanif Q, Cao Y, Yu Y, Lei C, Zhang G, Zhao Y. Whole genome scan and selection signatures for climate adaption in yanbian cattle. Front Genet. 2020;11:94.
Bhati M, Kadri NK, Crysnanto D, Pausch H. Assessing genomic diversity and signatures of selection in Original Braunvieh cattle using whole-genome sequencing data. BMC Genomics. 2020;21(1):1–14.
Xia X, Zhang S, Zhang H, Zhang Z, Chen N, Li Z, Sun H, Liu X, Lyu S, Wang X. Assessing genomic diversity and signatures of selection in Jiaxian Red cattle using whole-genome sequencing data. BMC Genomics. 2021;22(1):1–11.
Tijjani A, Utsunomiya YT, Ezekwe AG, Nashiru O, Hanotte O. Genome sequence analysis reveals selection signatures in endangered trypanotolerant West African Muturu cattle. Front Genet. 2019;10:442.
Yurchenko AA, Daetwyler HD, Yudin N, Schnabel RD, Vander Jagt CJ, Soloshenko V, Lhasaranov B, Popov R, Taylor JF, Larkin DM. Scans for signatures of selection in Russian cattle breed genomes reveal new candidate genes for environmental adaptation and acclimation. Sci Rep. 2018;8(1):1–16.
Weber JA, Aldana R, Gallagher BD, Edwards JS. Sentieon DNA pipeline for variant detection-Software-only solution, over 20x faster than GATK 3.3 with identical results. Peer J Pre Prints. 2016;4:e1672v1672.
Porto-Neto LR, Kijas JW, Reverter A. The extent of linkage disequilibrium in beef cattle breeds using high-density SNP genotypes. Genet Sel Evol. 2014;46(1):1–5.
Ma Y, Ding X, Qanbari S, Weigend S, Zhang Q, Simianer H. Properties of different selection signature statistics and a new strategy for combining them. Heredity (Edinb). 2015;115(5):426–36.
Liu WB, Liu J, Liang C, Guo X, Bao P, Chu M, Ding X, Wang H, Zhu X, Yan P. Associations of single nucleotide polymorphisms in candidate genes with the polled trait in D atong domestic yaks. Anim Genet. 2014;45(1):138–41.
Chartier-Harlin M-C, Dachsel JC, Vilariño-Güell C, Lincoln SJ, Leprêtre F, Hulihan MM, Kachergus J, Milnerwood AJ, Tapia L, Song M-S. Translation initiator EIF4G1 mutations in familial Parkinson disease. Am J Hum Genet. 2011;89(3):398–406.
Nandolo W, Mészáros G, Wurzinger M, Banda LJ, Gondwe TN, Mulindwa HA, Nakimbugwe HN, Clark EL, Woodward-Greene MJ, Liu M. Detection of copy number variants in African goats using whole genome sequence data. BMC Genomics. 2021;22(1):1–15.
Yang M, Lv J, Zhang L, Li M, Zhou Y, Lan X, Lei C, Chen H. Association study and expression analysis of CYP4A11 gene copy number variation in Chinese cattle. Sci Rep. 2017;7(1):1–8.
Stein U, Walther W, Arlt F, Schwabe H, Smith J, Fichtner I, Birchmeier W, Schlag PM. MACC1, a newly identified key regulator of HGF-MET signaling, predicts colon cancer metastasis. Nat Med. 2009;15(1):59–67.
Zheng X, Ju Z, Wang J, Li Q, Huang J, Zhang A, Zhong J, Wang C. Single nucleotide polymorphisms, haplotypes and combined genotypes of LAP3 gene in bovine and their association with milk production traits. Mol Biol Rep. 2011;38(6):4053–61.
He X, Huang Q, Qiu X, Liu X, Sun G, Guo J, Ding Z, Yang L, Ban N, Tao T. LAP3 promotes glioma progression by regulating proliferation, migration and invasion of glioma cells. Int J Biol Macromol. 2015;72:1081–9.
Flori L, Moazami-Goudarzi K, Alary V, Araba A, Boujenane I, Boushaba N, Casabianca F, Casu S, Ciampolini R, Coeur D’Acier A. A genomic map of climate adaptation in Mediterranean cattle breeds. Mol Ecol. 2019;28(5):1009–29.
Mamedova L, Robbins K, Johnson B, Bradford B. Tissue expression of angiopoietin-like protein 4 in cattle. J Anim Sci. 2010;88(1):124–30.
Berger I, Hershkovitz E, Shaag A, Edvardson S, Saada A, Elpeleg O. Mitochondrial complex I deficiency caused by a deleterious NDUFA11 mutation. Ann Neurol. 2008;63(3):405–8.
Jiang P, Zhang D. Maternal embryonic leucine zipper kinase (MELK): a novel regulator in cell cycle control, embryonic development, and cancer. Int J Mol Sci. 2013;14(11):21551–60.
Lange SS, Tomida J, Boulware KS, Bhetawal S, Wood RD. The polymerase activity of mammalian DNA pol ζ is specifically required for cell and embryonic viability. PLoS Genet. 2016;12(1):e1005759.
Santangelo L, Giurato G, Cicchini C, Montaldo C, Mancone C, Tarallo R, Battistelli C, Alonzi T, Weisz A, Tripodi M. The RNA-binding protein SYNCRIP is a component of the hepatocyte exosomal machinery controlling microRNA sorting. Cell Rep. 2016;17(3):799–808.
Scott EY, Woolard KD, Finno CJ, Murray JD. Cerebellar abiotrophy across domestic species. Cerebellum. 2018;17(3):372–9.
Martín-Satué M, Lavoie EG, Fausther M, Lecka J, Aliagas E, Kukulski F, Sévigny J. High expression and activity of ecto-5′-nucleotidase/CD73 in the male murine reproductive tract. Histochem Cell Biol. 2010;133(6):659–68.
Yamazaki K, Umeno J, Takahashi A, Hirano A, Johnson TA, Kumasaka N, Morizono T, Hosono N, Kawaguchi T, Takazoe M. A genome-wide association study identifies 2 susceptibility loci for Crohn’s disease in a Japanese population. Gastroenterology. 2013;144(4):781–8.
Sakurai N, Takahashi K, Emura N, Fujii T, Hirayama H, Kageyama S, Hashizume T, Sawai K. The necessity of OCT-4 and CDX2 for early development and gene expression involved in differentiation of inner cell mass and trophectoderm lineages in bovine embryos. Cellular Reprogram (Formerly" Cloning and Stem Cells"). 2016;18(5):309–18.
Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood J Am Soc Hemat. 2002;100(5):1532–42.
Takeo Y, Kurabayashi N, Nguyen MD, Sanada K. The G protein-coupled receptor GPR157 regulates neuronal differentiation of radial glial progenitors through the Gq-IP3 pathway. Sci Rep. 2016;6(1):1–12.
Liu T-T, Yang Q, Li M, Zhong B, Ran Y, Liu L-L, Yang Y, Wang Y-Y, Shu H-B. LSm14A plays a critical role in antiviral immune responses by regulating MITA level in a cell-specific manner. J Immunol. 2016;196(12):5101–11.
Sun C, Cheng M-C, Qin R, Liao D-L, Chen T-T, Koong F-J, Chen G, Chen C-H. Identification and functional characterization of rare mutations of the neuroligin-2 gene (NLGN2) associated with schizophrenia. Hum Mol Genet. 2011;20(15):3042–51.
Rato MG, Bexiga R, Nunes SF, Vilela CL, Santos-Sanches I. Human group A streptococci virulence genes in bovine group C streptococci. Emerg Infect Dis. 2010;16(1):116.
Zheng H, Stratton CJ, Morozumi K, Jin J, Yanagimachi R, Yan W. Lack of Spem1 causes aberrant cytoplasm removal, sperm deformation, and male infertility. Proc Natl Acad Sci. 2007;104(16):6852–7.
Wu X, Li M, Li Y, Deng Y, Ke S, Li F, Wang Y, Zhou S. Fibroblast growth factor 11 (FGF11) promotes non-small cell lung cancer (NSCLC) progression by regulating hypoxia signaling pathway. J Transl Med. 2021;19(1):1–14.
Agerholm JS, McEvoy FJ, Menzi F, Jagannathan V, Drögemüller C. A CHRNB1 frameshift mutation is associated with familial arthrogryposis multiplex congenita in Red dairy cattle. BMC Genomics. 2016;17(1):1–10.
Yx Ge, Wang Ch, Fy Hu, Lx P. Min J, Niu Ky, Zhang L, Li J, Xu T: New advances of TMEM88 in cancer initiation and progression, with special emphasis on Wnt signaling pathway. J Cell Physiol. 2018;233(1):79–87.
Gao Y, Tian S, Sha Y, Zha X, Cheng H, Wang A, Liu C, Lv M, Ni X, Li Q. Novel bi-allelic variants in DNAH2 cause severe asthenoteratozoospermia with multiple morphological abnormalities of the flagella. Reprod Biomed Online. 2021;42(5):963–72.
Chung N, Bogliotti YS, Ding W, Vilarino M, Takahashi K, Chitwood JL, Schultz RM, Ross PJ. Active H3K27me3 demethylation by KDM6B is required for normal development of bovine preimplantation embryos. Epigenetics. 2017;12(12):1048–56.
Cataldo S, Annoni GA, Marziliano N. The perfect storm? Histiocytoid cardiomyopathy and compound CACNA2D1 and RANGRF mutation in a baby. Cardiol Young. 2015;25(1):174–6.
Haitina T, Lindblom J, Renström T, Fredriksson R. Fourteen novel human members of mitochondrial solute carrier family 25 (SLC25) widely expressed in the central nervous system. Genomics. 2006;88(6):779–90.
Mucha S, Mrode R, Coffey M, Kizilaslan M, Desire S, Conington J. Genome-wide association study of conformation and milk yield in mixed-breed dairy goats. J Dairy Sci. 2018;101(3):2213–25.
Higgins MG, Fitzsimons C, McClure MC, McKenna C, Conroy S, Kenny DA, McGee M, Waters SM, Morris DW. GWAS and eQTL analysis identifies a SNP associated with both residual feed intake and GFRA2 expression in beef cattle. Sci Rep. 2018;8(1):1–12.
Sasaki S, Mori D, Toyo-Oka K, Chen A, Garrett-Beal L, Muramatsu M, Miyagawa S, Hiraiwa N, Yoshiki A, Wynshaw-Boris A. Complete loss of Ndel1 results in neuronal migration defects and early embryonic lethality. Mol Cell Biol. 2005;25(17):7812–27.
Zhuang Z, Xu L, Yang J, Gao H, Zhang L, Gao X, Li J, Zhu B. Weighted single-step genome-wide association study for growth traits in Chinese Simmental beef cattle. Genes. 2020;11(2):189.
Radner FP, Marrakchi S, Kirchmeier P, Kim G-J, Ribierre F, Kamoun B, Abid L, Leipoldt M, Turki H, Schempp W. Mutations in CERS3 cause autosomal recessive congenital ichthyosis in humans. PLoS Genet. 2013;9(6):e1003536.
Jungers KA, Le Goff C, Somerville RP, Apte SS. Adamts9 is widely expressed during mouse embryo development. Gene Expr Patterns. 2005;5(5):609–17.
Carayol J, Sacco R, Tores F, Rousseau F, Lewin P, Hager J, Persico AM. Converging evidence for an association of ATP2B2 allelic variants with autism in male subjects. Biol Psychiat. 2011;70(9):880–7.
Moreira TG, Zhang L, Shaulov L, Harel A, Kuss SK, Williams J, Shelton J, Somatilaka B, Seemann J, Yang J. Sec13 regulates expression of specific immune factors involved in inflammation in vivo. Sci Rep. 2015;5(1):1–14.
Hou Q, Huang J, Ju Z, Li Q, Li L, Wang C, Sun T, Wang L, Hou M, Hang S. Identification of splice variants, targeted microRNAs and functional single nucleotide polymorphisms of the BOLA-DQA2 gene in dairy cattle. DNA Cell Biol. 2012;31(5):739–44.
Twomey AJ, Berry DP, Evans RD, Doherty ML, Graham DA, Purfield DC. Genome-wide association study of endo-parasite phenotypes using imputed whole-genome sequence data in dairy and beef cattle. Genet Sel Evol. 2019;51(1):1–17.
Srirattana K, St John JC. Manipulating the mitochondrial genome to enhance cattle embryo development. G3. 2017;7(7):2065–80.
Melo TPD, De Camargo GMF, De Albuquerque LG, Carvalheiro R. Genome-wide association study provides strong evidence of genes affecting the reproductive performance of Nellore beef cows. PLoS One. 2017;12(5):e0178551.
Cochran SD, Cole JB, Null DJ, Hansen PJ. Single nucleotide polymorphisms in candidate genes associated with fertilizing ability of sperm and subsequent embryonic development in cattle. Biol Reprod. 2013;89(3):69 61-67.
Uemoto Y, Ohtake T, Sasago N, Takeda M, Abe T, Sakuma H, Kojima T, Sasaki S. Effect of two non-synonymous ecto-5′-nucleotidase variants on the genetic architecture of inosine 5′-monophosphate (IMP) and its degradation products in Japanese Black beef. BMC Genomics. 2017;18(1):1–15.
Wydooghe E, Vandaele L, Beek J, Favoreel H, Heindryckx B, De Sutter P, Van Soom A. Differential apoptotic staining of mammalian blastocysts based on double immunofluorescent CDX2 and active caspase-3 staining. Anal Biochem. 2011;416(2):228–30.
Zhenhua G, Rajput SK, Folger JK, Di L, Knott JG, Smith GW. Pre-and peri-/post-compaction follistatin treatment increases in vitro production of cattle embryos. PLoS ONE. 2017;12(1):e0170808.
Canovas S, Cibelli JB, Ross PJ. Jumonji domain-containing protein 3 regulates histone 3 lysine 27 methylation during bovine preimplantation development. Proc Natl Acad Sci. 2012;109(7):2400–5.
Glanzner WG, Rissi VB, de Macedo MP, Mujica LKS, Gutierrez K, Bridi A, de Souza JRM, Gonçalves PBD, Bordignon V. Histone 3 lysine 4, 9, and 27 demethylases expression profile in fertilized and cloned bovine and porcine embryos. Biol Reprod. 2018;98(6):742–51.
Liu DT, Hong WS, Chen SX, Zhu Y. Upregulation of adamts9 by gonadotropin in preovulatory follicles of zebrafish. Mol Cell Endocrinol. 2020;499:110608.
Carter NJ, Roach ZA, Byrnes MM, Zhu Y. Adamts9 is necessary for ovarian development in zebrafish. Gen Comp Endocrinol. 2019;277:130–40.
Fortune J, Willis E, Bridges P, Yang C. The periovulatory period in cattle: progesterone, prostaglandins, oxytocin and ADAMTS proteases. Anim Reprod. 2009;6(1):60.
Yoshida GM, Yáñez JM. Multi-trait GWAS using imputed high-density genotypes from whole-genome sequencing identifies genes associated with body traits in Nile tilapia. BMC Genomics. 2021;22(1):1–13.
Li Y, Chen R, Zhou Q, Xu Z, Li C, Wang S, Mao A, Zhang X, He W, Shu H-B. LSm14A is a processing body-associated sensor of viral nucleic acids that initiates cellular antiviral response in the early phase of viral infection. Proc Natl Acad Sci. 2012;109(29):11770–5.
Onouchi Y. Genetics of Kawasaki Disease-What We Know and Don’t Know. Circ J. 2012;76(7):1581–6.
Liu M, Fang L, Liu S, Pan MG, Seroussi E, Cole JB, Ma L, Chen H, Liu GE. Array CGH-based detection of CNV regions and their potential association with reproduction and other economic traits in Holsteins. BMC Genomics. 2019;20(1):1–10.
Hou Y, Bickhart DM, Chung H, Hutchison JL, Norman HD, Connor EE, Liu GE. Analysis of copy number variations in Holstein cows identify potential mechanisms contributing to differences in residual feed intake. Funct Integr Genomics. 2012;12(4):717–23.
Li B, VanRaden P, Null D, O’Connell J, Cole J. Major quantitative trait loci influencing milk production and conformation traits in Guernsey dairy cattle detected on Bos taurus autosome 19. J Dairy Sci. 2021;104(1):550–60.
Hogan B, Costantini F, Lacy E. Manipulating the mouse embryo: a laboratory manual. 1986.
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J. From FastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43(1):11.10.11-11.10.33.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164–e164.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, De Bakker PI, Daly MJ. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75.
Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2(12):e190.
Alexander DH, Lange K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinformatics. 2011;12(1):1–6.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870–4.
Nielsen R, Williamson S, Kim Y, Hubisz MJ, Clark AG, Bustamante C. Genomic scans for selective sweeps using SNP data. Genome Res. 2005;15(11):1566–75.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST. The variant call format and VCFtools. Bioinformatics. 2011;27(15):2156–8.
DeGiorgio M, Huber CD, Hubisz MJ, Hellmann I, Nielsen R. SweepFinder2: increased sensitivity, robustness and flexibility. Bioinformatics. 2016;32(12):1895–7.
Ayres DL, Darling A, Zwickl DJ, Beerli P, Holder MT, Lewis PO, Huelsenbeck JP, Ronquist F, Swofford DL, Cummings MP. BEAGLE: an application programming interface and high-performance computing library for statistical phylogenetics. Syst Biol. 2012;61(1):170–3.
Szpiech ZA, Hernandez RD. selscan: an efficient multithreaded program to perform EHH-based scans for positive selection. Mol Biol Evol. 2014;31(10):2824–7.
Peripolli E, Reimer C, Ha N-T, Geibel J, Machado MA. Panetto JCdC, do Egito AA, Baldi F, Simianer H, da Silva MVGB: Genome-wide detection of signatures of selection in indicine and Brazilian locally adapted taurine cattle breeds using whole-genome re-sequencing data. BMC Genomics. 2020;21(1):1–16.
Illa SK, Mukherjee S, Nath S, Mukherjee A. Genome-wide scanning for signatures of selection revealed the putative genomic regions and candidate genes controlling milk composition and coat color traits in Sahiwal cattle. Front Genet. 2021;12:699422.
Ghoreishifar SM, Eriksson S, Johansson AM, Khansefid M, Moghaddaszadeh-Ahrabi S, Parna N, Davoudi P, Javanmard A. Signatures of selection reveal candidate genes involved in economic traits and cold acclimation in five Swedish cattle breeds. Genet Sel Evol. 2020;52(1):1–15.
Verity R, Collins C, Card DC, Schaal SM, Wang L, Lotterhos KE. minotaur: A platform for the analysis and visualization of multivariate results from genome scans with R Shiny. Mol Ecol Resour. 2017;17(1):33–43.
Todorov V, Templ M, Filzmoser P. Detection of multivariate outliers in business survey data with incomplete information. Adv Data Anal Classif. 2011;5(1):37–56.
Venables WN, Ripley BD. Modern applied statistics with S. 4th ed. New York: Springer; 2002.
Acknowledgements
We would like to thank Yu Jiang for his technical support.
Funding
This work was supported by the China Agriculture Research System of MOF and MARA (Grant No. CARS-37), Henan Beef Cattle Industrial Technology System (No. S2013-08), Science-Technology Foundation for innovation and creativity of Henan Academy of Agricultural Sciences (2020CX09).
Author information
Authors and Affiliations
Contributions
YH and SZ conceived and designed the experiments. SZ and ZY performed the statistical analysis and data upload. Xinmiao L and PY performed the sample DNA extraction. NC and XX provided suggestions for the revision of the manuscript. YJ provided technical assistance. ZZ, XL, LX, SL, QS, EW, and BR contributed to the sample collections. YJ provides a data analysis platform. HC, CL, and YH provided the laboratories for DNA extraction and statistical analysis. SZ drafted the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All cattle were handled following the guidelines established by the Council for Animal Welfare of China. The protocols for sample collection and animal handling have been approved by the Faculty of Animal Policy and Welfare Committee of Northwest A&F University (FAPWCNWAFU, Protocol number, NWAFAC 1008).
The study was carried out in compliance with the ARRIVE guidelines.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Figure S1. Phylogenetic tree constructed by neighbor-joining method from Pinan cattle and some other breeds.
Additional file 2:
Table S1. Summary of sequencing data. Table S2. List of additional cattle samples for analysis of genetic background in Pinan cattle. Table S3. Distribution of SNPs identified in cattle breeds within various genomic regions annotated by ANNOVAR. Only the breeds with a sample size no less than 5 were calculated in the table. Table S4. CV error corresponding to different K values. Table S5. 92 selective signal candidate regions identified by DCMS.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, S., Yao, Z., Li, X. et al. Assessing genomic diversity and signatures of selection in Pinan cattle using whole-genome sequencing data. BMC Genomics 23, 460 (2022). https://doi.org/10.1186/s12864-022-08645-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-022-08645-y