
Abstract
Background
Rice sheath blight, caused by Rhizoctonia solani Kühn (teleomorph: Thanatephorus cucumeris), is one of the most severe diseases in rice (Oryza sativa L.) worldwide. Studies on resistance genes and resistance mechanisms of rice sheath blight have mainly focused on indica rice. Rice sheath blight is a growing threat to rice production with the increasing planting area of japonica rice in Northeast China, and it is therefore essential to explore the mechanism of sheath blight resistance in this rice subspecies.
Results
In this study, RNA-seq technology was used to analyse the gene expression changes of leaf sheath at 12, 24, 36, 48, and 72 h after inoculation of the resistant cultivar ‘Shennong 9819’ and susceptible cultivar ‘Koshihikari’ with R. solani. In the early stage of R. solani infection of rice leaf sheaths, the number of differentially expressed genes (DEGs) in the inoculated leaf sheaths of resistant and susceptible cultivars showed different regularity. After inoculation, the number of DEGs in the resistant cultivar fluctuated, while the number of DEGs in the susceptible cultivar increased first and then decreased. In addition, the number of DEGs in the susceptible cultivar was always higher than that in the resistant cultivar. After inoculation with R. solani, the overall transcriptome changes corresponding to multiple biological processes, molecular functions, and cell components were observed in both resistant and susceptible cultivars. These included metabolic process, stimulus response, biological regulation, catalytic activity, binding and membrane, and they were differentially regulated. The phenylalanine metabolic pathway; tropane, piperidine, and pyridine alkaloid biosynthesis pathways; and plant hormone signal transduction were significantly enriched in the early stage of inoculation of the resistant cultivar Shennong 9819, but not in the susceptible cultivar Koshihikari. This indicates that the response of the resistant cultivar Shennong 9819 to pathogen stress was faster than that of the susceptible cultivar. The expression of plant defense response marker PR1b gene, transcription factor OsWRKY30 and OsPAL1 and OsPAL6 genes that induce plant resistance were upregulated in the resistant cultivar. These data suggest that in the early stage of rice infection by R. solani, there is a pathogen-induced defence system in resistant rice cultivars, involving the expression of PR genes, key transcription factors, PAL genes, and the enrichment of defence-related pathways.
Conclusion
The transcriptome data revealed the molecular and biochemical differences between resistant and susceptible cultivars of rice after inoculation with R. solani, indicating that resistant cultivars have an immune response mechanism in the early stage of pathogen infection. Disease resistance is related to the overexpression of PR genes, key transcriptome factors, and PAL genes, which are potential targets for crop improvement.
Similar content being viewed by others

Background
Rice (Oryza sativa L.) is one of the three most important crops worldwide, and rice sheath blight is one of the most destructive diseases [1,2,3]. The annual loss of rice products caused by rice sheath blight is as high as 50% worldwide [4,5,6,7]. Rhizoctonia solani is a soil-borne fungal plant pathogen [8]. The host range of the pathogen is wide, and the sclerotium of the pathogen is strongly resistant to the external environment [9, 10]. Due to the lack of resistant donors in cultivated cultivars [11, 12], studies on the resistance mechanism of rice sheath blight are lacking [13, 14]. For a long time, studies on the resistance genes and mechanisms underlying rice sheath blight have focused mainly on indica rice [15,16,17]. However, the total area of japonica rice in China accounts for one-third of the total area of rice plantations in China. Furthermore, the japonica rice planting area in Northeast China accounts for 53.7% of the total area of japonica rice [18]. In recent years, sheath blight has severely affected the production of japonica rice in Northeast China [19]. Koshihikari, a high-quality japonica rice cultivar, was developed in Japan in 1956. It is well known for its high eating quality [20]. At the end of July 2007, Japanese Koshihikari brand rice entered the Chinese market, with a price of up to 99 yuan Renminbi per kg, which attracted the attention of Chinese rice breeders to high-quality rice breeding [21]. Although the Koshihikari cultivar has poor disease resistance and is susceptible to lodging, it is still an excellent parent for high-quality rice breeding. In this study, the susceptible cultivar Koshihikari and the resistant cultivar Shennong 9819 were used to analyse the early transcripts of R. solani infection and explore its disease resistance mechanism.
With the rapid development of molecular biology techniques and the wide application of various omics technology in the interaction between plants and pathogens, the identification of rice sheath blight resistance genes and the interaction mechanism between sheath blight pathogen and rice are becoming increasingly deep. Chitinases are the members of PR proteins responsible for the hydrolysis of chitin, a structural polysaccharide of the cell wall of many pathogens. Overexpression of the chitinase gene CHI11 enhanced resistance to rice sheath blight [22, 23]. OsOSM1, a gene mainly expressed in the leaf sheath at the booting stage in rice, encodes an osmotin protein belonging to the pathogenesis-related protein 5 family. Overexpression of this gene can enhance the resistance of rice to sheath blight [24]. Plant polygalacturonase-inhibiting protein (PGIP) is a structural protein that specifically recognise and bind to fungal polygalacturonase (PG). PGIP plays an important role in antifungal activity in plants. Overexpression of PGIP-related genes such as ZmPGIP3, OsPGIP1, and OsPGIP2 increases resistance to rice sheath blight in rice [25,26,27]. Lignin deposition can enhance plant cell walls against pathogens and provide structural barriers for pathogen infection [28]. Overexpression of the lignin-related gene OsPAL4 increases resistance to rice sheath blight [29]. OsWRKY4 is an important positive regulatory factor in the interaction between rice and pathogens. It participates in the defence response of rice sheath blight through the jasmonic acid (JA)/ethylene (ET)-dependent signalling pathway [30].
Due to its high throughput and sensitivity, RNA-sequencing (RNA-Seq) technology is increasingly being used in the research and analysis of gene function [31, 32]. Transcriptome sequencing technology has been successfully used to study plant–pathogen interactions [33, 34]. Bagnaresi et al. [35] used comparative transcriptome technology to analyse the early molecular interaction of resistant and susceptible rice cultivars infected with Magnaporthe grisea. They found that chitinase and WRKY transcription factors were involved in the resistance of rice blast. Strauss et al. [36] identified one major Bs4C candidate transcript from pepper by RNA-seq to regulate the transcription activator-like effector AvrBs4 of Xanthomonas. Kawahara et al. [37] analysed the mixed transcripts of rice and blast fungus in infected leaves at 24 h after inoculation using the RNA-Seq technique. It was found that in the interaction between host plants and pathogens, the transcripts of glycosyl hydrolase, cutinases, and LysM domain-containing proteins of M. grisea were up-regulated, including the pathogenesis-related and phytoalexin biosynthetic genes in rice. Xiao et al. used next-generation sequencing technology to study the gene expression profiles of Fusarium head blight-related genes in common wheat. It was found that pathogen-related proteins such as PR5, PR14, ABC transporter, and JA signalling pathway were the key to Fusarium head blight resistance [38].
Transcriptional changes in indica rice cultivar inoculated with R. solani were analysed using RNA-Seq technology [39]. The rice cultivar resistant to sheath blight used in our study was japonica rice cultivar from Northeast China. This is the first study to compare the gene expression patterns of resistant and susceptible japonica rice inoculated with R. solani. In this study, the transcripts of resistant and susceptible cultivars were compared at 12, 24, 36, 48, and 72 h after inoculation with R. solani. The results showed significant differences in the expression of differentially expressed genes and genes related to metabolic pathways. The expression characteristics of metabolic pathways related to disease resistance were defined. The key genes related to rice sheath blight resistance were identified, which provided gene resources for molecular-assisted breeding of japonica rice in Northeast China.
Results
Symptoms of leaf sheath after inoculation
The symptoms of leaf sheath after inoculation are shown in Fig. S1. After inoculation for 24 h, evident brown spots appeared on the leaf sheath of Koshihikari; 48 h after inoculation, they were grey. These spots appeared on the leaf sheath of Shennong 9819 at 36 h after inoculation. The expanded area of the spot of Koshihikari was significantly larger than that of Shennong 9819 at 72 h after inoculation.
The inoculated leaf sheath was decolorized by chloral hydrate, stained with aniline blue, and observed under a light microscope. At 12 h after inoculation, hyphae were observed in the leaf sheaths of both cultivars, and more hyphae were found in the leaf sheath of Koshihikari than in Shennong 9819. At 24 h after inoculation, infection cushions appeared in the leaf sheaths of both cultivars. The number and density of the infection cushions of Koshihikari were greater than those of Shennong 9819 (Fig. S2).
RNA-seq results of Transcriptome samples
To study the changes in gene expression of the leaf sheath of Shennong 9819 and Koshihikari at the initial infection stage of R. solani, we used high-throughput sequencing technology to measure the transcription in rice leaf sheaths after inoculation. The transcriptome analysis of each cultivar included five time points with three biological repeats at each time point. A total of 342.24 Gb clean data was obtained from 36 samples; the clean data of each sample reached 8.08 Gb, and the percentage of Q30 base was 92.49% or more (Table S1). A total of 2,294,868,004 single-end clean reads (total records) were obtained after pre-processing the reads (Table S2). The clean reads of each sample were sequenced with the designated reference genome, and the efficiency of alignment ranged from 80.43 to 92.33%. The correlation analysis among the samples showed that the three repeats of the two cultivars had a high correlation (Fig. S3).
Differential gene analysis of leaf sheath after inoculation
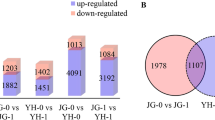
To determine which gene expression had changed and the stage of these changes, we counted the number of different genes between the two rice cultivars at each time point after inoculation with R. solani (Table 1) (SS for Shennong 9819; YY for Koshihikari). A total of 2275 differentially expressed genes (DEGs) were identified in our study (Table S3). After inoculation with R. solani, the number of upregulated genes in the leaf sheath of Koshihikari was higher than that of Shennong 9819 at all inoculation time points.
At 12 h, 2403 DEGs (1242 upregulated and 1161 downregulated) were identified in the leaf sheath of Shennong 9819, whereas 2817 DEGs (1419 upregulated and 1398 downregulated) were identified in Koshihikari, indicating that the Koshihikari was more susceptible to R. solani than Shennong 9819; the infection pressure on Koshihikari plants was thus higher than that on Shennong 9819. From 12 to 72 h, the number of DEGs in the Koshihikari leaf sheath was higher than Shennong 9819. The number of DEGs in the Koshihikari leaf sheath was highest at 36 h (4,873 DEGs; 2438 upregulated, 2435 downregulated) after inoculation. The data show that the number of DEGs in the susceptible cultivar was higher than that in the resistant cultivar.
The DEGs at 12, 24, 36, 48, and 72 h after inoculation of R. solani were analysed for the two cultivars. The DEGs related to the infection response of R. solani in rice were further analysed.
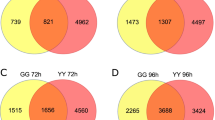
In this study, the DEGs of the two cultivars at the same time point (SS12-YY12, SS24-YY24, SS36-YY36, SS48-YY48, and SS72-YY72) were compared (Fig. 1), and the number of DEGs for each cultivar at different time points (SS12-SS24-SS36-SS48-SS72 or YY12-YY24-YY36-YY48-YY72h) were also compared (Fig. 2). As shown in Fig. 1, 1,347 and 1269 DEGs were identified in the two cultivars at 12 h and 36 h after inoculation, respectively, and the number of these DEGs was higher than that at other time points.

Venn diagram of DEGs discovered in both rice cultivars after inoculation with Rhizoctonia solani at the same time points. SS12, SS24, SS36, SS48, and SS72 represent DEG sets discovered from the leaf sheath of Shennong 9819 at 12, 24, 36, 48, and 72 h after inoculation; YY12, YY24, YY36, YY48, and YY72 represent DEG sets discovered from the leaf sheath of Koshihikari at 12, 24, 36, 48, and 72 h after inoculation

Venn diagram of DEGs in both rice cultivars after inoculation with Rhizoctonia solani at the different time points. SS12, SS24, SS36, SS48, and SS72 represent DEG sets discovered from the leaf sheath of Shennong 9819 at 12, 24, 36, 48, and 72 h after inoculation; YY12, YY24, YY36, YY48, and YY72 represent DEG sets discovered from the leaf sheath of Koshihikari at 12, 24, 36, 48, and 72 h after inoculation
In the leaf sheaths of Shennong 9819, 12 common DEGs were continuously expressed at 12, 24, 36, 48, and 72 h after inoculation, including four upregulated genes and seven downregulated genes. At 12 h, 832 specific DEGs were identified (575 upregulated and 268 downregulated). There were 23 specific DEGs (15 upregulated and 14 downregulated) discovered at 24 h, and 693 specific DEGs (284 upregulated and 419 downregulated) were identified at 36 h. In the leaf sheath of Koshihikari, 269 common DEGs were expressed at 12, 24, 36, 48, and 72 h after inoculation (132 upregulated and 137 downregulated). At 12 h, 537 specific DEGs (417 upregulated and 172 downregulated) were identified, and 925 specific DEGs (619 upregulated and 359 downregulated) were identified at 36 h. At 36 h, 1251 specific DEGs were identified (612 upregulated and 645 downregulated). In conclusion, the number of DEGs which were continuously expressed in the sheath of Koshihikari was higher than that in Shennong 9819 after inoculation.
Gene Ontology (GO) analysis of differentially expressed genes
GO annotation was used to classify the enriched DEGs between the control and inoculation treatments. The results showed that these enriched DEGs were involved in many biological activities (Fig. 3).

Functional classification of DEGs in the Shennong 9819 and Koshihikari. SS12, SS24, SS36, SS48, and SS72 represent DEG sets discovered from the leaf sheath of Shennong 9819 at 12, 24, 36, 48, and 72 h after inoculation; YY12, YY24, YY36, YY48, and YY72 represent DEG sets discovered from the leaf sheath of Koshihikari at 12, 24, 36, 48, and 72 h after inoculation. A Cellualr component B Molecular function C Biological process
After annotating the GO database, all the DEGs were classified into three main categories: biological process, molecular function, and cellular component. In the biological process, “metabolic process”, “cellular process”, “single-organism process”, “response to stimulus”, and “biological regulation” were the five processes with the highest degree of DEGs enrichment. In terms of molecular function, most DEGs were concentrated in two processes: “catalytic activity” and “binding”. For the cellular component, the five most common processes were “cell”, “membrane”, “organelle”, “organelle part” and “cell part”.
The results showed that the GO terms in different cultivars showed completely different patterns of expression. Taking “response to stimulation” as an example, in the leaf sheath of Shennong 9819, the number of DEGs related to this process reached the maximum at 12 h. The number of DEGs was the least at 24 h after inoculation, indicating that plants responded most strongly to external stimuli at 12 h and were closest to the uninoculated state at 24 h. The results showed that the plants could repair themselves in response to external stimuli; at 36 h, the number of DEGs increased, and again at 48 h, the number of DEGs decreased. Shennong 9819 exhibited a fluctuating response to external stress. However, the number of DEGs related to the process of “response to stimulation” increased rapidly in the leaf sheath of Koshihikari after inoculation, reached a maximum at 36 h and then decreased sharply. The genes related to this process in the leaf sheath of Koshihikari were always active after inoculation. The results showed that Shennong 9819 and Koshihikari had different resistance patterns to R. solani infection, and the genes in the leaf sheath of Koshihikari were always in a higher activity state.
Analysis of metabolic pathways of two rice cultivars after inoculation
To further study the specificity of pathways affected by R. solani infection in Shennong 9819 and Koshihikari, Kyoto Encyclopaedia of Genes and Genomes (KEGG) enrichment analysis was performed on the upregulated genes (|log2fc > 1|, FDR < 0.05) at different times after inoculation (Table S4 and S5). The results showed that alanine, aspartate and glutamate metabolism was significantly enriched in both cultivars at 12 h after inoculation, although Shennong 9819 was more significant. The phenylalanine metabolism, plant hormone signal transduction, tropane, piperidine and pyridine alkaloid biosynthesis pathways were significantly enriched in Shennong 9819 at 12 and 36 h after inoculation; however, no significant difference was found in Koshihikari. The tyrosine metabolism and isoquinoline alkaloid biosynthesis pathways were significantly enriched in Shennong 9819 at 24 and 36 h after inoculation, but not in Koshihikari. At 36 h after inoculation of Shennong 9819 and Koshihikari with R. solani, glycine, serine and threonine metabolism and beta-alanine metabolism pathways were significantly enriched, whereas Koshihikari was not.
Similarly, some pathways were specifically enriched in Koshihikari. For example, ascorbate and aldarate metabolism pathway was significantly enriched at 12 and 24 h after inoculation in Koshihikari. The linoleic acid metabolism pathway was significantly enriched 24 h after inoculation in Koshihikari. Valine, leucine and isoleucine degradation pathway was significantly enriched at 24 and 36 h in Koshihikari after inoculation. The arginine biosynthesis and 2-Oxocarboxylic acid metabolism pathways were significantly enriched at 36 h in Koshihikari after inoculation. The propanoate metabolism pathway was significantly enriched at 24, 36, 48, and 72 h in Koshihikari after inoculation. The above pathways were not significantly enriched at any time in Shennong 9819 after inoculation. At different times after inoculation, the upregulated genes of the two cultivars had specific enrichment pathways, suggesting that the resistance mechanisms of the two cultivars might be different.
To identify potential regulatory genes closely related to the phenylalanine metabolism pathway, DEGs involved in phenylalanine metabolism were identified by comparing the two cultivars. At 12 and 36 h, the expression levels of genes related to phenylalanine metabolism in the Shennong 9819 leaf sheath were significantly higher than those in Koshihikari. The difference between the two cultivars is shown in the heatmap (Fig. 4). Through gene analysis, a series of PAL genes was activated after inoculation with R. solani: OsPAL1 (LOC_Os02g41630), OsPAL2 (LOC_Os02g41650), OsPAL3 (LOC_Os02g41670), OsPAL4 (LOC_Os02g41680), OsPAL6 (LOC_Os04g43800), and OsPAL9 (LOC_Os12g33610). In this study, the expression of OsPAL1 and OsPAL6 in the leaf sheath of Shennong 9819 was higher than that in Koshihikari (Fig. 5). However, the expression of OsPAL4 and OsPAL9 in Koshihikari was higher than that in Shennong 9819 (Fig. 5).

Expression profiles of genes related to phenylalanine metabolism pathways

Expression of genes related to phenylalanine metabolism, plant–pathogen interaction, and plant signal transduction after inoculation based on transcripts per million (TPM)
We compared and analysed the genes involved in the plant–pathogen interaction pathway during early infection of Shennong 9819 and Koshihikari by R. solani. Pathogenesis-related proteins (PR) are defined as plant proteins that are induced in pathological or related situations. The expression of OsPR1a (LOC_Os07g03710) was upregulated in both resistant cultivar Shennong 9819 and susceptible cultivar Koshihikari. The expression of OsPR1a in Shennong 9819 was higher than that in Koshihikari at 72 h and lower than Koshihikari at other inoculation time points. The expression of OsPR1b was upregulated in Shennong 9819, and was always higher than that in Koshihikari. We detected that the gene expression changes of transcription factors OsWRKY24 (LOC_Os01g61080), OsWRKY30 (LOC_Os08g38990), OsWRKY53 (LOC_Os05g27730), and OsWRKY70 (LOC_Os05g39720) in Shennong 9819 were greater than that in Koshihikari at 36 h after inoculation (Table S6). The expression of OsWRKY30 in Shennong 9819 was always higher than that in Koshihikari (Fig. 5), and the expression of OsWRKY70 in Koshihikari was higher than that in Shennong 9819 (Fig. 5).
Furthermore, we compared the DEGs involved in the plant hormone signalling pathways between the two cultivars. We found that the expression of genes closely related to plant hormone signal transduction in the leaf sheaths of the two cultivars was similar to that of phenylalanine metabolism. At 12 and 36 h, the expression levels of genes related to plant hormone signal transduction in the leaf sheaths of Shennong 9819 were significantly higher than those in the leaf sheaths of Koshihikari (Fig. 6). The expression levels of resistance-related protein kinases OsSAPK9 (LOC_Os12g39630) [40] and OsSAPK10 (LOC_Os03g41460) [41] were upregulated in the leaf sheaths of Shennong 9819 and were higher than those in Koshihikari (Fig. 5). In addition, the NONEXPRESSOR OF PR1 (NPR1) (LOC_Os01g09800), a positive regulator related to resistance, was also detected, and its expression was upregulated in both cultivars (Fig. 5). The NPR1 homologous gene NPR3 and the transcription factor TGA2 were detected in this study. The negative regulator OsNPR3 (LOC_Os03g46440) was downregulated in the susceptible cultivar Koshihikari at 36 h, whereas the negative regulator rTGA2.1 (LOC_Os07g48820) was downregulated in the resistant cultivar at 12 h and 36 h (Fig. 5).

Expression profiles of genes related to plant hormone signal transduction pathways
As previously mentioned, the two cultivars participated in similar metabolic pathways after inoculation with R. solani; however, the upregulated differential expression pathways were different. Among the pathways related to disease resistance, plant hormone signal transduction and phenylalanine metabolism pathways were significantly enriched in Shennong 9819, except in Koshihikari. However, ascorbate and aldarate metabolism and linoleic acid metabolism were significantly enriched in Koshihikari, though not in Shennong 9819. It is necessary to further study the differentially expressed genes in the metabolic pathway to understand the different resistance mechanisms of the two cultivars after inoculation with R. solani.
Validation of DEGs by quantitative RT-PCR (qRT-PCR)
Real-time quantitative PCR (qRT-PCR) was used to validate RNA-seq data. OsPR1b and other genes were selected for further validation [42]. The Ct values obtained by qRT-PCR were normalised. The fold change in gene expression of the two rice cultivars inoculated with R. solani was calculated. The results showed that the expression trend of qRT-PCR was consistent with that of the RNA sequence, indicating that Illumina data were relatively reliable (Fig. S4).
Discussion
Gene expression changes in plants under the stress of Rhizoctonia solani
In the early stage of R. solani infection, the expression of DEGs in the inoculated leaf sheath of the resistant cultivar Shennong 9819 showed fluctuations. The number of DEGs observed in the leaf sheath inoculated with R. solani in Shennong 9819 was 2403 at 12 h, 190 at 24 h, 2127 at 36 h, 790 at 48 h, and 1120 at 72 h. The number of DEGs in the leaf sheath inoculated with R. solani in the susceptible cultivar Koshihikari first increased and then decreased; furthermore, the number of DEGs was significantly higher than that in the resistant cultivar. This is slightly different from the results reported by Zhang et al. [39]. Through a study on the early response to R. solani in inoculated leaves of the resistant indica rice cultivar Teqing and the susceptible japonica rice cultivar Lemont, Zhang found that the number of DEGs in inoculated leaves of both resistant and susceptible cultivars first increased and then decreased [39]. The responses were consistent with our results that the number of DEGs in the susceptible cultivar was significantly higher than that in the resistant cultivar. However, Kumari et al. [43] found that the number of DEGs in the resistant cultivar was slightly higher than that in the susceptible cultivar. These differences could be attributed to the use of different rice materials. The resistant cultivar Teqing selected in Zhang’s study is an indica rice cultivar developed in Southern China, whereas the susceptible cultivar Lemont is a conventional japonica rice cultivar developed in the United States. However, all the resistant and susceptible cultivars selected by Kumari are indica rice cultivars. We selected the japonica rice cultivar in Northeast China as the resistant cultivar and the Japanese japonica rice cultivar as the susceptible cultivar. In our study, the susceptible cultivar was more sensitive in the face of R. solani stress, while the differential gene expression of the resistant cultivar showed fluctuations.
Plant–pathogen interaction
PR proteins are markers of plant defence responses related to plant resistance to pathogens [44, 45]. PR1 proteins are the first pathogenesis-related proteins identified in the PR family [46]. In this study, OsPR1b and OsPR1a were detected, and their expression trends were quite different (Fig. 5). The expression level of OsPR1b in the resistant cultivar Shennong 9819 was always higher than that in the susceptible cultivar Koshihikari. In contrast, the expression level of OsPR1a in Koshihikari was higher than that in Shennong 9819 at most time points, indicating that they may have different resistance mechanisms.
After pathogen infection, timely transcriptional regulation of defence genes in plants is crucial [47, 48]. Proteins of the WRKY family are important regulators of this defence response pathway [49, 50]. The transcription factor WRKY30 participates in the expression of genes involved in the salicylic acid (SA) and JA immune signalling pathways; furthermore, it can enhance plant resistance to biotic and abiotic stresses [51]. Previous studies have shown that Magnaporthe grisea induced overexpression of OsWRKY30 is regulated by SA and/or JA [52], which can improve rice resistance to sheath blight and rice blast [53, 54]. In Arabidopsis, overexpression of WRKY30 can enhance its resistance to Peronospora parasitica and cucumber mosaic virus (CMV) [55, 56]. This is consistent with the results of our study. In the present study, the expression of OsWRKY30 in the resistant cultivar Shennong 9819 was always higher than that in the susceptible cultivar Koshihikari. This also verified that OsWRKY30 played an important role in improving the resistance of rice to R. solani. In addition, overexpression of OsWRKY30 in rice significantly increased drought tolerance [57]. OsWRKY70 is a transcription inhibitor of PR1 [58]. In the present study, expression of OsWRKY30 in the susceptible cultivar Koshihikari was higher than that in the resistant cultivar Shennong 9819. The lower resistance of Koshihikari to R. solani compared with the resistant cultivar Shennong 9819 may be related to the overexpression of the transcription factor OsWRKY70.
Plant signal transduction in plant disease resistance
In the natural environment, plants are constantly subjected to abiotic and biotic stresses, such as drought, salinity, and pathogen infection. Stress signals are recognised and transmitted to different cell compartments via specific signalling pathways, of which protein kinases and phosphatases are key components [59, 60]. Members of the sucrose nonfermenting1-related protein kinase2 (SnRK2) gene family are plant-specific serine/threonine kinases involved in plant responses to abiotic stresses [40, 61]. All members of the SnRK2 protein kinase gene family encoded by the rice genome are activated by hyperosmotic stress and have been designated as stress-activated protein kinases (SAPKs). We detected the upregulated resistance-related protein kinases OsSAPK9 and OsSAPK10 in the resistant cultivar Shennong 9819. Their expression in the resistant cultivar Shennong 9819 was higher than that in Koshihikari. Previous studies have shown that in rice plants carrying the non-host resistance gene Rxo1, the expression of OsSAPK9 was significantly upregulated after infection with Xanthomonas oryzae pv. oryzicola [40] In addition, OsSAPK9 has a positive regulatory effect on resistance to bacterial blight in rice [62]. Furthermore, it has been reported that OsSAPK10-mediated phosphorylation on Thr 129 of WRKY72 weakened its DNA binding ability with AOS1, promoted the endogenous JA level of rice, and enhanced the resistance to bacterial blight [41]. These results are consistent with the results of the present study. OsSAPK9 and OsSAPK10 play a role in improving the disease resistance of rice in the early stages of pathogen infection. In addition, overexpression of OsSAPK9 can significantly improve crop drought resistance [63] and NH+ tolerance [64]. Overexpression of OsSAPK10 can promote the growth of root hair [65] and induce closure of stomata [66] in rice.
NPR1 functions as a master regulator of SA signalling and plays an essential role in plant immunity [58]. Previous studies have confirmed that the overexpression of NPR1 leads to increased host resistance to various pathogens [67, 68]. In the present study, OsNPR1 was detected in the two cultivars; furthermore, the expression of OsNPR1 was upregulated in both cultivars. This indicated that both cultivars initiated the NPR1 resistance mechanism to resist infection from pathogens. rTGA2.1 is a negative regulator of the plant defence response [69]. At 12 and 36 h, the expression of rTGA2.1 was downregulated in the resistant cultivar. This result is consistent with previous findings [69]. Transcription factor rTGA2.1 negatively regulated plant resistance, and its downregulation increased the resistance to R. solani in the resistant cultivar Shennong 9819. OsWRKY70 is an inhibitor of the NPR1 induced resistance gene [70]. In this study, its expression in the susceptible cultivar Koshihikari was higher than that in the resistant cultivar Shennong 9819. This indicates that Shennong 9819 may induce plant resistance by regulating the expression of the NPR1 gene in the early stage of R. solani infection.
Expression of OsPAL in plant disease resistance
Phenylpropanoid metabolism is an important metabolic pathway in the secondary metabolism of plant disease resistance [71]. It leads to the biosynthesis of a wide range of plant natural products including hydroxycinnamic acids, flavonoids, coumarins, lignin, condensed tannins, and stilbenes, which have various biological functions as UV protectants, signal molecules, phytoalexins, and flower pigments [72]. L-phenylalanine ammonia-lyase (PAL; EC 4.3.1.5) is a key enzyme involved in phenylpropanoid metabolism [73]. The activity of PAL provides precursors for the biosynthesis of lignin and other phenolics such as SA [74], which accumulate when infected [75, 76]. PAL is an important regulatory defence gene. In the present study, at 12 and 36 h, the expression level of the phenylalanine metabolic pathway in the resistant cultivar Shennong 9819 was significantly higher than that in the susceptible cultivar Koshihikari. After inoculation with R. solani, six PAL genes were activated; the expression of OsPAL1 and OsPAL6 in the resistant cultivar was significantly higher than that in the susceptible cultivar. Previous studies have reported that PAL is involved in inducing plant disease resistance response [77, 78], and transcripts of the PAL gene accumulate in incompatible host pathogen combinations [79]. Transgenic tobacco with suppressed expression of PAL genes showed reduced basal resistance to Cercospora nicotianae [80]. In rice and cassava, overexpression of PAL1 endows wild-type rice with resistance to rice blast [81] and cassava brown streak disease resistant cultivars with resistance to cassava brown stripe virus [82]. OsPAL6 regulates biosynthesis of SA and lignin [29, 76]. Lignin accumulation is considered to be a response to plant incompatibility with pathogens [83], thereby increasing plant resistance to pathogens [84, 85]. The decrease in lignin content reduced the resistance of Malus hupehensis to Botryosphaeria dothidea [86]. In the present study, the overexpression of OsPAL1 and OsPAL6 in the resistant cultivar suggests that the resistant cultivar Shennong 9819 may improve its resistance to R. solani through the overexpression of PAL genes.
Conclusion
In summary, the early transcriptome analysis of resistant and susceptible cultivars infected by R. solani revealed that the resistant cultivar has a conservative and unique defence mechanism. The differential expression of resistance-related genes is associated with the early resistance of rice to R. solani. In the early stage of R. solani infection, it was found that the number of DEGs in the inoculated leaf sheath of resistant and susceptible cultivars showed different regularity, and the expression of DEGs in resistant cultivars fluctuated. Phenylalanine metabolic pathway, plant hormone signal transduction pathway, and tropane, piperidine and pyridine alkaloid biosynthesis pathways were differentially expressed in response to resistance. In the early stage of R. solani stress, resistant cultivars initiated a response defence system involving overexpression of PR genes, PAL genes, and key transcription factors, as well as enrichment of defence-related pathways. In conclusion, this study provides new insights into the mechanism of rice resistance to R. solani. The data obtained in this study can be used to screen suitable candidate genes for genetic improvement of susceptible rice cultivars and the development of cultivars of japonica rice that are resistant to R. solani.
Materials and methods
The experimental flow chart is shown in Fig. 7.

Experimental flow chart
Plant growth
Shennong 9819, a japonica rice cultivar from the Rice Research Institute of Shenyang Agricultural University (Shenyang, China), is a rice cultivar resistant to rice sheath blight. Resistance was identified by the Rice Disease Research Office of the Shenyang Agricultural University (Shenyang, China) [19].
Koshihikari is a japonica rice cultivar from Japanese, a cultivar susceptible to rice sheath blight.
All resistant and susceptible cultivars were planted at the experimental base of Jiamus Branch of Heilongjiang Academy of Agricultural Sciences (Jiamusi, China).
Pathogen inoculation
The strain of R. solani used in this experiment, R-36, was provided by the Rice Disease Research Office of Shenyang Agricultural University.
The inoculation method was carried out according to the method described by Zuo et al. [17]. A short toothpick (1 mm diameter, 1.0–1.2 cm long) colonised by R-36 was used as the inoculum for pathogen infection. Sterilised short toothpicks were placed on non-coagulated PDA plates. After the culture medium solidified, the activated mycelium was inoculated in the culture dish, cultured at 28 °C for 5 d, and a short toothpick with mycelium was selected for inoculation. At the late tillering stage, inoculation was performed using a short toothpick with mycelium. The inoculation site was the third leaf sheath from the top of the plant. Forceps were used as the inoculation tool. When inoculated, the original state of the leaf sheath should be maintained. To maintain the same temperature and humidity as the inoculated leaf sheath, the inoculated leaf sheath was wrapped with a cling film.
Sample and method
The inoculated leaf sheaths were cut from the plants 12, 24, 36, 48, and 72 h after inoculation, and uninoculated rice leaf sheaths were collected at 0 h after inoculation as a control. The sample collection time for each cultivar was six time points, and each time point was repeated thrice. A total of 36 samples were obtained from the two cultivars. Samples collected from plants were placed in cryotubes separately, immediately frozen in liquid nitrogen, and stored at − 80 °C. The transcriptome sequencing and cDNA library construction of all samples were completed by Beijing Biomarker Technology, Inc. (Beijing, China).
Optical microscope observation
The staining method that was used was proposed by Lux et al. [87]. Alcohol solution (95%) and glacial acetic acid were mixed in a 1:1 ratio to prepare fixative solution. Chloral hydrate (5 g) was dissolved in 2 ml distilled water to prepare saturated chloral hydrate. Aniline blue (1 g) was added to 100 ml distilled water to prepare the aniline blue staining solution.
At 12, 24, 36, 48, and 72 h after inoculation, the inoculated leaf sheaths were cut for microscopic observation. The leaf sheath tissue inoculated with R. solani was cut into small blocks (3 × 5 mm) with a bimodal blade. The small blocks were fixed in the fixative for 24 h and then soaked in saturated chloral hydrate aqueous solution for transparency. After the tissue was transparent, it was removed, washed with water, stained with aniline blue staining solution for 5–10 min, examined with a microscope, and photographed.
Genome sequence and expression
Raw reads in fast format were first processed using internal Perl scripts. Clean reads were obtained by removing the reads containing adapter, poly-n, and low-quality reads from the raw reads. Q20, Q30, GC-content, and sequence duplication levels of the clean reads were calculated. All downstream analyses were based on high-quality, clean reads. These clean reads were mapped to the reference genome sequence. The reference genome used was Nipponbare MSU_v7.0. The programme Hisat2 was used to compare the reads [88], and stringties were used to assemble, evaluate, and quantify the reads [89]. Fragments per kilobase of transcript per million fragments mapped (FPKM) was used to calculate gene expression [90].
Screening and functional annotation of DEGs
Differential expression analysis of the differential groups was performed using DEseq [91]. The resulting P values were adjusted using Benjamini and Hochberg’s approach to control the false discovery rate. Genes with an adjusted P-value < 0.01 found by DEseq, were assigned as differentially expressed. The GO enrichment analysis of the DEGs was implemented using the GOseq R package [92], which can adjust for gene length bias in DEGs. We used KOBAS software to test the statistical enrichment of DEGs in the KEGG pathways [93].
Real-time PCR analysis
Eight genes that were co-expressed in the two cultivars were selected for real-time quantitative PCR. Specific primers were designed using Primer-BLAST of NCBI and are listed in Table S7.
The RNA was extracted using the TaKaRa MiniBEST Plant RNA Extraction Kit, and the 1st Strand cDNA Synthesis Kit (TaKaRa, Tokyo, Japan) was used for reverse transcription into cDNA. qRT-PCR experiments were performed using a Bio-Rad CFX96 Real-Time PCR System (Bio-Rad, Hercules, CAUSA) according to the manufacturer’s instructions. Reactions were prepared using 20 μL of total volume, 10 μL of SYBR® Premix Ex Taq™ II, 1.0 μL of gene-specific primers (0.5 μL each primer), and 0.5 μl of cDNA.
The reaction procedure was run as follows: (1) 95 °C for 30 s; (2) 95 °C for 5 s, 60 °C for 30 s, for 40 cycles. (3) 95 °C for 15 s, 60 °C for 60 s, and 95 °C for 15 s. The actin gene was used as an internal control to normalise the data. Gene expression levels were calculated using the 2-ΔΔCT algorithm [94].
Availability of data and materials
All datasets generated or analysed during this study are included in this published article and its supplementary information files.
Abbreviations
- DEGs:
-
Differentially expressed genes
- ET:
-
Ethylene
- GO:
-
Gene ontology
- JA:
-
Jasmonic acid
- KEGG:
-
Kyoto encyclopaedia of genes and genomes
- NPR1 :
-
Nonexpressor of PR1 genes
- PAL:
-
L-phenylalanine ammonialyase
- PG:
-
Polygalacturonase
- PGIP:
-
Plant polygalacturonase-inhibiting protein
- PR:
-
Pathogenic-related
- qRT-PCR:
-
Real-time quantitative PCR
- RNA-Seq:
-
RNA-sequencing
- SA:
-
Salicylic acid
- SAR:
-
Systemic acquired resistance
- TPM:
-
Transcripts per million
- Xoo :
-
Xanthomonas oryzae pv. oryzae
- Xoc :
-
Xanthomonas oryzae pv. Oryzicola
References
Telesh-Sasani S, Soltani BM, Behmanesh M, Safaie N. A magnaporthe avr-pita gene orthologous in Rhizoctonia solani AG1-IA shows characteristic of an effector protein. Austr Plant Path. 2015;44(5):567–74. https://doi.org/10.1007/s13313-015-0372-5.
Swain DM, Sahoo RK, Chandan RK, Ghosh S, Kumar R, Jha G, et al. Concurrent overexpression of rice G-protein β and γ subunits provide enhanced tolerance to sheath blight disease and abiotic stress in rice. Planta. 2019;250(5):1505–20. https://doi.org/10.1007/s00425-019-03241-z.
Choudhary P, Rai P, Yadav J, Verma S, Chakda H, Goswami SK, et al. A rapid colorimetric LAMP assay for detection of Rhizoctonia solani AG-1 IA causing sheath blight of rice. Sci Rep. 2020;10(1):22022. https://doi.org/10.1038/s41598-020-79117-0.
Lee FN, Rush MC. Rice sheath blight: a major Rice disease. Plant Dis. 1983;67(7):829–32. https://doi.org/10.1094/PD-67-829.
Liu G, Jia Y, Correa-Victoria FJ, Prado GA, Yeater KM, McClung A, et al. Mapping quantitative trait loci responsible for resistance to sheath blight in rice. Phytopathology. 2009;99:1078–84. https://doi.org/10.1094/PHYTO-99-9-1078.
Zheng A, Lin R, Zhang D, Qin P, Xu L, Ai P, et al. The evolution and pathogenic mechanisms of the rice sheath blight pathogen. Nat Commun. 2013;4:1424. https://doi.org/10.1038/ncomms2427.
Zhou Y, Bao J, Zhang D, Li Y, Li H, He H. Effect of heterocystous nitrogen-fixing cyanobacteria against rice sheath blight and the underlying mechanism. Appl Soil Ecol. 2020;153(5):103580. https://doi.org/10.1016/j.apsoil.2020.103580.
Okubara PA, Dickman MB, Blechl AE. Molecular and genetic aspects of controlling the soilborne necrotrophic pathogens Rhizoctonia and Pythium. Plant Sci. 2014;228:61–70. https://doi.org/10.1016/j.plantsci.2014.02.001.
Lin R, He L, He J, Qin P, Wang Y, Deng Q, et al. Comprehensive analysis of microRNA-Seq and target mRNAs of rice sheath blight pathogen provides new insights into pathogenic regulatory mechanisms. DNA Res. 2016;23:415–25. https://doi.org/10.1093/dnares/dsw024.
Singh P, Mazumdar P, Harikrishna JA, Babu S. Sheath blight of rice: a review and identification of priorities for future research. Planta. 2019;250(5):1387–407. https://doi.org/10.1007/s00425-019-03246-8.
Maeda S, Dubouzet JG, Kondou Y, Jikumaru Y, Seo S, Oda K, et al. The rice CYP78A gene BSR2 confers resistance to Rhizoctonia solani and affects seed size and growth in Arabidopsis and rice. Sci Rep. 2019;9(1):587. https://doi.org/10.1038/s41598-018-37365-1.
Rao TB, Chopperla R, Methre R, Punniakotti E, Venkatesh V, Sailaja B, et al. Pectin induced transcriptome of a Rhizoctonia solani strain causing sheath blight disease in rice reveals insights on key genes and RNAi machinery for development of pathogen derived resistance. Plant Mol Biol. 2019;100:59–71. https://doi.org/10.1007/s11103-019-00843-9.
Ghosh S, Kanwar P, Jha G. Alterations in rice chloroplast integrity, photosynthesis and metabolome associated with pathogenesis of Rhizoctonia solani. Sci Rep. 2017;7:41610. https://doi.org/10.1038/srep41610.
Zhao C, Wang A, Shi Y, Wang L, Liu W, Wang Z, et al. Identification of defense-related genes in rice responding to challenge by Rhizoctonia solani. Theor Appl Genet. 2008;116:501–16. https://doi.org/10.1007/s00122-007-0686-y.
Zou JH, Pan XB, Chen ZX, Xu JY, Lu JF, Zhai WX, et al. Mapping quantitative trait loci controlling sheath blight resistance in two rice cultivars (Oryza sativa L.). Theor Appl Genet. 2000;101(4):569–73. https://doi.org/10.1007/s001220051517.
Pinson SRM, Capdevielle FM, Oard JH. Confirming QTLs and finding additional loci conditioning sheath blight resistance in rice using recombinant inbred lines. Crop Sci. 2005;45(2):503. https://doi.org/10.2135/cropsci2005.0503.
Zuo S, Yin Y, Pan C, Chen Z, Zhang Y, Gu S, et al. Fine mapping of qSB-11LE, the QTL that confers partial resistance to rice sheath blight. Theor Appl Genet. 2013;126(5):1257–72. https://doi.org/10.1007/s00122-013-2051-7.
Tang L, Chen W. Development trend and prospect of Geng Rice in Northeast China. China Rice. 2021;27(5):1–4 (in Chinese with English summary).
Yang X, Wei S, Gu X, Yao L, Gao X, Shen H, et al. A preliminary report on resistance of rice germplasm resources in the northeast of China to rice sheath blight disease. Plant Prot. 2020;46(6):205–8 (in Chinese with an English Abstract).
Kobayashi A, Hori K, Yamamoto T, Yamamoto T, Yana M. Koshihikari: a premium short-grain rice cultivar-its expansion and breeding in Japan. Rice. 2018;11(1):15. https://doi.org/10.1186/s12284-018-0207-4.
Li D, Zhang J, Wang H, Li M, Piao Z. Differences in amylopectin structure and grain quality of rice between some high-quality japonica rice cultivars from the lower Yangtze River region, China and Koshihikari from Niigata, Japan. Chin J Rice Sci. 2010;24(4):379–84 (in Chinese with English summary).
Richa K, Tiwari IM, Devanna BN, Botella JR, Sharma V, Sharma TR. Novel chitinase gene LOC_Os11g47510 from indica rice tetep provides enhanced resistance against sheath blight pathogen Rhizoctonia solani in rice. Front Plant Sci. 2017;8:596. https://doi.org/10.3389/fpls.2017.00596.
Zhang C, Huang M, Sang X, Li P, Ling Y, Zhao F, et al. Association between sheath blight resistance and chitinase activity in transgenic rice plants expressing McCHIT1 from bitter melon. Transgenic Res. 2019;28:381–90. https://doi.org/10.1007/s11248-019-00158-x.
Xue X, Cao ZX, Zhang XT, Wang Y, Zhang YF, Chen ZX, et al. Overexpression of OsOSM1 enhances resistance to rice sheath blight. Plant Dis. 2016;100(8):1634–42. https://doi.org/10.1094/PDIS-11-15-1372-RE.
Zhu G, Liang E, Lan X, Li Q, Qian J, Tao H, et al. ZmPGIP3 gene encodes a polygalacturonase-inhibiting protein that enhances resistance to sheath blight in rice. Phytopathology. 2019;109(10):1732–40. https://doi.org/10.1094/PHYTO-01-19-0008-R.
Wang R, Lu L, Pan X, Hu Z, Ling F, Yan Y, et al. Functional analysis of OsPGIP1 in rice sheath blight resistance. Plant Mol Biol Rep. 2015;87:181–91. https://doi.org/10.1007/s11103-014-0269-7.
Chen X, Chen Y, Zhang L, He Z, Huang B, Chen C, et al. Amino acid substitutions in a polygalacturonase inhibiting protein (OsPGIP2) increases sheath blight resistance in rice. Rice. 2019;12(1):56. https://doi.org/10.1186/s12284-019-0318-6.
Kawasaki T, Koita H, Nakatsubo T, Hansegawa K, Wakabayashi K, Takahashi H, et al. Cinnamoyl-CoA reductase, a key enzyme in lignin biosynthesis, is an effector of small GTPase Rac in defense signaling in rice. Proc Natl Acad Sci U S A. 2006;103(1):230–5. https://doi.org/10.1073/pnas.0509875103.
Tonnessen BW, Manosalva P, Lang JM, Baraoida M, Bordeos A, Mauleon R, et al. Rice phenylalanine ammonia-lyase gene OsPAL4 is associated with broad spectrum disease resistance. Plant Mol Biol. 2015;87(3):273–86. https://doi.org/10.1007/s11103-014-0275-9.
Wang H, Meng J, Peng X, Tang X, Zhou P, Xiang J, et al. Rice WRKY4 acts as a transcriptional activator mediating defense responses toward Rhizoctonia solani, the causing agent of rice sheath blight. Plant Mol Biol Report. 2015;89:157–71. https://doi.org/10.1007/s11103-015-0360-8.
Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10(1):57–63. https://doi.org/10.1038/nrg2484.
Metzker ML. Sequencing technologies-the next generation. Nat Rev Genet. 2010;11:31–46. https://doi.org/10.1201/9781420008876.pt1.
Zhang H, Yang Y, Wang C, Liu M, Li H, Fu Y, et al. Large-scale transcriptome comparison reveals distinct gene activations in wheat responding to stripe rust and powdery mildew. BMC Genomics. 2014;15(1):898. https://doi.org/10.1186/1471-2164-15-898.
Han Y, Zhang K, Yang J, Zhang N, Fang A, Zhang Y, et al. Differential expression profiling of the early response to Ustilaginoidea virens, between false smut resistant and susceptible rice varieties. BMC Genomics. 2015;16(1):955. https://doi.org/10.1186/s12864-015-2193-x.
Bagnaresi P, Biselli C, Orrù L, Urso S, Crispino L, Abbruscato P, et al. Comparative transcriptome profiling of the early response to Magnaporthe oryzae in durable resistant vs susceptible rice (Oryza sativa L.) genotypes. PLoS One. 2012;7(12):e51609. https://doi.org/10.1371/journal.pone.0051609.
Strauß T, Poecke RMP, Strauß A, Römer P, Minsavage G, Singh S, et al. RNA-seq pinpoints a Xanthomonas TAL-effector activated resistance gene in a large-crop genome. Proc Natl Acad Sci U S A. 2012;109:19480–5. https://doi.org/10.1073/pnas.1212415109.
Kawahara Y, Oono Y, Kanamori H, Matsumoto T, Itoh T, Minami E. Simultaneous RNA-Seq analysis of a mixed transcriptome of rice and blast fungus interaction. PLoS One. 2012;7:e49423. https://doi.org/10.1371/journal.pone.0049423.
Xiao J, Jin X, Jia X, Wang H, Cao A, Zhao W, et al. Transcriptome-based discovery of pathways and genes related to resistance against Fusarium head blight in wheat landrace Wangshuibai. BMC Genomics. 2013;14(1):1–19. https://doi.org/10.1186/1471-2164-14-197.
Zhang J, Chen L, Fu C, Wang L, Liu H, Cheng Y, et al. Comparative transcriptome analyses of gene expression changes triggered by Rhizoctonia solani AG1 IA infection in resistant and susceptible rice varieties. Front Plant Sci. 2017;8:1422. https://doi.org/10.3389/fpls.2017.01422.
Xu MR, Huang LY, Zhang F, Zhu LH, Zhou YL, LI ZK. Genome-wide phylogenetic analysis of stress-activated protein kinase genes in rice (OsSAPKs) and expression profiling in response to Xanthomonas oryzae pv. Oryzicola infection. Plant Mol Biol Rep. 2013;31(4):877–85. https://doi.org/10.1007/s11105-013-0559-2.
Hou Y, Wang Y, Tang L, Tong X, Wang L, Liu L, et al. SAPK10-mediated phosphorylation on WRKY72 releases its suppression on jasmonic acid biosynthesis and bacterial blight resistance. iScience. 2019;16:499–510. https://doi.org/10.1016/j.isci.2019.06.009.
Agrawal GK, Rakwal R, Jwa NS. Rice (Oryza sativa L.) OsPR1b gene is phytohormonally regualted in close interaction with light signals. Biochem Biophys Res Commun. 2000;278(2):290–8. https://doi.org/10.1006/bbrc.2000.3781.
Kumari A, Jain P, Kamboj R, Sharma TR. RNASeq reveals differential regulation of genes in resistant and susceptible rice varieties during early phases of infection with Rhizoctonia solani. Indian J Genet Pl Br. 2017;77(4):475–84. https://doi.org/10.5958/0975-6906.2017.00063.3.
Mitsuhara I, Iwai T, Seo S, Yanagawa Y, Kawahigasi H, Hirose S, et al. Characteristic expression of twelve rice PR1 family genes in response to pathogen infection, wounding, and defense-related signal compounds (121/180). Mol Gen Genomics. 2008;279(4):415–27. https://doi.org/10.1007/s00438-008-0322-9.
Wu Q, Hou MM, Li LY, Liu LJ, Hou YX, Liu GZ. Induction of pathogenesis-related proteins in rice bacterial blight resistant gene XA21-mediated interactions with Aanthomonas oryzae pv. Oryzea. J Plant Pathol. 2011;93(2):455–9.
Showmy KS, Yusuf A. Characterization of disease resistance in nine traditional rice (Oryza sativa L.) cultivars and expression of chennellu PR1 gene in response to Xanthomonas oryzae pv. Oryzae. Indian Phytopathol. 2020;73(1). https://doi.org/10.1007/s42360-020-00220-3.
Nimchuk Z, Eulgem T, Holt BF, Dangl JL. Recognition and response in the plant immune system. Annu Rev Genet. 2003;37(1):579–609. https://doi.org/10.1146/annurev.genet.37.110801.142628.
Jones DA, Takemoto D. Plant innate immunity-direct and indirect recognition of general and specific pathogen-associated molecules. Curr Opin Immunol. 2004;16:48–62. https://doi.org/10.1016/j.coi.2003.11.016.
Ülker B. Somssich IE.WRKY transcription factors: from DNA binding towards biological function. Curr Opin Plant Biol. 2004;7(5):491–8. https://doi.org/10.1016/j.pbi.2004.07.012.
Zhang YJ, Wang L. The WRKY transcription factor superfamily: its origin in eukaryotes and expansion in plants. BMC Evol Biol. 2005;5:1. https://doi.org/10.1186/1471-2148-5-1.
Scarpeci TE, Zanor MI, Mueller-Roeber B, Valle EM. Overexpression of AtWRKY30 enhances abiotic stress tolerance during early growth stages in Arabidopsis thaliana. Plant Mol Biol. 2013;83(3):265–77. https://doi.org/10.1007/s11103-013-0090-8.
Ryu HS, Han M, Lee SK, Cho JI, Ryoo N, Heu S, et al. A comprehensive expression analysis of the WRKY gene superfamily in rice plants during defense response. Plant Cell Rep. 2006;25(8):836–47. https://doi.org/10.1007/s00299-006-0138-1.
Peng X, Hu Y, Tang X, Zhou P, Deng X, Wang H, et al. Constitutive expression of rice WRKY30 gene increases the endogenous jasmonic acid accumulation, PR gene expression and resistance to fungal pathogens in rice. Planta. 2012;236(5):1485–98. https://doi.org/10.1007/s00425-012-1698-7.
Jalmi SK, andSinha A K. Functional involvement of a mitogen activated protein kinase module, OsMKK3-OsMPK7-OsWRK30 in mediating resistance against Xanthomonas oryzae in Rice. Sci Rep. 2016;6:37974. https://doi.org/10.1038/srep37974.
Jiang W, Wu J, Zhang Y, Yin L, Lu J. Isolation of a WRKY30 gene from Muscadinia rotundifolia (Michx) and validation of its function under biotic and abiotic stresses. Protoplasma. 2015;252(5):1361–74. https://doi.org/10.1007/s00709-015-0769-6.
Zou L, Yang F, Ma Y, Wu Q, Yi K, Zhang D. Transcription factor WRKY30 mediates resistance to cucumber mosaic virus in Arabidopsis. Biochem Bioph Res Co. 2019;517(1):118–24. https://doi.org/10.1016/j.bbrc.2019.07.030.
Shen H, Liu C, Zhang Y, Meng X, Zhou X, Chu C, et al. OsWRKY30 is activated by MAP kinases to confer drought tolerance in rice. Plant Mol Biol. 2012;80(3):241–53. https://doi.org/10.1007/s11103-012-9941-y.
Chen J, Mohan R, Zhang Y, Li M, Chen H, Palmer IA, et al. NPR1 promotes its own and target gene expression in plant defense by recruiting CDK8. Plant Physiol. 2019;181(1):289–304. https://doi.org/10.1104/pp.19.00124.
Stone JM, Walker JC. Plant protein kinase families and signal transduction. Plant Physiol. 1995;108:451–7. https://doi.org/10.1104/pp.108.2.451.
Agueci F, Rutten T, Demidov D, Houben A. Arabidopsis AtNek2 kinase is essential and associates with microtubules. Plant Mol Biol Rep. 2012;30(2):339–48. https://doi.org/10.1007/s11105-011-0342-1.
Wang P, Xue L, Batelli G, Lee S, Hou Y, Oosten MJV, et al. Quantitative phosphoproteomics identifies SnRK2 protein kinase substrates and reveals the effectors of abscisic acid action. Proc Natl Acad Sci U S A. 2013;110(27):11205–10. https://doi.org/10.1073/pnas.1308974110.
Zhang F, Zeng D, Huang L, Shi Y, Chen T, Zhang F, et al. Stress-activated protein kinase OsSAPK9 regulates tolerance to salt stress and resistance to bacterial blight in rice. Rice. 2019;12(1):80. https://doi.org/10.1186/s12284-019-0338-2.
Dey A, Samanta MK, Gayen S, Maiti MK. The sucrose non-fermenting 1-related kinase 2 gene SAPK9 improves drought tolerance and grain yield in rice by modulating cellular osmotic potential, stomatal closure and stress-responsive gene expression. BMC Plant Biol. 2016;16(1):158. https://doi.org/10.1186/s12870-016-0845-x.
Sun L, Di DW, Li G, Kronzucker HJ, Wu X, Shi W. Endogenous ABA alleviates rice ammonium toxicity by reducing ROS and free ammonium via regulation of the SAPK9–bZIP20 pathway. J Exp Bot. 2020;75(15):4562–77. https://doi.org/10.1093/jxb/eraa076.
Wang T, Li C, Wu Z, Jia Y, Wang H, Sun S, et al. Abscisic acid regulates auxin homeostasis in rice root tips to promote root hair elongation. Front Plant Sci. 2017;8:1121. https://doi.org/10.3389/fpls.2017.01121.
Min MK, Choi EH, Kim JA, Yoon IS, Han S, Lee Y, et al. Two clade a phosphatase 2Cs expressed in guard cells physically interact with abscisic acid signaling components to induce stomatal closure in rice. Rice. 2019;12(1):37. https://doi.org/10.1186/s12284-019-0297-7.
Liu G, Holub EB, Alonso JM, Ecker JR, Fobert PR. An Arabidopsis NPR1-like gene, NPR4, is required for disease resistance. Plant J. 2005;41:304–18. https://doi.org/10.1111/j.1365-313X.2004.02296.x.
Li H, Wu J, Shang X, Geng M, Gao J, Zhao S, et al. WRKY transcription factors shared by BTH-induced resistance and NPR1-mediated acquired resistance improve broad-spectrum disease resistance in wheat. Mol Plant Microbe In. 2020;33(3):433–43. https://doi.org/10.1094/MPMI-09-19-0257-R.
Zhang Y, Cheng YT, Qu N, Zhao Q, Bi D, Li X. Negative regulation of defense responses in Arabidopsis by two NPR1 paralogs. Plant J. 2006;48:647–56. https://doi.org/10.1111/j.1365-313X.2006.02903.x.
Saleh A, Withers J, Mohan R, Marqués J, Gu Y, Yan S, et al. Posttranslational modifications of the master transcriptional regulator NPR1 enable dynamic but tight control of plant immune responses. Cell Host Microbe. 2015;18(2):169–82. https://doi.org/10.1016/j.chom.2015.07.005.
Dixon RA, Achnine L, Kota P, Liu C, Reddy MSS, Wang L. The phenylpropanoid pathway and plant defence- a genomics perspective. Mol Plant Pathol. 2002;3(5):371–90. https://doi.org/10.1046/j.1364-3703.2002.00131.x.
Reichert AI, He XZ, Dixon RA. Phenylalanine ammonia-lyase (PAL) from tobacco (Nicotiana tabacum): characterization of the four tobacco PAL genes and active heterotetrameric enzymes. Biochem J. 2009;424(2):233–42. https://doi.org/10.1042/BJ20090620.
Liu Y, Liu L, Yang S, Zeng Q, He Z, Liu Y. Cloning, characterization and expression of the phenylalanine Ammonia-Lyase gene (PaPAL) from spruce Picea asperata. Forests. 2019;10(8):613. https://doi.org/10.3390/f10080613.
Mauch-Mani B, Slusarenko AJ. Production of salicylic acid precursors is a major function of phenylalanine ammonia-lyase in the resistance of Arabidopsis to Peronospora parasitica. Plant Cell. 1996;8(2):203–12. https://doi.org/10.2307/3870265.
Duan L, Liu H, Li X, Xiao J, Wang S. Multiple phytohormones and phytoalexins are involved in disease resistance to Magnaporthe oryzae invaded from roots in rice. Physiol Plant. 2014;152(3):486–500. https://doi.org/10.1111/ppl.12192.
He J, Liu Y, Yuan D, Duan M, Liu Y, Shen Z, et al. An R2R3 MYB transcription factor confers brown planthopper resistance by regulating the phenylalanine ammonia-lyase pathway in rice. Proc Natl Acad Sci. 2019;117(1):201902771. https://doi.org/10.1073/pnas.1902771116.
Tanaka N, Che FS, Watanabe N, Fujiwara S, Takayama S, Lsogai A. Flagellin from an incompatible strain of Acidovorax avenae mediates H2O2 generation accompanying hypersensitive cell death and expression of PAL, Cht-1, and PBZ1, but not of lox in rice. Mol Plant-Microbe Inter. 2003;16(5):422–8. https://doi.org/10.1094/MPMI.2003.16.5.422.
Kim DS, Hwang BK. An important role of the pepper phenylalanine ammonia-lyase gene (PAL1) in salicylic acid-dependent signalling of the defence response to microbial pathogens. J Exp Bot. 2014;65(9):2295–306. https://doi.org/10.1093/jxb/eru109.
Edwards K, Cramer CL, Bolwell GP, Dixon RA, Schuch W, Lamb C. Rapid transient induction of phenylalanine ammonia-lyase mRNA in elicitor-treated bean cells. Proc Natl Acad Sci U S A. 1985;82(20):6731–5. https://doi.org/10.1073/pnas.82.20.6731.
Maher EA, Bate NJ, Ni W, Elkind Y, Dixon RA, Lamb CJ. Increased disease susceptibility of transgenic tobacco plants with suppressed levels of preformed phenylpropanoid products. Proc Natl Acad Sci U S A. 1994;91(16):7802–6.
Zhou X, Liao H, Chern M, Yin J, Chen Y, Wang J, et al. Loss of function of a rice TPR-domain RNA-binding protein confers broad-spectrum disease resistance. Proc Natl Acad Sci U S A. 2018;115:12. https://doi.org/10.1073/pnas.1705927115.
Kavil S, Otti G, Bouvaine S, Armitage A, Maruthi MN. PAL1 gene of the phenylpropanoid pathway increases resistance to the cassava brown streak virus in cassava. Virol J. 2021;18(1):184. https://doi.org/10.1186/s12985-021-01649-2.
Lee MH, Jeon HS, Kim SH, Chung JH, Roppolo D, Lee HJ, et al. Lignin-based barrier restricts pathogens to the infection site and confers resistance in plants. EMBO J. 2019;38(23):e101948. https://doi.org/10.15252/embj.2019101948.
Li C, He Q, Zhang F, Yu J, Li C, Zhao T, et al. Melatonin enhances cotton immunity to Verticillium wilt via manipulating lignin and gossypol biosynthesis. Plant J. 2019;100(4):784–800. https://doi.org/10.1111/tpj.14477.
Tronchet M, Balagué C, Kroj T, Jouanin L, Roby D. Cinnamyl alcohol dehydrogenases-C and D, key enzymes in lignin biosynthesis, play an essential role in disease resistance in Arabidopsis. Mol Plant Pathol. 2010;11(1):83–92. https://doi.org/10.1111/j.1364-3703.2009.00578.x.
Yu X, Gong H, Cao L, Hou Y, Qu S. MicroRNA397b negatively regulates resistance of Malus hupehensis to Botryosphaeria dothidea by modulating MhLAC7 involved in lignin biosynthesis. Plant Sci. 2020;292:110390. https://doi.org/10.1016/j.plantsci.2019.110390.
Lux A, Morita S, Abe J, Ito K. An improved method for clearing and staining free-hand sections and whole-mount samples. Ann Bot. 2005;96(6):989–96. https://doi.org/10.1093/aob/mci266.
Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357–60. https://doi.org/10.1038/nmeth.3317.
Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33(3):290–5. https://doi.org/10.1038/nbt.3122.
Florea L, Song L, Salzberg SL. Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues. F1000. Research. 2013;2:188. https://doi.org/10.12688/f1000research.2-188.v1.
Wang L, Feng Z, Wang X, Wang X, Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26(1):136–8. https://doi.org/10.1093/bioinformatics/btp612.
Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNAseq: accounting for selection bias. Genome Biol. 2010;11(2):1–14. https://doi.org/10.1186/gb-2010-11-2-r14.
Mao X, Cai T, Olyarchuk JG, Wei L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics. 2005;21(19):3787–93. https://doi.org/10.1093/bioinformatics/bti430.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25(4):402–8. https://doi.org/10.1006/meth.2001.1262.
Acknowledgments
We would like to thank Editage (www.editage.cn) for English language editing.
Funding
This work was supported by the earmarked fund for China Agriculture Research System (No. CARS-01-34), the Scientific Research Fund of Liaoning (No. LSNZD201902), the Applied Technology Research and Development Plan of Heilongjiang Province (No. GA20B104), and the Agricultural Science and Technology Innovation Leaping Project of Heilongjiang Academy of Agricultural Sciences (HNK2019CX14). The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
YXH participated in the conception and design of the study, performed the experimental work and data analysis, and drafted the manuscript. WSH and FJF supervised the study and wrote the commentary. GX, and MQY participated in the experimental design. YLL, and DJJ participated in the conception and experimental design of the study, and assisted in biological interpretation. GXD, and ZMM assisted with computational data analysis and biological interpretation, and revised the manuscript. All of the authors reviewed and accepted the final version of the manuscript. The author(s) read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
With the permission to collect, all materials of Oryza sativa accessions were identified and collected from China, and now deposited at Shenyang Agricultural University in Shenyang, China. The study comply with relevant institutional, national, and international guidelines and legislation. This research did not involve any human subjects, human material, or human data. Oryza sativa in current research did not belong to the endangered.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
Symptoms of sheath blight disease detected in Shennong 9819 and Koshihikari.
Additional file 2: Fig. S2.
Infection of hyphae in inoculated leaf sheaths at different time points.
Additional file 3: Fig. S3.
Overall relatedness of transcriptomes at different times. (SS for Shennong 9819; YY for Koshihikari).
Additional file 4: Fig. S4.
RT-qPCR validation of differentially expressed genes identified by Illumina sequencing. Histogram: Relative expression, detection results of real-time fluorescent quantitative PCR; Line graph: log2FC, fold change in differentially expressed genes in the transcriptome.
Additional file 5: Table S1.
Statistics of Illumina sequencing data. (SS for Shennong 9819; YY for Koshihikari).
Additional file 6: Table S2.
Summary for clean read mapping to the Oryza sativa Nipponbare reference genomeclean reads. (SS for Shennong 9819; YY for Koshihikari).
Additional file 7: Table S3.
The 2275 co-regulated genes in both rice cultivars at different time points (SS for Shennong 9819; YY for Koshihikari).
Additional file 8: Table S4.
Analysis of pathways involving upregulated genes after inoculation with Rhizoctonia solani in Shennong 9819. (SS for Shennong 9819).
Additional file 9: Table S5.
Analysis of pathways involving upregulated genes after inoculation with Rhizoctonia solani in Koshihikari. (YY for Koshihikari).
Additional file 10: Table S6.
DEGs associated with plant–pathogen interaction.
Additional file 11: Table S7.
Specific primers for differential gene sequences for qRT-PCR.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
Cite this article
Yang, X., Gu, X., Ding, J. et al. Gene expression analysis of resistant and susceptible rice cultivars to sheath blight after inoculation with Rhizoctonia solani. BMC Genomics 23, 278 (2022). https://doi.org/10.1186/s12864-022-08524-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-022-08524-6