
Abstract
Background
Understanding the population structure and genetic bases of well-adapted cattle breeds to local environments is one of the most essential tasks to develop appropriate genetic improvement programs.
Results
We performed a comprehensive study to investigate the population structure, divergence and selection signatures at genome-wide level in diverse Chinese local cattle using Bovine HD SNPs array, including two breeds from North China, one breed from Northwest China, three breeds from Southwest China and two breeds from South China. Population genetic analyses revealed the genetic structures of these populations were mostly related to the geographic locations. Notably, we detected 294 and 1263 candidate regions under selection using the di and iHS approaches, respectively. A series of group-specific and breed-specific candidate genes were identified, which are involved in immune response, sexual maturation, stature related, birth and bone weight, embryonic development, coat colors and adaptation. Furthermore, haplotype diversity and network pattern for candidate genes, including LPGAT1, LCORL, PPP1R8, RXFP2 and FANCA, suggest that these genes have been under differential selection pressures in various environmental conditions.
Conclusions
Our results shed insights into diverse selection during breed formation in Chinese local cattle. These findings may promote the application of genome-assisted breeding for well-adapted local breeds with economic and ecological importance.
Similar content being viewed by others

Background
Cattle, as an important domesticated farm animal, is raised for many purposes, including meat, dairy, leather, and labor. Most of the modern cattle can be categorized into two subspecies: taurine cattle (humpless) and indicine cattle (humped) with dramatic phenotypic differences. The process of domestication was estimated to start approximately 11,000 years ago separately in the Fertile Crescent for Bos t. taurus and the Indian Subcontinent for Bos t. indicus [1, 2]. Selection (natural and human-imposed) and nonselective forces (introgression and the demographic events) drove the changes of cattle genome. Their combined effects have generated remarkable phenotypic diversity and genetic adaptation to local environment in global cattle populations [3, 4].
China has rich cattle genetic resources, and these breeds can be divided into different groups based on their locations, morphologies and sex chromosome polymorphisms [5,6,7]. Previous genetic analyses using mtDNA and Y microsatellite (Y-STR) data indicated that Chinese cattle breeds are influenced by the two above subspecies, with cattle in the north and northeast China are primarily of Bos t. taurus ancestry and cattle in southern China are predominantly of Bos t. indicus [5,6,7,8,9,10,11]. In contrast, cattle lived between North and South regions are mostly of Bos t. taurus, Bos t. indicus hybrids [12]. Modern farm animals are a result of selective breeding for many traits of economic and adaptive importance since domestication. Accurate identification of genomic regions involved with selection signatures is essential for illustrating the mechanism underling genetic variation contributing to phenotypic diversity [13]. Therefore, Chinese cattle represent geographically and biologically diverse cattle breeds and can offer valuable genetic resources for investigating the population structure, admixture and selection signatures of the cattle genome. Recently, high density SNP array and next-generation sequencing have markedly expedited the studies of the genetic bases underline important economic and adaptive traits in domestic animals. These technologies and related approaches have been utilized to explore process of domestication, and identify natural and artificial selection signatures in the divergent populations of dogs [14, 15], pigs [16], chickens [17] and horses [18].
Selection signature analyses can help to identify numerous candidate genes associated with production traits in domesticated animals. For example, using selection sweep analysis, Rubin et al. revealed that several genes (IGF1, TSHR, PMCH, etc) are associated with growth and metabolic regulation in chicken, and another study demonstrated that NR6A1, PLAG1, and LCORL are associated with morphological changes in pigs during domestication [16, 17]. Also, a recent genome-wide investigation of world-wide sheep breeds suggested a list of candidate genes under recent selection that were related to body size, growth, skeletal morphology, coat pigmentation, reproduction and adaptations to local climates [19,20,21,22]. In dairy and beef cattle, many studies have been conducted to explore the selection signatures in commercial breeding populations and revealed a list of candidate genes and SNPs, which were related to milk and meat production, fertility, disease resistance traits [23,24,25,26,27,28,29,30,31,32]. Increasing productivity and efficiency will be important, but maintenance of genetic diversity will also be crucial. To better understand cattle genome and its function in adaptation, many other studies have been conducted to investigate the genome selection signatures, and obtain a large numbers of genes which play a pivotal role in adaptation during domestication process [3, 4, 30, 33, 34].
Many studies have explored the population structure and selection signatures in world-wide and local cattle populations [3, 4, 25,26,27, 30, 33,34,35,36]. The high density SNP array has previously been shown to improve the detection of positive selection and to reduce false discoveries [4, 25, 37, 38]. To comprehensively uncover genetic differentiation in cattle genome, we performed a comprehensive study using high density SNP array to investigate the genetic structure, divergence and selection signatures in diverse Chinese cattle across a broad latitudinal range. The primary aims of current study were to 1) investigate the population structure within Chinese local cattle by sampling eight populations across a broad latitudinal range; 2) identify the genomic selection signatures involved in natural and artificial selection for local cattle population; and 3) explore the potential genetic bases underline adaptive traits in diverse range of environments.
Methods
Sample selection and genotyping
We genotyped a total of 179 samples using Illumina BovineHD SNPs array (contains a total of 777,962 SNPs) from eight cattle breeds including Yanhuang cattle (YHC), Menggu cattle (MGC), Caidamu cattle (CDM), Liangshan cattle (LSC), Pingwu cattle (PWC), Zhaotong cattle (ZTC), Wenshan cattle (WSC) and Nandan cattle (NDC). The full name, associated abbreviation for each breed and additional sampling information were presented in Additional file 1: Table S1. All samples were divided into four groups (North, Northwest, Southwest and South) based on their geographical distribution. We utilized PLINK v1.07 and custom R scripts for file conversion and SNPs quality control [39]. All samples showed a genotyping call rate of more than 95%. Closely related individuals (the PI-HAT value was greater than 0.25) were removed. Only autosomal SNPs were used for subsequently analyses. Also, the SNPs were filtered with the criterion of geno < 0.1 and MAF < 0.05.
Genetic diversity, heterozygosity and current effective population size
Analyses of genetic diversity and effective population size were conducted for eight diverse breeds in China. These include two breeds (YHC, MGC) from Northern China, one breed from Northwest China (CDM), two breed from Southwest China (LSC and PWC), three breeds from South China (ZTC, WSC and NDC) (Additional file 1: Table S1 lists the breed names, breed codes, and their sampling location information including geographic origin, latitude, longitude and altitude). Genetic diversity was estimated using the distributions of minor allele frequency (MAF) and the average MAF across breeds. The observed heterozygosity (Ho) was estimated using PLINK v1.07 with option –hardy. Ne was calculated using the software SNeP v1.1 as described before [40]. The method inferred Ne based on LD against past t generations, where t = 1/2c and c is the distance between SNPs in Morgans (100 Mb = 1 Morgan was assumed) [41].
Population structure and phylogenetic analysis
We conducted Multidimensional scaling (MDS) analysis using 78,865 SNPs after LD filter (r2 > 0.2). Pairwise genome-wide identity-by-state (IBS) pairwise distances were estimated for samples cluster using PLINK v1.07 (−mds -plot 4). Population admixture was examined using 12,478 SNPs after strict LD-based filter (r2 > 0.02) in STRUCTURE 2.3.4 [42, 43]. Each process was implemented using 10,000 replicates and 10,000 burn-in cycles under admixture and correlated allele frequencies models. We estimated the genetic distance (D) between pairwise combination of individuals using approaches implemented in PLINK v1.07 [39], where D = 1-(IBS2 + 0.5IBS1)/N, and IBS1 and IBS2 were the number of loci that share either one or two alleles identical by state, respectively and the N was the number of loci [44]. We next built Neighbor-joining phylogenetic tree using PHYLIP v3.69. The phylogenetic tree was visualized with Figtree 1.3.1, as reported before [4]. Patterns of splits and mixtures history of the populations were evaluated using TreeMix v1.13 [45]. To further test for evidence of admixture across populations, ancestry graph, three-population (f3) and four-population (f4) tests implemented in TreeMix were utilized to examine the presence of admixture [45, 46].
Genome-wide LD estimation
We estimated LD across each breed as our previously reported [4]. In brief, pairwise LD (r2) for all retained SNPs were estimated using the PLINK “-ld” option with the default window size of 1 Mb, and the “–ld-window-r2” was set to 0 to generate all pairwise results. The LD decay was further evaluated and fitted against genomic distance using smooth.spline function in R (v3.2.4).
Identification of selection signatures
To explore differences among groups from different geographic zones, we estimated the di statistics based on the unbiased estimates of pairwise FST. We then combined individual pairwise FST between groups (North, Northwest, Southwest and South) and averaged across all SNPs to obtain the standardized summary di statistic [15]. Then, we estimated the di value for each group in a nonoverlapping sliding window with 50 neighboring SNPs. Selection regions were defined as those ranking in the top 1% windows with highest average di values [4, 15, 47].
To infer recent selection sweeps, we further utilized the Integrated Haplotype score (iHS) method to measure structure of haplotype, which essentially reflected unusually long haplotypes across genome [47]. iHS was estimated using selscan with default settings (except for the maximum gap was set to 800,000) [48]. To identify genomic regions with signals of selection, we considered 100-kb nonoverlapping windows, the density of signal in each region was evaluated according to the proportion of SNPs with |iHS| > 2. Finally, we defined candidate regions under selection as those top 1% in the empirical distribution (regions with < 10 SNPs were removed) [47].
Gene annotation of selection regions
Based on the bovine UMD3.1 reference genome assembly, we retrieved genes within the intervals spanning the candidate regions from the UCSC genome browser. The gene enrichment analyses were performed using Database for Annotation, Visualization and Integrated Discovery (DAVID) functional annotation tools [49]. Only clusters with enrichment scores more than 1 (P-value < 0.05 after Bonferroni multiple test) were considered.
Haplotype diversity and haplotype network analysis of candidate genes
To explore the diversity of haplotypes and evolutionary patterns of candidate genes cross groups/breeds, we first extracted SNP genotypes in each gene as unphased information, then the haplotypes were constructed and their frequencies were estimated using PHASE v2.1 [50]. To obtain reliable results, we also performed inference with 10,000 iterations and 10,000 burn-ins, and the final results were obtained using option -X 100 in PHASE v2.1. Haplotype networks was constructed and evaluated for genes (LCORL, LPGAT1, PPP1R8, RXFP2 and FANCA) using the functions haplotype and haploNet from the pegas package [51].
Results
Genetic diversity, heterozygosity and current effective population size
After quality control, we obtained a total of 643,772 autosome SNPs for subsequent analyses. The call rates of SNP were generally high across samples (call rate > 0.95). We observed the distributions of minor allele frequency (MAF) display distinct patterns across breeds, which may reflect the diverse population history, genetic structure and geographic origin of breed formation. We also observed the average MAF ranged from 0.16 to 0.26 (Additional file 1: Table S1). Among them, WSC and NDC from southern China, which are indicine-derived population, had low MAF compared to taurine-derived populations in north and northwest China. To investigate diversity across our data set, we further calculated observed heterozygosity (Ho) within breeds (Additional file 1: Table S1). We observed the average Ho ranged from 0.22 to 0.35, and animals from South China displayed the lowest heterozygosity. In addition, we found the estimated Ne at 13 generations ago in Chinese naive cattle ranged from 85 to 132, which were consistent with previous current Ne estimation in the majority of breeds of domestic cattle (a current Ne of 150 or less) (Additional file 4: Figure S1).
Population structure, admixture and phylogenetic analysis
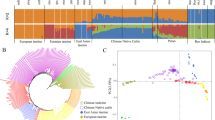
All samples were divided into four groups including North, Northwest, Southwest and South. Seven of them are representative of Chinese local cattle, while YHC shows nearly 75%~ 80% of genetic pool contributed by Yanbian cattle in Northern China, with the remaining contribution from Limousin cattle of France. MDS analysis presented a clear genetic structure among the eight breeds from different geographical areas. Our result revealed the first dimension separates the North breeds (primarily originate from taurine) and South breeds (primarily originate from indicine), while the second dimension separates the breeds from highland (CDM) and breed from lowland (MGC) (Fig. 1a). The separated clusters indicated that the studied population was reasonable to investigate the genomic characteristics of these breeds. To identify admixture level, we performed admixture analysis using 12,478 LD-filtered SNPs with the K (number of clusters) varied from 2 to 8. When K = 2 or 3, the clustering pattern implied the remarkable division of cattle from North and South China. When K = 4, we observed the three breeds LSC, PWC and ZTC displayed expected levels of admixture, agreeing with their breed history (Fig. 1b). While for K = 6 to 8, we did not found any additional breed was separated.

Population genetic analyses of eight diverse breeds. a. MDS analysis results of 179 individuals. Individuals are plotted according to their coordinates on the first two components. b. Clustering of 179 individuals based on LD filtered SNPs when K = 2 to 8. Individuals were shown as a thin vertical line colored in proportion to their estimated ancestry. c. Maximum likelihood tree inferred from 8 populations with two migration edges. The scale bar depicts ten times the average standard error of the estimated entries in the sample covariance matrix
We constructed a neighbor-joining tree using pairwise nucleotide genetic distances as described before [39, 52]. We observed animals from the same breed almost clustered together, while slight difference was found in the internal branches within a breed (Additional file 5: Figure S2). The pattern was consistent with previous report that modern breeds in China were derived from both taurine and indicine, and the formation of them may happen in a short period of time [7, 9]. We next estimated patterns of LD decay among them, and we found the average r2 generally decayed rapidly against distances as previously described [3, 4, 24, 53]. The pattern of LD and the decay trend differed between breeds and could be distinguished according to the breed type (Additional file 6: Figure S3). In agreement with previous studies, we also observed indicine breeds (South and Southwest) had shorter LD compared to taurine (North and Northwest) and the composite (hybrid) breeds [3, 53].
To further test for evidences of admixture across populations, we assessed the population history with admixture events using maximum likelihood approaches implemented in TreeMix. Our findings also confirmed several features already detected by STRUCTURE (Fig. 1c). As migration edges showed introgression between groups, we colored migration edges according to percent ancestry received from the donor population. Vectors 1–2 denoted gene flow from domestic North and Northwest into South and Southwest groups, mirroring the K = 2 results from STRUCTURE (Fig. 1b). The first vector indicated gene flow from the Northern group (MGC, YHC and CDM) to ZTC (Fig. 1c). The second vector connected PWC to the North group (MGC and YHC), indicating gene flow from MGC and YHC to PWC. To evaluate the presence of admixture, we further utilized the threepop and fourpop program implemented in the TreeMix v1.13 package to compute f3 and f4 statistics. The significant negative f3 statistic values indicated that population A was admixed population and related to B and C. In this study, f3 test results confirmed the admixture of PWC and ZTC. Our results suggested the three most extreme f3 scores (− 34.22, − 31.96 and − 32.22) to the geographically proximate populations of YHC|NDC, MGC|NDC and CDM|NDC as sources of admixture for PWC. The similar introgression pattern with extreme f3 scores (− 34.41, − 33.09 and − 32.44) was observed for ZTC from three populations (MGC, YHC and CDM). In addition, we did not observe significant result for the f4 test.
Candidate selection signatures and genes under positive selection
Signatures of selection—the d i approach
Based on the results from MDS analysis, we divided eight populations into four groups: North (YHC and MGC), Northwest (CDM), Southwest (LSC, PWC and ZTC) and South (WSC and NDC). Totally, we identified the top 1% with highest average di values in the empirical distribution as candidate selection regions for each group. The genome-wide distribution of the standardized di values were generated for these groups (Fig. 2). We detected a total of 127 windows using top 1% criteria for each group. To identify shared and group-specific selection regions, we generated a Venn diagram based on these selection regions across four groups (Fig. 3), and estimated the numbers of overlapping regions among them. We obtained 294 candidate regions with 169 of them (~ 57.5%) uniquely detected in only one group (61 for North, 15 for Southwest, 34 for Northwest, and 59 for South). Moreover, we found 125 regions (~ 42.5%) were shared by two or more groups, 65 regions (~ 22.1%) were shared by three, and 24 (8.2%) regions were shared by four groups (Additional file 2: Table S2).

Genomic distribution of selection regions in four cattle groups (North, Northwest, Southwest and South). The distribution of average di value for each 50-SNP windows across auto chromosomes is plotted for each breed. Breeds are abbreviated as described in Additional file 1: Table S1

Venn diagram for shared versus group-specific selection events for top 1% (127) windows among four cattle groups (North, Northwest, Southwest and South).
In this study, we identified 107, 103, 100, and 100 regions overlapping with genes in North, Northwest, Southwest and South group, respectively. Based on these regions, we totally detected 901 unique genes overlapped with the top 1% regions. Among them, there were 188, 71, 61 and 125 unique genes for each of North, Northwest, Southwest and South groups. We further performed DAVID analyses on identified genes in group-specific regions for each group. Notably, we found several functional clusters that were significantly enriched into functional importance processes among these breeds. In North group, we observed top three enriched clusters, including aromatic compound catabolic process, arylesterase activity, and response to toxic substance. In Northwest group, we discovered enrichments for ATP binding. In South group, we found most genes were enriched in lipid catabolic process, while no functional enrichment was observed in Southwest group.
In present study, we detected many candidate genes under positive selection which have been identified in previous reports, and three genes (WIF1, HSPA9 and UBE2D2) were identified under positive selection in four groups (North, Northwest, Southwest and South) (see Table 1). Twelve genes were detected in more than two groups, including RXFP2, LYN, TGS1, NPR2, SUOX, HBEGF, NMNAT1, SAR1B and DGAT1, RAB5B, PMEL and CDK2. Notably, we observed several genes displayed group-specific selection which may potentially imply the adaption for local environments. For instance, LCORL was detected in the North group, four genes PPP1R8, CPNE7, FANCA and SPG7 in the Northwest group, BMP2 in Southwest group, and LPGAT1 in the South group.
Signatures of selection—the iHS approach
To explore the genomic regions under recent selection in diverse Chinese local cattle, we further carried out integrated haplotype score (iHS) analysis, because this approach could provide important insights into recent selections. We identified a total of 2315 (1263 unique) candidate regions across multiple chromosomes in eight breeds. Among them, we identified 294, 286, 285, 306,298, 289, 305 and 252 candidate regions in YHC, MGC, CDM, PWC, LSC, ZTC, WSC, and NDC cattle, respectively. Totally, we found 1425 genes under selection in 1263 nonredundant regions (Additional file 3: Table S3). Moreover, we found 548 unique genes under selection based on iHS values in two or more breeds, and 131, 120, 144, 98, 104, 88, 105, 87 were uniquely detected in YHC, MGC, CDM, PWC, LSC, ZTC, WSC and NDC, respectively. Additionally, we found some genes (e.g. lysozymes) were enriched in breakdown of pathogen’s cell wall macromolecules in MGC, while several genes were observed enriched in regulation of appetite and RNA phosphodiester bond hydrolysis in WSC and NDC, respectively.
Haplotype network analyses for candidate genes
We further investigated the evolution history of several genes under selection by estimating haplotype diversity and constructing haplotype network. We found that the most common haplotype showed distinct frequency distribution among groups. Interestingly, we obtained 20 and 73 haplotypes within the 151 kb and 121 kb haploblock regions harboring LCORL and LPGAT1, and these two genes contained North and Northwest dominant haplotypes. Also, we found 23, 11 and 23 haplotypes within the 61 kb, 18 kb and 38 kb regions near RXFP2, PPP1R8, FANCA, and these genes had South and Southwest dominant haplotypes. For LCORL, the most common haplotype H1 (with frequency of 30.82%), was mainly found in North and Northwest cattle (YHC, MGC and CDM) and only minor portions occur in Southwest and South groups (PWC, LSC, ZTC, WSC and NDC) (Fig. 4a). H2 (with frequency of 18.40%) included a large proportion of Northern cattle (YHC and MGC). We also observed two haplotypes (H3 and H4) exclusive to indicine cattle (South and Southwest) with a frequency of (12.88 and 8.28%). Thus, this pattern may imply that separate haplotypes were clustered only for the indicine cattle (WSC and NDC), while other common haplotypes were identified for taurine cattle (YHC and MGC). PWC, LSC and ZTC were the only exceptions, as these breeds were derived from both taurine and indicine cattle. This was not unexpected, as it might mirror the complex ancestral backgrounds of breeds, i.e. these cattle are known as indicine breeds with taurine influences. For FANCA, we observed South and Southwest dominant haplotypes with high frequencies, H1 (21.39%) and H2 (19.22%), while three haplotypes (H1, H3, and H5) were shared by multiple groups from the North, Northwest, South and Southwest (Fig. 4b). We also found similar haplotype network pattern for LPGAT1, PPP1R8 and RXFP2 (Additional file 7: Figure S4).

Analyses of haplotype networks for two genes. a LCORL and (b) FANCA for four cattle groups. Each node represents a different haplotype, with the size of the circle proportional to frequency. Circles are color coded according to group (red: North; blue: Northwest; cyan: Southwest; and green: South)
Discussion
We have performed population structure, admixture and phylogenetic analyses on diverse local cattle across a broad latitudinal range in China. Our analyses revealed the population structure was mostly correlated with the geographic distribution. These results were consistent with previous analyses using mitochondrial D-loop sequences and microsatellites in Chinese cattle, which also implied the geographical segregation of local breeds during breed formation [5, 7, 11]. Our findings also generally agreed with the history that the cattle from Northern and Southern China originate from two different sub-species Bos t. taurus and Bos t. indicus (zebu), followed by the recent introgression of European taurine cattle [9, 10]. In addition, we have shown that a certain degree of mixture among breeds from adjacent locations, suggesting the potential migration and introgression among them [54].
Based on population structure estimation, we conducted selection signature analyses for four groups in Chinese local cattle, including the North, Northwest, Southwest and South groups. To our knowledge, this is one of the first comprehensive studies on selection signatures using the high density genome-wide SNP array in Chinese local cattle. Our findings may offer valuable insights to understand the genetic mechanism of adaption in different local environmental conditions. In current study, on the group level, we used di to detect the locus-specific divergence derived from ancient selection events. On the other hand, on the breed level, we utilized iHS based on extend haplotype homozygosity to infer recent selection sweeps. An integrated strategy combining both could facilitate the detection of both ancient and recent selection events [55]. We totally detected 294 and 1263 candidate regions using the di and iHS approaches, respectively. When compared the identified regions from two methods, we found the 13.5 Mb overlapped regions, corresponding to 221 common genes. However, these two methods are targeting at the different levels (groups or breeds) at different time scales (ancient vs. recent events), therefore the overlaps do not necessarily imply the strongest selection signatures but may reflect the long-lasting selection events. Our combined data suggest that while Chinese cattle breeds share some selected regions, they also retain unique signatures of adaptation in responses to local environments.
Using the di method, we observed three genes (WIF1, HSPA9 and UBE2D2) under selection in all four groups (North, Northwest, Southwest and South). WIF1 and HSPA9, related to immune response, were implicated in previous investigations for selection evidences in multiple cattle populations [4, 23, 25, 30, 56]. UBE2D2, which may function as ligase activity and acid-amino acid ligase activity, has also been reported in East African Shorthorn Zebu [33]. Previous studies suggested common genomic regions across various populations may indicate historical selection shared between them, most likely due to ancestral (within archetypes), geographical similarities [30]. In present study, the identified shared genes may be associated with metabolic homeostasis or other common traits like disease resistance and behavior, and these shared selection genes may also reflect the pleiotropic effects of them on traits [57]. Several genes were detected in more than two groups, these genes include reduced bone mass and sexual maturation related genes (RXFP2) [19, 34, 58], stature related, birth and bone weight, and embryonic development related genes (LYN, TGS1, NPR2, SUOX, HBEGF, and NMNAT1) [4, 19, 21, 23, 30, 36, 59,60,61,62,63], milk production related genes (SAR1B and DGAT1) [4, 30, 31], and immune, coat colors and hypoxia adaptation related genes (RAB5B, PMEL and CDK2) [36, 64,65,66,67]. In addition, we found some candidate genes were previously reported in multiple species, for instance, genes for statue and development (LCORL, NPR2, and BMP2) and horns (RXFP2) have been identified in cattle, pig, horse and sheep, which could be due to parallel evolution because of similar nature and/or human-imposed selection [55].
Our study also revealed a list of genes within group-specific selection regions. These unique genes under selection can be responsible in shaping particular characteristics of the population, thus resulting in the origin and maintenance of that breed. For instance, in the North group, LCORL were identified as a candidate, and this gene has been previously reported in human, cattle, horse and pig and other domestic animals [4, 16, 18, 27, 68]. LCORL has been associated with height or stature in human genome-wide association study [68], as well as with body size in cattle [69], horses [70] and dogs [71]. As North group cattle normally have larger morphology and stature, this might be explained that local environment can influences body size with obvious correlations between cold and aridity, and body size [72, 73]. In the Northwest group, we detected four genes PPP1R8, CPNE7, FANCA and SPG7 displaying groups-specific selection pattern which were related to reproduction, lipid metabolic, process cardiac system and nervous system development [25, 34, 74]. In the Southwest group, we found that BMP2 gene showed selection signatures in cattle genome [4, 25, 55], and a previous study has suggested this gene may have critical role in controlling body dimension and muscle development [75]. In the South group, LPGAT1 gene was identified as group-specific, which is supported by the previous evidence [28]. A recent study suggested that LPGAT1 play an essential role in lipid synthesis in mice [76]. Also, LPGAT1 may play a significant role in influencing BMI, percent body fat and hepatic triacylglycerol secretion in humans [77, 78]. Thus, based on the selection genes across genome, we suspected that cattle from Southwest group have been subjected for a longer time to tropical and humid environments and are well adapted to specific conditions. Additionally, these cattle may have interbred with more recently introduced European taurine individuals. These results generally agree with the recent findings from genomic analysis of climate adaptation in Mediterranean cattle breeds [73].
In addition, using the iHS approach, we totally found 1425 genes under selection in 1263 nonredundant regions (Additional file 3: Table S3). Moreover, 548 genes were identified in two or more breeds. Some genes have been previously described by previous studies in various populations. The iHS approach further identified several common genes within the shared regions that were most likely undergo similar selection pressure. For instance, we found ZFHX4 and LPCAT1 (related to milk production, somatic cell score and conformation) [79,80,81] in YHC and MGC within the North group. We detected TNNI2, EPHX1 and ROBO1 (associated with growth, meat quality and immune response, neuron differentiation) [35, 82,83,84,85] and UBE2E1 and GNA14 (related to hematological traits and high-altitude adaptation) in PWC, LSC and ZTC within Southwest groups, respectively [86, 87]. These genes can speculatively be interpreted as resulting from local adaption for mountain areas in the Southwest of China.
Notably, we observed numerous candidate genes displaying breed-specific selection in Chinese local cattle. For instance, we detected 28 candidate regions with the high iHS values on multiple chromosomes in the CDM breed from Northwest (Additional file 3: Table S3). These regions contained some functional important genes (SLC16A7, PTPN4, NFE2L3, ENPP2, KITLG, CAPN2, KLHL29, ABHD15, FRMD4A, ALDH5A1, and AREG), which are involved in growth, morphology, metabolism, meat quality and immune response. These identified genes have previously been suggested under selection in cattle or other farm animals [26, 29, 36, 60, 88,89,90,91,92,93]. Moreover, these selection genes detected in CMD may be important for physiological adaptations required to endure drought and hash in the areas of Caidamu Basin.
Our haplotype network analyses revealed a subset of candidate genes under selection. We found that common haplotypes were often present within the North and Northwest (taurine-derived) cattle, while separate distinct haplotypes were detected in the Southwest and South (indicine-derived) cattle, suggesting different evolutionary history for these populations. Also, haplotype diversity and their network pattern for positive selection genes, including LPGAT1, LCORL, PPP1R8, RXFP2 and FANCA, also implied that these genes have been under different selection pressures in distinct environmental conditions. Continued efforts to sequence and analyze the genomes of local cattle populations, with a focus on under-sampled areas of the world, will provide a more comprehensive insight of genetic diversity, population structure and selection of diverse cattle population. Also, further attention is required to elucidate the mechanism underlying observed differences of morphological feature across populations under different environmental conditions [54, 73]. As many traits seem to be highly complex and may be influenced by many loci across the genome, it is probable that a considerable component of selection in cattle is polygenic and is yet to be discovered by exploring the genomic regions under selection using advanced statistics methods [94]. Also, new technology platforms like next generation sequencing may shed valuable insights for understanding the evolutionary history of cattle genome during the domestication process [95, 96]. In addition, the analysis bias caused by the genome assembly should be also been considered, and more accurate detection of selection signatures in diverse cattle population could be improved by analysis based on their own genome assemblies.
Conclusions
Our results provided an important glimpse into diverse genomic selection during breed formation in Chinese local cattle. We identified a series of group-specific and breed-specific candidate genes, which are involved in immune response, sexual maturation, stature related, birth and bone weight, embryonic development, coat colors and adaptation. Furthermore, haplotype diversity and their network pattern for positive selection genes, including LPGAT1, LCORL, PPP1R8, RXFP2 and FANCA, suggest that these genes have been under differential selection pressures in various environmental conditions.
Availability of data and materials
The genotype data reported in this article are available upon request for research purpose.
Abbreviations
- BTA:
-
Bos Taurus autosomes
- CDM:
-
Caidamu cattle
- Ho :
-
Observed heterozygosity
- IBS:
-
Identity-by-state
- iHS:
-
Integrated Haplotype Score
- LD:
-
Linkage disequilibrium
- LSC:
-
Liangshan cattle
- MAF:
-
Minor allele frequency
- MDS:
-
Multidimensional scaling
- MGC:
-
Menggu cattle
- NDC:
-
Nandan cattle
- PWC:
-
Pingwu cattle
- QTL:
-
Quantitative trait loci
- SNP:
-
Single nucleotide polymorphism
- WSC:
-
Wenshan cattle
- YHC:
-
Yanhuang cattle
- ZTC:
-
Zhaotong cattle
References
Zeder MA. Domestication and early agriculture in the Mediterranean Basin: origins, diffusion, and impact. Proc Natl Acad Sci U S A. 2008;105:11597–604.
Bradley DG, MacHugh DE, Cunningham P, Loftus RT. Mitochondrial diversity and the origins of African and European cattle. Proc Natl Acad Sci U S A. 1996;93:5131–5.
Gibbs RA, Taylor JF, Van Tassell CP, Barendse W, Eversole KA, Gill CA, Green RD, Hamernik DL, Kappes SM, Lien S, et al. Genome-wide survey of SNP variation uncovers the genetic structure of cattle breeds. Science. 2009;324:528–32.
Xu L, Bickhart DM, Cole JB, Schroeder SG, Song J, Tassell CP, Sonstegard TS, Liu GE. Genomic signatures reveal new evidences for selection of important traits in domestic cattle. Mol Biol Evol. 2015;32:711–25.
Lei CZ, Chen H, Zhang HC, Cai X, Liu RY, Luo LY, Wang CF, Zhang W, Ge QL, Zhang RF, et al. Origin and phylogeographical structure of Chinese cattle. Anim Genet. 2006;37:579–82.
Lai SJ, Liu YP, Liu YX, Li XW, Yao YG. Genetic diversity and origin of Chinese cattle revealed by mtDNA D-loop sequence variation. Mol Phylogenet Evol. 2006;38:146–54.
Cai X, Chen H, Wang S, Xue K, Lei C. Polymorphisms of two Y chromosome microsatellites in Chinese cattle. Genet Sel Evol. 2006;38:525–34.
Mannen H, Kohno M, Nagata Y, Tsuji S, Bradley DG, Yeo JS, Nyamsamba D, Zagdsuren Y, Yokohama M, Nomura K, Amano T. Independent mitochondrial origin and historical genetic differentiation in north eastern Asian cattle. Mol Phylogenet Evol. 2004;32:539–44.
Cai X, Chen H, Lei C, Wang S, Xue K, Zhang B. mtDNA diversity and genetic lineages of eighteen cattle breeds from Bos taurus and Bos indicus in China. Genetica. 2007;131:175–83.
Li R, Zhang XM, Campana MG, Huang JP, Chang ZH, Qi XB, Shi H, Su B, Zhang RF, Lan XY, et al. Paternal origins of Chinese cattle. Anim Genet. 2013;44:446–9.
Zhang GX, Wang ZG, Chen WS, Wu CX, Han X, Chang H, Zan LS, Li RL, Wang JH, Song WT, et al. Genetic diversity and population structure of indigenous yellow cattle breeds of China using 30 microsatellite markers. Anim Genet. 2007;38:550–9.
Decker JE, McKay SD, Rolf MM, Kim J, Molina Alcala A, Sonstegard TS, Hanotte O, Gotherstrom A, Seabury CM, Praharani L, et al. Worldwide patterns of ancestry, divergence, and admixture in domesticated cattle. PLoS Genet. 2014;10:e1004254.
Andersson L, Georges M. Domestic-animal genomics: deciphering the genetics of complex traits. Nat Rev Genet. 2004;5:202–12.
Axelsson E, Ratnakumar A, Arendt ML, Maqbool K, Webster MT, Perloski M, Liberg O, Arnemo JM, Hedhammar A, Lindblad-Toh K. The genomic signature of dog domestication reveals adaptation to a starch-rich diet. Nature. 2013;495:360–4.
Akey JM, Ruhe AL, Akey DT, Wong AK, Connelly CF, Madeoy J, Nicholas TJ, Neff MW. Tracking footprints of artificial selection in the dog genome. Proc Natl Acad Sci U S A. 2010;107:1160–5.
Rubin CJ, Megens HJ, Barrio AM, Maqbool K, Sayyab S, Schwochow D, Wang C, Carlborg O, Jern P, Jorgensen CB, et al. Strong signatures of selection in the domestic pig genome. Proc Natl Acad Sci U S A. 2012;109:19529–36.
Rubin CJ, Zody MC, Eriksson J, Meadows JR, Sherwood E, Webster MT, Jiang L, Ingman M, Sharpe T, Ka S, et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature. 2010;464:587–91.
Petersen JL, Mickelson JR, Rendahl AK, Valberg SJ, Andersson LS, Axelsson J, Bailey E, Bannasch D, Binns MM, Borges AS, et al. Genome-wide analysis reveals selection for important traits in domestic horse breeds. PLoS Genet. 2013;9:e1003211.
Kijas JW, Lenstra JA, Hayes B, Boitard S, Porto Neto LR, San Cristobal M, Servin B, McCulloch R, Whan V, Gietzen K, et al. Genome-wide analysis of the world's sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS Biol. 2012;10:e1001258.
Lv FH, Agha S, Kantanen J, Colli L, Stucki S, Kijas JW, Joost S, Li MH, Ajmone Marsan P. Adaptations to climate-mediated selective pressures in sheep. Mol Biol Evol. 2014;31:3324–43.
Fariello MI, Servin B, Tosser-Klopp G, Rupp R, Moreno C, International Sheep Genomics C, San Cristobal M, Boitard S. Selection signatures in worldwide sheep populations. PLoS One. 2014;9:e103813.
Decker JE, Vasco DA, McKay SD, McClure MC, Rolf MM, Kim J, Northcutt SL, Bauck S, Woodward BW, Schnabel RD, Taylor JF. A novel analytical method, birth date selection mapping, detects response of the Angus (Bos taurus) genome to selection on complex traits. BMC Genomics. 2012;13:606.
Qanbari S, Gianola D, Hayes B, Schenkel F, Miller S, Moore S, Thaller G, Simianer H. Application of site and haplotype-frequency based approaches for detecting selection signatures in cattle. BMC Genomics. 2011;12:318.
Qanbari S, Pausch H, Jansen S, Somel M, Strom TM, Fries R, Nielsen R, Simianer H. Classic selective sweeps revealed by massive sequencing in cattle. PLoS Genet. 2014;10:e1004148.
Zhao F, McParland S, Kearney F, Du L, Berry DP. Detection of selection signatures in dairy and beef cattle using high-density genomic information. Genet Sel Evol. 2015;47:49.
Lee T, Cho S, Seo KS, Chang J, Kim H, Yoon D. Genetic variants and signatures of selective sweep of Hanwoo population (Korean native cattle). BMB Rep. 2013;46:346–51.
Flori L, Fritz S, Jaffrezic F, Boussaha M, Gut I, Heath S, Foulley JL, Gautier M. The genome response to artificial selection: a case study in dairy cattle. PLoS One. 2009;4:e6595.
Qanbari S, Pimentel EC, Tetens J, Thaller G, Lichtner P, Sharifi AR, Simianer H. A genome-wide scan for signatures of recent selection in Holstein cattle. Anim Genet. 2010;41:377–89.
Gurgul A, Szmatola T, Ropka-Molik K, Jasielczuk I, Pawlina K, Semik E, Bugno-Poniewierska M. Identification of genome-wide selection signatures in the Limousin beef cattle breed. J Anim Breed Genet. 2016;133:264–76.
Randhawa IA, Khatkar MS, Thomson PC, Raadsma HW. A meta-assembly of selection signatures in cattle. PLoS One. 2016;11:e0153013.
Larkin DM, Daetwyler HD, Hernandez AG, Wright CL, Hetrick LA, Boucek L, Bachman SL, Band MR, Akraiko TV, Cohen-Zinder M, et al. Whole-genome resequencing of two elite sires for the detection of haplotypes under selection in dairy cattle. Proc Natl Acad Sci U S A. 2012;109:7693–8.
Stella A, Ajmone-Marsan P, Lazzari B, Boettcher P. Identification of selection signatures in cattle breeds selected for dairy production. Genetics. 2010;185:1451–61.
Bahbahani H, Clifford H, Wragg D, Mbole-Kariuki MN, Van Tassell C, Sonstegard T, Woolhouse M, Hanotte O. Signatures of positive selection in east African shorthorn zebu: a genome-wide single nucleotide polymorphism analysis. Sci Rep. 2015;5:11729.
Gautier M, Naves M. Footprints of selection in the ancestral admixture of a New World creole cattle breed. Mol Ecol. 2011;20:3128–43.
Boitard S, Boussaha M, Capitan A, Rocha D, Servin B. Uncovering adaptation from sequence data: lessons from genome resequencing of four cattle breeds. Genetics. 2016;203:433–50.
Urbinati I, Stafuzza NB, Oliveira MT, Chud TC, Higa RH, Regitano LC, de Alencar MM, Buzanskas ME, Munari DP. Selection signatures in Canchim beef cattle. J Anim Sci Biotechnol. 2016;7:29.
O'Brien AMP, Utsunomiya YT, Meszaros G, Bickhart DM, Liu GE, Van Tassell CP, Sonstegard TS, Da Silva MVB, Garcia JF, Solkner J. Assessing signatures of selection through variation in linkage disequilibrium between taurine and indicine cattle. Genet Sel Evol. 2014;46:19.
Gonzalez-Rodriguez A, Munilla S, Mouresan EF, Canas-Alvarez JJ, Diaz C, Piedrafita J, Altarriba J, Baro JA, Molina A, Varona L. On the performance of tests for the detection of signatures of selection: a case study with the Spanish autochthonous beef cattle populations. Genet Sel Evol. 2016;48:81.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Barbato M, Orozco-terWengel P, Tapio M, Bruford MW. SNeP: a tool to estimate trends in recent effective population size trajectories using genome-wide SNP data. Front Genet. 2015;6:109.
Barbato M, Hailer F, Orozco-terWengel P, Kijas J, Mereu P, Cabras P, Mazza R, Pirastru M, Bruford MW. Genomic signatures of adaptive introgression from European mouflon into domestic sheep. Sci Rep. 2017;7:7623.
Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–59.
Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164:1567–87.
Stevens EL, Heckenberg G, Roberson ED, Baugher JD, Downey TJ, Pevsner J. Inference of relationships in population data using identity-by-descent and identity-by-state. PLoS Genet. 2011;7:e1002287.
Pickrell JK, Pritchard JK. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 2012;8:e1002967.
Reich D, Thangaraj K, Patterson N, Price AL, Singh L. Reconstructing Indian population history. Nature. 2009;461:489–94.
Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72.
Szpiech ZA, Hernandez RD. Selscan: an efficient multithreaded program to perform EHH-based scans for positive selection. Mol Biol Evol. 2014;31:2824–7.
Huang d W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57.
Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68:978–89.
Paradis E. Pegas: an R package for population genetics with an integrated-modular approach. Bioinformatics. 2010;26:419–20.
Retief JD. Phylogenetic analysis using PHYLIP. Methods Mol Biol. 2000;132:243–58.
Porto-Neto LR, Kijas JW, Reverter A. The extent of linkage disequilibrium in beef cattle breeds using high-density SNP genotypes. Genet Sel Evol. 2014;46:22.
Gao Y, Gautier M, Ding X, Zhang H, Wang Y, Wang X, Faruque MO, Li J, Ye S, Gou X, et al. Species composition and environmental adaptation of indigenous Chinese cattle. Sci Rep. 2017;7:16196.
Kim ES, Elbeltagy AR, Aboul-Naga AM, Rischkowsky B, Sayre B, Mwacharo JM, Rothschild MF. Multiple genomic signatures of selection in goats and sheep indigenous to a hot arid environment. Heredity (Edinb). 2016;116:255–64.
Porto-Neto LR, Sonstegard TS, Liu GE, Bickhart DM, Da Silva MV, Machado MA, Utsunomiya YT, Garcia JF, Gondro C, Van Tassell CP. Genomic divergence of zebu and taurine cattle identified through high-density SNP genotyping. BMC Genomics. 2013;14:876.
Gutierrez-Gil B, Arranz JJ, Wiener P. An interpretive review of selective sweep studies in Bos taurus cattle populations: identification of unique and shared selection signals across breeds. Front Genet. 2015;6:167.
Edea Z, Dadi H, Kim SW, Park JH, Shin GH, Dessie T, Kim KS. Linkage disequilibrium and genomic scan to detect selective loci in cattle populations adapted to different ecological conditions in Ethiopia. J Anim Breed Genet. 2014;131:358–66.
Utsunomiya YT, do Carmo AS, Carvalheiro R, Neves HH, Matos MC, Zavarez LB, Perez O'Brien AM, Solkner J, McEwan JC, Cole JB, et al. Genome-wide association study for birth weight in Nellore cattle points to previously described orthologous genes affecting human and bovine height. BMC Genet. 2013;14:52.
Magalhaes AF, de Camargo GM, Fernandes GAJ, Gordo DG, Tonussi RL, Costa RB, Espigolan R, Silva RM, Bresolin T, de Andrade WB, et al. Genome-wide association study of meat quality traits in Nellore cattle. PLoS One. 2016;11:e0157845.
Nishimura S, Watanabe T, Mizoshita K, Tatsuda K, Fujita T, Watanabe N, Sugimoto Y, Takasuga A. Genome-wide association study identified three major QTL for carcass weight including the PLAG1-CHCHD7 QTN for stature in Japanese Black cattle. BMC Genet. 2012;13:40.
Drogemuller C, Tetens J, Sigurdsson S, Gentile A, Testoni S, Lindblad-Toh K, Leeb T. Identification of the bovine Arachnomelia mutation by massively parallel sequencing implicates sulfite oxidase (SUOX) in bone development. PLoS Genet. 2010;6:e1001079.
Zhao LH, Cui XZ, Yuan HJ, Liang B, Zheng LL, Liu YX, Luo MJ, Tan JH. Restraint stress inhibits mouse implantation: temporal window and the involvement of HB-EGF, estrogen and progesterone. PLoS One. 2013;8:e80472.
Ezougou CN, Ben-Rached F, Moss DK, Lin JW, Black S, Knuepfer E, Green JL, Khan SM, Mukhopadhyay A, Janse CJ, et al. Plasmodium falciparum Rab5B is an N-terminally myristoylated Rab GTPase that is targeted to the parasite's plasma and food vacuole membranes. PLoS One. 2014;9:e87695.
Schmutz SM, Dreger DL. Interaction of MC1R and PMEL alleles on solid coat colors in Highland cattle. Anim Genet. 2013;44:9–13.
Li Y, Wu DD, Boyko AR, Wang GD, Wu SF, Irwin DM, Zhang YP. Population variation revealed high-altitude adaptation of Tibetan mastiffs. Mol Biol Evol. 2014;31:1200–5.
Adachi S, Ito H, Tamamori-Adachi M, Ono Y, Nozato T, Abe S, Ikeda M, Marumo F, Hiroe M. Cyclin a/cdk2 activation is involved in hypoxia-induced apoptosis in cardiomyocytes. Circ Res. 2001;88:408–14.
Lango Allen H, Estrada K, Lettre G, Berndt SI, Weedon MN, Rivadeneira F, Willer CJ, Jackson AU, Vedantam S, Raychaudhuri S, et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature. 2010;467:832–8.
Pryce JE, Hayes BJ, Bolormaa S, Goddard ME. Polymorphic regions affecting human height also control stature in cattle. Genetics. 2011;187:981–4.
Signer-Hasler H, Flury C, Haase B, Burger D, Simianer H, Leeb T, Rieder S. A genome-wide association study reveals loci influencing height and other conformation traits in horses. PLoS One. 2012;7:e37282.
Vaysse A, Ratnakumar A, Derrien T, Axelsson E, Rosengren Pielberg G, Sigurdsson S, Fall T, Seppala EH, Hansen MS, Lawley CT, et al. Identification of genomic regions associated with phenotypic variation between dog breeds using selection mapping. PLoS Genet. 2011;7:e1002316.
Franks SJ, Hoffmann AA. Genetics of climate change adaptation. Annu Rev Genet. 2012;46:185–208.
Flori L, Moazami-Goudarzi K, Alary V, Araba A, Boujenane I, Boushaba N, Casabianca F, Casu S, Ciampolini R, Coeur D'Acier A, et al. A genomic map of climate adaptation in Mediterranean cattle breeds. Mol Ecol. 2019;28:1009–29.
Schubert M, Jonsson H, Chang D, Der Sarkissian C, Ermini L, Ginolhac A, Albrechtsen A, Dupanloup I, Foucal A, Petersen B, et al. Prehistoric genomes reveal the genetic foundation and cost of horse domestication. Proc Natl Acad Sci U S A. 2014;111:E5661–9.
Fan B, Onteru SK, Du ZQ, Garrick DJ, Stalder KJ, Rothschild MF. Genome-wide association study identifies loci for body composition and structural soundness traits in pigs. PLoS One. 2011;6:e14726.
Soh J, Iqbal J, Queiroz J, Fernandez-Hernando C, Hussain MM. MicroRNA-30c reduces hyperlipidemia and atherosclerosis in mice by decreasing lipid synthesis and lipoprotein secretion. Nat Med. 2013;19:892–900.
Traurig MT, Orczewska JI, Ortiz DJ, Bian L, Marinelarena AM, Kobes S, Malhotra A, Hanson RL, Mason CC, Knowler WC, et al. Evidence for a role of LPGAT1 in influencing BMI and percent body fat in native Americans. Obesity (Silver Spring). 2013;21:193–202.
Hiramine Y, Emoto H, Takasuga S, Hiramatsu R. Novel acyl-coenzyme a:monoacylglycerol acyltransferase plays an important role in hepatic triacylglycerol secretion. J Lipid Res. 2010;51:1424–31.
Kolbehdari D, Wang Z, Grant JR, Murdoch B, Prasad A, Xiu Z, Marques E, Stothard P, Moore SS. A whole genome scan to map QTL for milk production traits and somatic cell score in Canadian Holstein bulls. J Anim Breed Genet. 2009;126:216–27.
Kolbehdari D, Wang Z, Grant JR, Murdoch B, Prasad A, Xiu Z, Marques E, Stothard P, Moore SS. A whole-genome scan to map quantitative trait loci for conformation and functional traits in Canadian Holstein bulls. J Dairy Sci. 2008;91:2844–56.
Duran Aguilar M, Roman Ponce SI, Ruiz Lopez FJ, Gonzalez Padilla E, Vasquez Pelaez CG, Bagnato A, Strillacci MG. Genome-wide association study for milk somatic cell score in Holstein cattle using copy number variation as markers. J Anim Breed Genet. 2017;134:49–59.
Strillacci MG, Frigo E, Schiavini F, Samore AB, Canavesi F, Vevey M, Cozzi MC, Soller M, Lipkin E, Bagnato A. Genome-wide association study for somatic cell score in Valdostana red pied cattle breed using pooled DNA. BMC Genet. 2014;15:106.
Mao X, Sahana G, De Koning DJ, Guldbrandtsen B. Genome-wide association studies of growth traits in three dairy cattle breeds using whole-genome sequence data. J Anim Sci. 2016;94:1426–37.
Cho HR, Ha J, Kwon SG, Hwang JH, Park DH, Kim TW, Lee HK, Song KD, Kim SW, Kim CW. Single-nucleotide polymorphisms in pig EPHX1 gene are associated with pork quality traits. Anim Biotechnol. 2015;26:237–42.
Ai H, Fang X, Yang B, Huang Z, Chen H, Mao L, Zhang F, Zhang L, Cui L, He W, et al. Adaptation and possible ancient interspecies introgression in pigs identified by whole-genome sequencing. Nat Genet. 2015;47:217–25.
Zhang F, Zhang Z, Yan X, Chen H, Zhang W, Hong Y, Huang L. Genome-wide association studies for hematological traits in Chinese Sutai pigs. BMC Genet. 2014;15:41.
Dong K, Yao N, Pu Y, He X, Zhao Q, Luan Y, Guan W, Rao S, Ma Y. Genomic scan reveals loci under altitude adaptation in Tibetan and Dahe pigs. PLoS One. 2014;9:e110520.
An XP, Hou JX, Li G, Song YX, Wang JG, Chen QJ, Cui YH, Wang YF, Cao BY. Polymorphism identification in the goat KITLG gene and association analysis with litter size. Anim Genet. 2012;43:104–7.
Yuan Z, Liu E, Liu Z, Kijas JW, Zhu C, Hu S, Ma X, Zhang L, Du L, Wang H, Wei C. Selection signature analysis reveals genes associated with tail type in Chinese indigenous sheep. Anim Genet. 2017;48:55–66.
Yang H, Huang X, Zeng Z, Zhang W, Liu C, Fang S, Huang L, Chen C. Genome-wide association analysis for blood lipid traits measured in three pig populations reveals a substantial level of genetic heterogeneity. PLoS One. 2015;10:e0131667.
Thompson-Crispi KA, Sargolzaei M, Ventura R, Abo-Ismail M, Miglior F, Schenkel F, Mallard BA. A genome-wide association study of immune response traits in Canadian Holstein cattle. BMC Genomics. 2014;15:559.
Baik M, Nguyen TH, Jeong JY, Piao MY, Kang HJ. Effects of castration on expression of lipid metabolism genes in the liver of korean cattle. Asian-Australas J Anim Sci. 2015;28:127–34.
Guo Z, Gonzalez JF, Hernandez JN, McNeilly TN, Corripio-Miyar Y, Frew D, Morrison T, Yu P, Li RW. Possible mechanisms of host resistance to Haemonchus contortus infection in sheep breeds native to the Canary Islands. Sci Rep. 2016;6:26200.
Roesti M, Gavrilets S, Hendry AP, Salzburger W, Berner D. The genomic signature of parallel adaptation from shared genetic variation. Mol Ecol. 2014;23:3944–56.
Kim J, Hanotte O, Mwai OA, Dessie T, Bashir S, Diallo B, Agaba M, Kim K, Kwak W, Sung S, et al. The genome landscape of indigenous African cattle. Genome Biol. 2017;18:34.
Mei C, Wang H, Liao Q, Wang L, Cheng G, Wang H, Zhao C, Zhao S, Song J, Guang X, et al. Genetic architecture and selection of Chinese cattle revealed by whole genome resequencing. Mol Biol Evol. 2017;35:688–99.
Acknowledgements
The authors would like to thank Prof. Shangzhong Xu and Prof. Ming Zhang for sample collection.
Funding
This study was supported by the Basic R&D Fund for the Central Level Scientific Research Institute (2016ywf-yb-6), the Agricultural Science and Technology Innovation Program in Chinese Academy of Agricultural Sciences (CAAS-XTCX2016010, CAAS-ZDXT2018006, ASTIP-IAS03, ASTIP-IAS-TS-9 and ASTIP-IAS-TS-16) for the design of the study and collection. Also, it was supported by the Elite Youth Program in Chinese Academy of Agricultural Sciences for the data analysis and interpretation of the study.
Author information
Authors and Affiliations
Contributions
LX and JYL drafted the manuscript and performed the experiments. XG, HJG and LPZ collected the blood samples and exacted DNA samples. LYX, LY, WGZ and BZ performed data analysis. LYX, LY, GEL, and JYL conceived of the study, and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
No ethics approval was required for any aspect of this study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. The full names, associated abbreviation for each breeds and additional information on the locations of the sampling areas for eight populations. (XLSX 12 kb)
Additional file 2:
Table S2. Candidate selective signature and genes under positive Selection using di approach for four groups including North, Northwest, South and Southwest. (XLSX 31 kb)
Additional file 3:
Table S3. Candidate selective signature and genes under positive Selection using iHS approach for eight populations including YHC, MGC, CDM, PWC, LSC, ZTC, WSC and NDC. (XLSX 83 kb)
Additional file 4:
Figure S1. The effective population sizes (Ne) of Chinese native cattle analyzed in this study. Ne of YHC, MGC, CDM, LSC, PWC, ZTC, WSC and NDC is plotted separately. X and Y axis represents generations and Ne respectively. (TIF 1662 kb)
Additional file 5:
Figure S2. Neighbor-joining tree of eight populations. The tree was constructed using genetic sharing distances. (TIF 2514 kb)
Additional file 6:
Figure S3. LD decay patterns in the eight breeds. Pairwise LD measures for all retained SNPs were performed using the PLINK “-ld” option. We used the default window size of 1 Mb, and set “–ld-window-r2” to 0 in order to get all pairs reported. The LD decay along genomic distance was fitted by smooth.spline function in R. (TIF 1673 kb)
Additional file 7:
Figure S4. Haplotype networks of two loci. (A) LPGAT1 and (B) PPP1R8, (C) RXFP2 for four cattle groups. Each node represents a different haplotype, with the size of the circle proportional to frequency. Branch lengths are proportional to the number of nucleotide differences. Circles are color coded according to group (red: North, blue: Northwest, cyan: Southwest, and green: West). (TIF 1362 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Xu, L., Yang, L., Zhu, B. et al. Genome-wide scan reveals genetic divergence and diverse adaptive selection in Chinese local cattle. BMC Genomics 20, 494 (2019). https://doi.org/10.1186/s12864-019-5822-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-019-5822-y