
Abstract
Background
An important aspect of studying evolution is to understand how new species are formed and their uniqueness is maintained. Hybridization can lead to the formation of new species through reorganization of the adaptive system and significant changes in phenotype. Interestingly, eight stable strains of 2nNCRC derived from interspecies hybridization have been established in our laboratory. To examine the phylogeographical pattern of the widely distributed genus Carassius across Eurasia and investigate the possible homoploid hybrid origin of the Carassius auratus complex lineage in light of past climatic events, the mitochondrial genome (mtDNA) and one nuclear DNA were used to reconstruct the phylogenetic relationship between the C. auratus complex and 2nNCRC and to assess how demographic history, dispersal and barriers to gene flow have led to the current distribution of the C. auratus complex.
Results
As expected, 2nNCRC had a very close relationship with the C. auratus complex and similar morphological characteristics to those of the C. auratus complex, which is genetically distinct from the other three species of Carassius. The estimation of divergence time and ancestral state demonstrated that the C. auratus complex possibly originated from the Yangtze River basin in China. There were seven sublineages of the C. auratus complex across Eurasia and at least four mtDNA lineages endemic to particular geographical regions in China. The primary colonization route from China to Mongolia and the Far East (Russia) occurred during the Late Pliocene, and the diversification of other sublineages of the C. auratus complex specifically coincided with the interglacial stage during the Early and Mid-Pleistocene in China.
Conclusion
Our results support the origin of the C. auratus complex in China, and its wide distribution across Eurasia was mainly due to natural Pleistocene dispersal and recent anthropogenic translocation. The sympatric distribution of the ancestral area for both parents of 2nNCRC and the C. auratus complex, as well as the significant changes in the structure of pharyngeal teeth and morphological characteristics between 2nNCRC and its parents, imply that homoploid hybrid speciation (HHS) for C. auratus could likely have occurred in nature. The diversification pattern indicated an independent evolutionary history of the C. auratus complex, which was not separated from the most recent common ancestor of C. carassius or C. cuvieri. Considering that the paleoclimate oscillation and the development of an eastward-flowing drainage system during the Pliocene and Pleistocene in China provided an opportunity for hybridization between divergent lineages, the formation of 2nNCRC in our laboratory could be a good candidate for explaining the HHS of C. auratus in nature.
Similar content being viewed by others

Background
A fundamental question in evolutionary biology is whether speciation is gradual or punctuated [1]. Hybridization is regarded as an ‘evolutionary catalyst’ that may be an important source for the origination of a new species or an increase the amount of genetic variability. Hybridization can result in the reorganization of adaptive systems and lead to the formation of new species [2]. Hybridization between species can trigger vast genetic and genomic imbalances, including a high rate of DNA mutations and combinations [3], often resulting in significant changes in phenotypes and genotypes of hybrid offspring, which may facilitate speciation and adaptive radiation.
Drastic climatic and geological oscillations, such as glacial expansion and megadroughts, forced freshwater species to contract their distribution ranges and reside in small refuges in many cases. Potential gene exchange and hybrid speciation likely occurred owing to the close contact between distant species. Increasingly, studies suggest that interspecies hybridization and gene introgression are not uncommon in fish, such as in the families Poeciliidae, Atherinidae, Cyprinidae, Cobitidae, and even Carcharhinidae [4,5,6], especially East African cichlids, which have attracted much attention in recent years [7,8,9]. As the most common species of vertebrates, more than 34,000 fish species have been recorded (https://www.fishbase.de/). The formation of many fishes has been determined to be associated with hybridization [10, 11]. Ancestral hybridization has been suggested to have played a key role in facilitating species diversification of cichlid fish in Lake Malawi, Lake Victoria, and Lake Tanganyika [7,8,9].
However, for a long time, there has been a lack of enough direct evidence to prove that fish hybridization will produce new species. Distant hybridization can generate allotetraploid and autotetraploid fish, supplying important and direct evidence for hybridization speciation in fish [12]. It was reported that a newly born homodiploid raw fish (2nNCRC, 2n =100), which is very similar to the crucian carp with no beard, was derived from interspecies hybridization between Cyprinus carpio (female, 2n = 100) and Megalobrama amblycephala (male, 2n = 48) by artificial propagation [13]. Fortunately, we repeated this experiment three times with similar results in our laboratory. In addition, eight stable generations of 2nNCRC with a very clear genetic background have been founded in our laboratory since 2014, supplying a valuable model system for studying hybrid speciation in fish.
It is widely believed that five species exist in the genus Carassius, including Carassius carassius, Carassius cuvieri, Carassius langsdorfii, Carassius gibelio and Carassius auratus, which are generally accepted as valid species and they can be found in FishBase (https://www.fishbase.de/). Our previous study showed that the hybrid speciation would be a possible route of formation of C. auratus [13]. It is particularly interesting to find out whether the origin of C. auratus is identical to that of 2nNCRC and whether C. auratus evolved from 2nNCRC. First and foremost, it is necessary to clarify the historical evolutionary processes of C. auratus. Phylogeographic studies might address this issue because they are usually used to determine the historical processes that affect vicariance, dispersal, extinction and radiation that then lead to the geographic distributions of genetic lineages [14, 15].
The recent climatic oscillation and geological events during the Pliocene-Pleistocene have played a fundamental role in shaping contemporary patterns of biodiversity and species diversification and distribution [16, 17]. Pleistocene glaciations are known to have had a far-reaching influence on the evolution of organisms in the Northern Hemisphere [18, 19]. For example, paleontological and genogeographical studies indicate that European and North American species experienced repeated episodes of contraction and expansion of their ranges due to major climatic oscillations [20]. The Carassius species complex has a wide distribution across the Eurasian continent and neighboring islands [21, 22]. According to the fossil record of Carassius species in the Pliocene epoch (5.3–2.6 million years ago, Mya) discovered in northern China [23], Quaternary paleoenvironmental changes in East Asia and Europe had a great influence on the speciation and diversification of Carassius species. In freshwater fishes, the dynamics of recolonization are tightly linked to the history of river drainage systems [24]. During glacial melt periods, ephemeral rivers and periglacial lakes could arise, and the reconfiguration of the landscape caused by drastic climatic change and geological events may have allowed species to disperse into new habitats, providing opportunities for colonization and new species interactions.
These processes have resulted in complicated recolonization scenarios for the Carassius species complex in East Asia, where the haplotypes of the mitochondrial control region and tf alleles are clustered into four and three major lineages, respectively, and based on this information, it has been speculated that the Yangtze River basin was the potential origination center for the Carassius species complex then radiated across East Asia [22, 25]. Gao et al. [21] deduced that the close relationship between the C. auratus complex from eastern mainland China and south-central Ryukyus was the result of natural Pleistocene dispersal. However, the existence of two distinct lineages of C. carassius in Europe [24] was mainly due to the Danubian catchment being separated from other river systems by the Alps, the Sudetes Mountains and the Carpathian Mountains.
According to the formation of the newly born 2nNCRC, combined with the close phylogenetic relationship between the C. carpio and Carassius species complex [26, 27], we speculated that the existence of a possible route through distant hybridization under natural conditions may have generated C. auratus, and this hybridization likely occurred during the Pliocene–Pleistocene based on the following hypotheses: (1) 2nNCRC has a very close relationship with the C. auratus complex in terms of phenotype and genotype; (2) several geographic lineages of Carassius are endemic to specific regions of Eurasia, especially the C. auratus complex in China; and (3) the most recent common ancestor (TMRCA) of both Cyprinus and Megalobrama had a much earlier divergence time than that of the C. auratus complex, and all had a sympatric distribution in history. According to the distribution of current fish species and fossil data [23, 28], the speciation of C. auratus in China might have been punctuated and not derived from the processing of lineage differentiation with other species of Carassius. Thus, establishing a robust time-calibrated phylogeny is the first requirement for tracing the possible origin and diversification patterns of the Carassius species complex.
In the present study, the mitochondrial genome and nuclear DNA were used to reconstruct the phylogenetic relationship between 2nNCRC and the C. auratus complex. The cytb was the largest data set including the Genbank data, including the population information for the species of Carassius, were obtained and used to investigate the evolutionary history of the C. auratus complex.
Results
Morphological traits
Twelve to twenty samples of each generation of 2nNCRC were randomly selected to summarize the morphological characteristics of qualitative and quantitative traits (Table S1 in Supplementary Information). Both 2nNCRC and C. auratus have no barbells and almost the same quantitative traits. In particular, they have the same morphotype and pattern of pharyngeal teeth and four tabular teeth on each side of the pharyngeal bone, which are very different from those of C. carpio and M. amblycephala (Fig. 1). 2nNCRC (n = 30), C. auratus (n = 27), C. carpio (n = 38) and M. amblycephala (n = 28) were used to examine shape variation through principal component analysis (PCA) and canonical variance analysis (CVA). The results showed that the first three PCs explained 94.13% of the variation in shape morphology (Table S2 in Supplementary Information), while the first two CVs explained 97.94% of the variation (Table S3 in Supplementary Information). The scatter plot of CV1 and CV2 showed clear differentiation between 2nNCRC and its parents (Fig. 1) but a close relationship and partial overlap between 2nNCRC and C. auratus. The Mahalanobis distances based on geometrical morphology data showed a significant difference among 2nNCRC and the three species (P < 0.0001) (Table S4 in Supplementary Information), while the distance values between 2nNCRC and C. auratus were much lower than those between 2nNCRC and its parents.

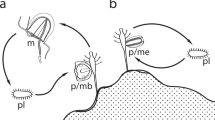
Scatter plots from the Canonical Variate Analysis (CVA) for C. auratus, 2nNCRC, C. carpio and M. amblycephala, and number (N) of each species was shown. A The pharyngeal teeth of C. auratus possessed four compressed teeth in each side. Bar = 0.25 cm. B The pharyngeal teeth of 2nNCRC same as (A). Bar = 0.25 cm (C) The pharyngeal teeth of C. carpio possessed rounded and ridged molariform teeth. Bar = 0.28 cm. D The pharyngeal teeth of M. amblycephala possesses recurved and uncinate teeth. Bar = 0.33 cm
Phylogenetic tree and haplotype network
The aligned and concatenated sequences (14 mitochondrial genes) were 13,634 bp with 5841 variable sites. Of these, 816 were parsimony-informative. The maximum likelihood (ML) and Bayesian tree showed a clear identification of the species of Carassius and most species of Cyprinidae (Fig. 2a). All species from Carassius containing 2nNCRC formed a monophyletic clade with high support values, and three valid species of Carassius were observed (Fig. 2a, in bold). However, 2nNCRC, together with C. gibelio, was unable to be separated from C. auratus but formed a single genetic cluster, and the genetic distances among 2nNCRC, C. auratus and C. gibelio were much lower than those among 2nNCRC and other species of Carassius (Table S5 in Supplementary Information). Consequently, C. auratus and C. gibelio are named the C. auratus complex in the present study.

a ML and Bayesian tree of Carassius with 2nNCRC and some representative Cyprinidae fish (including 29 specimens of Carassius, 19 of Cyprinus, 14 of Megalobrama and 23 of other cyprinid fish) using complete mitochondrial genomes based on the concatenated dataset (twelve proteins and two rRNAs); b ML and Bayesian tree of 2nNCRC and Cyprinidae (40 specimens representing 21 species) using the partial cds of HoxA2b. Numbers at nodes are maximum likelihood bootstrap values (up the branch) and Bayesian posterior probabilities (down the branch)
Another phylogenetic tree using HoxA2b (nuclear DNA) also clearly identified most species of Cyprinidae and two distinct clusters in the C. auratus complex with 2nNCRC, which were monophyletic and sister to Procypris merus and C. carpio. However, it was also impossible to distinguish between 2nNCRC and C. auratus (Fig. 2b).
The median joining network of cytb showed that there was no shared haplotype among the C. carassius, C. cuvieri, C. langsdorfii and C. auratus complexes (Fig. 3a). The haplotype of C. carassius was distributed in Europe with two distinct sublineages. The haplotype of C. cuvieri had a narrow distribution in Japan, and most of the haplotypes of C. langsdorfii were distributed in Japan with very few shared haplotypes (Fig. 3a). In addition, there were two shared haplotypes (H1 and H3) between 2nNCRC and C. auratus. However, the C. auratus complex had an admixture distribution across Eurasia with many shared haplotypes, such as H2, H4, H6 and H7, which could be found in Europe, China, Western Asia and Siberia (Fig. 3a). Surprisingly, a significant phylogeographical pattern with four specific regions was found for the C. auratus complex in China, two in Southeast China (Sublineage B1 in Fujian Province, Sublineage B4 in Taiwan Island), one in North China (Sublineage B2 in Amur River Basin), and one in the middle of the Yangtze River basin (Sublineage B5) (Fig. 3b).

Median-joining haplotype network based on a) 831 sequences of cytb including 15 2nNCRC and 816 specimens of Carassius (see Additional file 3 in Supplementary Information) derived from GenBank, where the specimens of Carassius in the network were found: A the Europe, except for the south of Alpes mountains and Danube River basin, B the south of Alpes mountains and Danube River basin in Europe, C Western Asia, D Siberia in Russia and Mongolia, E China, F Southeast Asia, G the Japan and Ryukyu Islands; and b) 128 sequences of cytb of C. auratus complex just distributed in China. The size of the circles represents haplotype frequency. Each connecting line represents a single nucleotide substitution, and each little short line represents mutated position. The haplotypes of different geographic samples were represented as different colors
Divergence time estimation
The fossil-calibrated molecular clock using the 14 incorporated genes from mitochondria showed that most clades were estimated to approach the earliest known fossils in time; for example, the TMRCA of Cyprinidae in the present study was approximately 44.07 Mya (Fig. 4a), close to the oldest and reliable fossils of Cyprinidae that are from the mid-Eocene (48.6 Mya, Fig. 4a) (Patterson, 1993). TMRCA of Schizothoracinae was dated at approximately 23.99 Mya (Fig. 4a), coinciding with the origin time of schizothoracine fishes estimated by Ruber et al. [29] and Wang et al. [30], in the Oligocene-Miocene boundary (approximately 23 Ma, Fig. 4a). The divergence between Cyprinus and Carassius was approximately 20.36 Mya (95% HPD: 21.69–19.04 Mya) (Fig. 4a). TMRCA of Megalobrama and Cyprinus was dated 6.5 Mya (95% HPD: 7.08–6.88 Mya) and 10.84 Mya (95% HPD: 11.68–9.98 Mya), respectively (Fig. 4a). Both of these values were much earlier than that of the C. auratus complex (3.12 Mya, Fig. 4a, b). The molecular data estimated by the incorporated 14 genes and by cytb only yielded similar results for the first divergence of C. carassius from the Carassius, at approximately 7.85 Mya (95% HPD: 8.43–7.30 Mya, Fig. 4a) and 8.08 Mya (95% HPD: 8.84–7.23 Mya, Fig. 4b), respectively. They both dated TMRCA of the C. auratus complex during the Late Pliocene (at approximately 3.12 Mya) with clear consistency, although the approaches used different datasets and fossil calibrations. However, there also existed a subtle difference between the two dates C. cuvier was separated at approximately 5.23 Mya (95% HPD: 5.42–5.04 Mya, Fig. 4a) using the 14 incorporated genes, while the TMRCA of C. cuvier and C. langsdorfi dated to approximately 4.67 Mya (95% HPD: 5.41–3.84 Mya, Fig. 4b) using only cytb; in addition, C. cuvier split from C. langsdorfi at approximately 4.03 Mya (95% HPD: 4.82–3.26 Mya, Fig. 4b).

a Dated phylogeny using the concatenated dataset (twelve proteins and two rRNAs) with three calibration points (red dots indicate time-calibration markers, ①: Cavender, 1986 [31]; ②: Liu and Chang, 2009 [32]; ③: Harrison, 1992 [33]), and b using cytb gene with two calibration points (red dots indicate time-calibration markers, ①: Liu and Su, 1962 [23]; ②: Jeffries et al., 2016 [24]) to estimate the divergence of Carassius. Node ages are shown together with 95% highest posterior density bars indicating a range of age estimates
Within the widely distributed C. auratus complex, several sublineages were found in Eurasia, and most were consistent with the six sublineages (sublineages B1-B6) distributed in China, except for sublineage A from the Far East (Russia) and Mongolia, which first diverged from the C. auratus complex at approximately 3.12 Mya (95% HPD: 3.81–2.44 Mya, Fig. 4b). Sublineage B1 from Fujian in China and Vietnam was estimated to have separated at approximately 1.6 Mya (95% HPD: 1.89–1.25 Mya, Fig. 4b), followed shortly thereafter by the divergence of sublineage B2, which was mostly distributed in the Amur River basin and Eurasia at approximately 1.49 Mya. Sublineage B3 was distributed in mainland China and Ryukyus and split at approximately 1.26 Mya. Two specific sublineages, B4 (on Taiwan Island) and B5 (in the Yangtze River basin), separated from the others at approximately 1.16 Mya (95% HPD: 1.39–0.85 Mya, Fig. 4b) and 0.86 Mya (95% HPD: 1.08–0.59 Mya, Fig. 4b), respectively. Sublineage B6 (widely distributed in mainland China and Europe) diverged at approximately 0.47 Mya (Fig. 4b).

Ancestral range reconstruction for C. auratus complex, Cyprinus and Megalobrama just distributed in China using 26 mtDNA. The colors of the charts correspond to the most likely ancestral areas inferred, and the black color means the unknown area. Letters represent the biogeographic regions for C. auratus complex: A the Yangtze River basin, B The Pearl River basin, C the Yellow River basin, D the River Basins of Southeastern China, E the Amur River basin, F River Basins of Southwestern China. The blue curves in the distribution map of Carassius mean the river systems
Ancestral range reconstruction
The results of the ancestral range reconstruction are depicted in Fig. 5 and Fig. S2 (in Supplementary Information). Among the six models of geographic-range evolution compared in the likelihood framework in Bio-GeoBEARS, the DIVA + j model was chosen for both 24 cytb sequences and 26 concatenated dataset (twelve proteins and two rRNAs in mitogenomes) according to the best likelihood and AICc associated scores (Table S6 in Supplementary Information). According to the ancestral area reconstruction using the 24 cytb gene across Eurasia, 41 dispersal and 21 vicariance events occurred within the evolution of the studied Carassius. TMRCA of Carassius probably diverged into two distinct clades (Fig. S2) in the late Miocene. One clade included C. carassius, which was mainly distributed in Europe, while the other clade further split into two separate lineages through two dispersal events and one vicariance event; the dispersal events corresponding to TMRCA of C. cuvieri, which was likely to disperse from Eurasia during early Pliocene according to the molecular dating (Fig. 4b). And then it diverged in two lineages through a vicariance event, the first one corresponding to lineages distributed in Japan (C. cuvieri and C. langsdorfii, node 43, G: 65.86%), and the second one corresponding to the lineage of C. auratus complex, whose ancestor was most likely to live in China (node 40). Three vicariance events occurred within the first lineage: a first one about 4.03 Ma separating C. cuvieri from C. langsdorfii, and other events during Pleistocene leading to different sublineages of C. langsdorfii across Eurasia. Within the second lineage, fifteen diversification events occurred: a vicariance event between Far East (Sulineage A) and China (Sublineage B) about 3.12 Mya, and fourteen events between 1.6–0.34 Mya leading to sublineages B1–B6. The dispersal events from the China to the Southeast Asia (Vietnam) explain the actual distribution of Sublineage B1 in both areas. The same condition can be found for the Sublineage B2 co-occur in the North China (Amur River Basin) and Far East, as well as the Sublineage B3 co-occur in China and Ryukyus and Japanese Islands. According to the ancestral area reconstruction using the 26 concatenated dataset of the C. auratus complex, Cyprinus and Megalobrama were distributed in China, and both TMRCAs of Cyprinus (node 42) and Megalobrama (node 50) were mainly distributed in the Pearl River basin, while TMRCA of the C. auratus complex was mainly distributed in the Yangtze River basin (node 31, A: 80.28%) (Fig. 5).
Discussion
This study represents the first attempt to better understand the relationship of artificial hybrid fish species and naturally evolving fish. 2nNCRC is very similar to C. auratus in terms of morphological characteristics based on qualitative and quantitative traits, as well as the geometric shape of fish. Our phylogenetic analysis showed that C. auratus and C. gibelio were unable to be separated from each other based on mtDNA and HoxA2b but were distinct from the other three species of Carassius. The intraspecies relationship between 2nNCRC and C. auratus complex was uncovered by both mtDNA and HoxA2b. The C. auratus complex was likely to have originated in China, and the admixture of mtDNA haplotype lineages across Eurasia was mainly due to natural Pleistocene dispersal and/or anthropogenic translocation.
Prerequisite conditions and ecological selection for the HHS of 2nNCRC
2nNCRC has two sets of chromosomes, one each from the maternal C. carpio, and the great morphologic and genetic variation in the parents can be seen as HHS [13]. Compared with allopolyploid hybrid speciation, HHS has at least two issues. First, reproductive isolation can easily be generated between the allopolyploid hybrid species and their parents due to the inaccuracy of chromosomal pairing and the inability to formulate germ cells [34], while reproductive isolation cannot be immediately generated between HHS and parents, such as 2nNCRC and C. carpio. Second, two complete genomes from their parents can be found in allopolyploid hybrid species; they possess a protective mechanism for the genome, while HHS lacks this mechanism. However, the F1 2nNCRC females and males are fertile, and eight stable strains of the new homoploid species have been established in our laboratory since 2014, suggesting that HHS not only is common in plants but also occurs in fish.
The prerequisite condition for HHS is that the distribution of parents should overlap in history, making hybrid speciation possible [35]. Both TMRCAs of Cyprinus and Megalobrama had a sympatric distribution in the Pearl River basin and Yangtze River basin (Fig. 5), and currently, they have a wide distribution with the C. auratus complex in these river systems. In particular, the connectivity between the Yangtze River and Pearl River system before the early Pleistocene would have facilitated the dispersion of freshwater species between the two river systems [36, 37]. Consequently, the connectivity of river systems and flooding mediated dispersion and encounters among freshwater species.
In addition, occupying a different and suitable ecological niche from parents is very important for HHS [7, 37], and this scenario which may be lost during continuous backcrossing with parents. Fortunately, the reproductive trait of 2nNCRC was the same as that of the C. auratus complex, both reaching sexual maturity at the age of 1 year [13]. C. carpio and M. amblycephala reach sexual maturity at least 2 years old. Furthermore, the same shape and dental pattern of pharyngeal teeth were found for 2nNCRC and the C. auratus complex (Fig. 1). The same compressed teeth in C. auratus and 2nNCRC suggested that they had the same omnivorous feeding habit but mainly fed on plant food (the stem and leaf of aquatic plants). However, rounded and ridged molariform teeth are found in C. carpio, which feeds on omnivores but mainly on animal food (freshwater snails and aquatic insects). M. amblycephala possesses recurved and uncinate teeth and mainly feeds on aquatic plants. In terms of freshwater aquatic resources, in comparison to C. carpio and M. amblycephala, the homoploid 2nNCRC and C. auratus have stronger adaptability, as they have a wider range of food in the aquatic ecosystem. Ecological selection is a major factor promoting HHS, and the foundation of a novel ecological niche supplies an opportunity for the evolution of new species while maintaining its own unique phenotype [37]. The family Cyprinidae exhibits species-specific numbers and arrangements of pharyngeal teeth, which are an important feeding organ and taxonomic characteristic, and extensive variation in tooth shape with high hereditary stability [38]. The same pharyngeal teeth were found among the C. auratus complex and different generations of 2nNCRC. Although the homoploid 2nNCRC and C. auratus showed very similar phenotypes, there were also significant differences between them, including the qualitative and quantitative traits found by Wang et al. [13] and the geometrical morphological shapes, which exhibited a significant humped back in 2nNCRC, found in the present study (Fig. 1). The morphological variation between 2nNCRC and C. auratus might be attributed to long-term morphological evolution.
Evolutionary relationships of the C. auratus complex with the homoploid 2nNCRC
In the phylogenetic tree, ML and Bayesian analyses based on the concatenated sequence produced essentially the same tree topology with high ML bootstrap values and Bayesian posterior probabilities. We determined that the robust tree topology adequately reflected the phylogeny of the species in Carassius and Cyprinus, as well as other species in Cyprinidae. Carassius clustered with Cyprinus first and then clustered with other species in Cyprinidae, Barbinae, Schizothoracinae and Labeoninae, all of which belong to Barbini, a monophyletic group in Cyprinidae, and this result was consistent with those of previous phylogenetic studies and the morphological characteristics of Cyprinidae [39, 40]. Carassius, as a monophyletic clade, included four definite interspecies clusters, and there was no shared haplotype among C. carassius, C. cuvieri, C. langsdorfii and other species, including C. gibelio and C. auratus (Fig. 3a). The haplotypes of C. carassius observed in Europe were separated into two clades without shared haplotypes, in accordance with the existence of two C. carassius lineages in Europe [24]. The haplotypes of C. cuvieri were restricted to mainland Japan, and the major haplotypes of C. langsdorfii observed across Eurasia were distributed on Japanese islands. Considering that the distribution of haplotypes was restricted to specific areas, we considered that the three species evolved independently for a considerable period of time on the main Japanese islands and Europe. However, the widely distributed species C. gibelio and C. auratus were paraphyletic, and they had many shared haplotypes. It has been documented that the triploid C. gibelio has a more shared haplotype with the sympatric diploid C. auratus than with allopatric triploids [41]. Previous studies have proposed that an autopolyploidy event might have resulted in the formation of C. gibelio (3n = 150) [42, 43]. Liu et al. [22] considered that the polyploid C. gibelio might have originated from the sympatric C. auratus (2n = 100) via autopolyploidy. However, it was reported that the autopolyploidy has not contributed to the separation between diploid C. auratus and triploid C. gibelio as they were both intermixed in the same lineages and share the same alleles [22, 25]. Although C. gibelio is naturally gynogenic, many phenomena similar to those in normal sexual diploid species, such as normal meiosis, multiple modes of unisexual gynogenesis and sexual reproduction, have been identified in some strains of C. gibelio [44, 45]. Hence, there exist the possibility of the breeding between diploid C. auratus and triploid C. gibelio in theory. Furthermore, the shared haplotype between the sympatric C. gibelio and C. auratus found at present and in previous studies confirmed the existence of gene flow between them. Consequently, it is likely that extensive gene flow and no substantial genetic separation exist between C. gibelio and C. auratus. Though the lack of genetic separation between them could be also the result of recent divergence, TMRCA of C. auratus complex and divergence between sublineage A and B, as well as among B1-B6 could be traced back to as early as the later Pliocene (Fig. 4b) indicating a complex evolutionary history for C. auratus complex.
The preliminary analysis of the nuclear genome of the homoploid 2nNCRC suggested that no characteristic exists that distinguishes the homoploid 2nNCRC from the diploid C. auratus [13]. The homoploid 2nNCRC was clustered with C. auratus based on mtDNA and nuclear DNA (HoxA2b) data. However, the genus Carassius was not a monophyletic group in the HoxA2b tree and showed a closer relationship between the C. auratus complex and C. carpio (Fig. 2b). The nuclear–mitochondrial discordance in the phylogenetic placement of C. auratus and C. cuvieri suggested a possible complex evolutionary history of the genus Carassius. Certainly, more nuclear markers and specimens are needed to further investigate the phylogenetic relationship in Carassius, and further studies including genomic data are required. We found six sublineages, and four had high regional specificity that were composed of endemic populations indigenous to each region in China (Fig. 3b), including Southeast China (Fujian), North China (Aumer River basin), Taiwan Island, and the middle and lower Yangtze River basin. However, a perplexing but concurrent event occurred in which the C. auratus complex found in Japan was not related to the Japanese crucian carp (C. langsdorfii or C. cuvieri) and the C. auratus complex widely distributed in Europe was not related to European crucian carp (C. carassius). The C. cuvieri was the monophyletic group (Fig. 2a) and was native to Japan [21]. However, the C. auratus complex in Japan was clustered with Chinese lineages. Similarily, most of the C. auratus complex across Europe examined in this study was nested in native Chinese lineages, and there were many shared haplotypes between Europe and China (Figs. 3a, 4b). The C. auratus complex distributed in Europe possibly originated via artificial introduction from Asia [41]. In contrast, genealogical analyses clustered the haplotype of C. gibelio distributed in Far East Russia (H29) with strong support, as well as two haplotypes with typical regions (H12 and H16) found in Ryukyus (Fig. 3a) and three specific haplotypes (H81, H82 and H83) found in Vietnam, suggesting that the C. auratus complex rapidly radiated from mainland China rather being distributed through recent anthropogenic translocation [41].
Biogeographic history of the C. auratus complex
Patterns of divergence suggest historical biogeographic events. Sublineage A from Far East Russia and Mongolia first split from the C. auratus complex at approximately 3.12 Mya (Fig. 3b), far preceding the Pleistocene glacial epoch in China, suggesting that dispersal from China rather than vicariance created the current distribution pattern of the C. auratus complex in Asia (Fig. 4a). With the humid and warm climate during the early Pliocene in North China [46], which supplied a suitable habitat for freshwater species, the well-dated Carassius fossil in North China has been recorded in the Pliocene epoch. Furthermore, recurrent glaciations during the Pleistocene were another contributor to species diversification in East Asia. The early separation of sublineage B1 at approximately 1.6 Mya exactly coincided with the Poyang Glaciation (1.8 ~ 1.5 Mya) in China [47, 48] and Gonzi Glaciation in Europe. Sublineage B1 only existed in warm and moist tropical or subtropical areas (Vietnam and Fujian, China), which provided important refugia for freshwater species during glaciations [21, 49]. Many haplotypes found in sublineage B2 and sublineage A were C. gibelio, while sublineage B2 has a closer genealogical relationship with the C. auratus complex in China; the separation of sublineage B2 in the Amur River basin of China occurred at approximately 1.49 Mya, far postdating the divergence of sublineage A, suggesting that sublineage B2 was the result of another dispersal event during the Poyang-Dagu interglacial stage (1.5–1.1 Mya) [47, 48]. Furthermore, during interglacial times, the warm, wet East Asian summer monsoons intensified, which also promoted the dispersion of the C. auratus complex from China to Ryukyus [21]. In sublineage B3, the divergence between the C. auratus complex in Ryukyus and China dated to approximately 0.84 Mya, exactly coinciding with the Poyang-Dagu interglacial stage (0.9 ~ 0.4 Mya) in the Mid-Pleistocene [47, 48], and this result was consistent with our previous speculation that natural dispersed from mainland China to Ryukyus occurred in the Pleistocene. Sublineages B3 and B6 distributed across mainland China may be the result of repeated episodes of range contraction and expansion during glacial cycling. Most of the eastward river systems in mainland China are known to have repeatedly anastomosed with one another during the postglacial and Holocene due to erosion and stream capture [50], especially with recent anthropogenic translocations. In turn, these events likely had a dramatic effect on the evolution of many freshwater species, and might have promoted genetic exchange between previously isolated C. auratus complex lineages and even removed any geographic signatures. A similar situation can be found in other aquatic organisms, such as freshwater snails and frogs [49, 51]. However, a specific sublineage, B5, was found in the middle and lower Yangtze River basin, as well as three other sublineages (B1, B2 and B4) in China with specific regions, suggesting that the genetic structure in the C. auratus complex has not been completely changed by recent human-mediated translocations [21].
C. auratus complex not a lineage differentiated from the common ancestor of C. cuvieri or C. carassius
Combined with the reconstruction of ancestral state and haplotype networks in the present study and a previous study on the population genetics of the C. auratus complex in China [21, 25], we concluded that the C. auratus complex originated in the Yangtze River basin in China (Fig. 5). TMRCA of C. carassius was mostly distributed in Europe, and both C. langsdorfii and C. cuvieri were on the main islands of Japan, essentially correspond to the results of a previous report on the distribution of some lineages of Carassius endemic to particular geographical regions [52,53,54,55,56].
It was believed that the freshwater ichthyofaunas from the islands of Japan and China resembled each other during the Miocene and Pliocene, as the main islands of Japan and mainland China are known to have formed a contiguous land mass in the late Pliocene [57]. Fossils indicate that in the late Pliocene, C. auratus was distributed in North China and Japan [23, 58], coinciding with the dating of TMRCA of the C. auratus complex in this study. Thus, we hypothesized that the C. auratus complex lineage should be separated from the C. cuvieri lineage based on molecular data and that a recent common ancestor existed for those distributed in Asia. The divergent and current distribution patterns of Carassius were mainly caused by dispersal that was aided by the formation of land bridges between Japanese archipelago islands and the continent [59, 60]. The common ancestor of the C. cuvieri and C. auratus complex likely dispersed from the Eurasian continent to the islands of Japan rather than in the reverse direction. Consequently, TMRCAs of C. cuvieri and C. auratus should be distributed on mainland China, which is incompatible with the present study (Fig. S2, node 44). Hence, there are reasons to believe that C. auratus did not diverge from TMRCA of C. cuvieri.
Similarly, TMRCA of C. carassius was distributed in Europe. However, the C. auratus complex currently distributed in Europe has been verified as dispersing from Asia by anthropogenic translocation. For instance, recent genetic evidence supports the anthropogenic introduction of the crucian carp to the UK during the fifteenth century [61]. C. carassius is native to parts of central, eastern and northern Europe and almost exclusively restricted to lentic ecosystems [56, 62]. A strong geographic structure was found because of the susceptibility of C. carassius to genetic isolation and bottlenecks due to their small population sizes and especially their low dispersal rates [24]. Two genetically distinct sublineages of C. carassius distributed in Europe were found in the present study and in a previous study [24]; therefore, the Carpathian Mountains and the Central European Highlands are inferred to have acted as barriers to the colonization of C. carassius in northern European drainages during the Pleistocene. According to the reconstructed ancestral areas, C. carassius was mainly distributed in Europe and restricted to its native areas (Fig. S2). Consequently, very few records of C. carassius distributed in Asia were found, except in West Siberia, Kazakhstan, Uzbekistan and Turkey, all of which are very close to Europe with no significant geographical barrier with Europe. However, the Central Siberian Plateau, Mongolian Plateau, Altay Mountains, Tianshan Mountains, Pamir Mountains, Himalayas, Iran Plateau and Great Caucasus, with elevations greater than 1000 m, formed natural dispersal barriers in the watersheds impeding freshwater species exchange. Furthermore, the separation of C. carassius from other species of Carassius far postdated the formation of high-elevation orogens such as the Tibetan Plateau, Mongolian Plateau and Tian-Shan, which occurred during the Miocene and even earlier [63]; in addition, well-dated Carassius fossils have been found in North China [23] that have far postdated the formation of these mountains, suggesting that C. auratus is not a lineage that diverged from TMRCA of C. carassius.
Paleoclimate and geological events facilitated distant hybridization
Based on the above analysis and the homoploid 2nNCRC derived from distant hybridization in our laboratory [13], our results also reinforce a possible naturally occurring hybrid origin of C. auratus. Rapid hybrid speciation was recently shown to occur by Lamichhaney et al. [64], and many species of cichlid fishes in East Africa have been verified as experiencing ancestral distant hybridization that has been suggested to play a key role in facilitating species diversification [7,8,9]. Our findings further supported that this mechanism was very likely to have occurred in the Cyprinidae family.
The robust, time-calibrated molecular phylogeny suggested that the separation of C. carpio and Carassius occurred at approximately 11.86 Mya (Fig. 4b), close to the divergence between goldfish and common carp (10.0 Mya) [65], and the origin of the common ancestor of common carp and crucian carp (11.4–8.1 Mya) [66], all of which occurred close to the time of the hybrid speciation of allotetraploid C. carpio (12.4 Mya) as estimated by Xu et al. [27]. Our results showed that extreme climate oscillations and geological events that occurred during the Pliocene and Pleistocene might have had a great influence on the speciation and radiation of the C. auratus complex in China.
Landscape features and geological history are hypothesized to strongly affect biogeographical processes, and this can be especially true in freshwater systems [67, 68]. One major consequence of extreme climatic changes and geological activities is ecosystem and landscape reconfigurations. A specifically large event was the drastic uplift of the Tibetan Plateau that which ultimately led to the formation of eastward-flowing drainage systems in China, such as the Yangtze River, which formed between the Oligocene and Miocene, but the unification of the upper and middle Yangtze River in the Three Gorges Mountain region occurred between the Late Pliocene and Early Pleistocene [69, 70]. The Yangtze River and Han River flowed into the middle of the Jianghan basin. The connection of the upper and middle Yangtze River facilitated species dispersal from west to east, similar to what was observed for the frog Quasipaa boulengeri [49] and the freshwater snail Bellamya aeruginosa [51, 71]. Therefore, the establishment of the eastward-flowing Yangtze River and the changes in river systems could have provided opportunities for dispersal, gene flow and even hybridization between closely related freshwater species, especially fish. Furthermore, the intensified tectonic movement and glacial cycling in the Pleistocene, as well as the prevailing monsoon, had major impacts on East Asian biota starting in the late Pliocene [72, 73]. As a result, the weather was cold and dry, and rivers and lakes experienced frequent range expansions and regressions in the Jianghan and Dongting Lake basins [74, 75]. The regression of lakes and rivers could dramatically reduce the habitats available to fish, and this precarious situation was further exacerbated in the glacial period. Consequently, restricted habitat regions caused hybridization between close and even divergent lineages. An example of a specifically large event is in the LGM, where the sea level was 130–150 m lower than that today, and this event resulted in strengthening downcutting processes of rivers. Most lakes in the plains region of eastern China have opened and dried, and this scenario has been verified in Taihu Lake, Boyang Lake and Dongting Lake [76]. The cooler and drier climates intensified during this period, and the fish habitats in these lakes were fragmented or even lost. Hybridization between divergent lineages would have been common when habitats were repeatedly lost, as the propagation of vast majority of freshwater fishes is oviparity, creating opportunities for new speciation and diversification, especially for sympatric species possessing similar ecological and breeding habits, such as the parents of 2nNCRC in the Yangtze River basin. Both C. carpio and M. amblycephala with sympatric distribution in history (Fig. 5) mostly occupy the middle and lower layers in freshwater, and they usually breed during May–June in every year [77]. A typical example of this scenario is the repeated range expansions and regressions of lakes that likely contributed to the high diversity of African cichlids [78, 79]. Since East Africa underwent dramatic climatic and geological changes in the Pleistocene over the past few million years, the constant expansion and regression of the great East African lakes have led to the repeated loss of habitats or the formation of new habitats [51, 79]. For example, hybridization might have facilitated speciation bursts for the cichlids in Lake Tanganyika, and time-calibrated trees support the concept that the radiation of Tanganyika cichlids coincided with lake formation [8]. Specially, the ancient hybridization of Lake Victoria cichlids is considered to be related to the capture of Malagarasi (Congo) tributaries and East African mega-droughts, providing an opportunity for the Congolese lineage to colonize the Lake Victoria region and the Congo-Nilotic admixture event to occur [7]. It has been speculated that hybridization between distant relatives, when coincident with ecological opportunity, may facilitate rapid adaptive radiation through recombination and sorting of admixture-derived polymorphisms by natural and sexual selection [7]. The C. auratus complex exhibits remarkable morphological and genetic diversity, and the different ploidy forms, unisexual gynogenesis and bisexual reproduction [21, 80, 81], might also benefit from hybridization-derived genetic variants. Future investigation will test for evidence of ancient admixture among the C. auratus complex, C. carpio, and M. amblycephala based on whole-genome sequencing data and the effect of ancestral hybridization on adaptive radiation, ecology and life history.
Conclusions
The newly born homodiploid 2nNCRC derived from the hybridization of C. carpio and M. amblycephala showed very similar phenotypic and genetic characteristics with the diploid C. auratus complex in nature. The very similar morphological characteristics and identical structures of the pharyngeal teeth between 2nNCRC and the C. auratus complex were totally different from those of C. carpio and M. amblycephala. The new structure of pharyngeal teeth suggested that 2nNCRC occupied a novel and suitable ecological niche from its parents, suggesting typical HHS in fish. Molecular phylogenetic analyses revealed an intraspecies relationship between 2nNCRC and the C. auratus complex. According to the reconstruction of ancestral areas and molecular data, the C. auratus complex lineage most likely originated from the Yangtze River basin in China during the Pliocene. The divergence pattern of the C. auratus complex in China was attributed to Pleistocene radiation, and the molecular data exactly coincided with the interglacial periods that occurred during the early and mid-Pleistocene in China. The diversification in the Carassius indicated that the C. auratus complex was impossible to derive from the recent common ancestor of C. carassius or C. cuvieri. HHS for 2nNCRC provides a good candidate speciation route for C. auratus, as the prerequisite condition of HHS and the hybridization between divergence lineages caused by the oscillation of the geologic climate during the Pliocene and Pleistocene could be satisfied in China.
Materials and methods
Samples and sequence data preparation
All of the 2nNCRC samples, including F1 to F5 generations used in this study, were cultured in ponds at the Protection Station of Polyploidy Fish, Hunan Normal University, and fed artificial feed. The F1 generation of 2nNCRC was denoted as 2nNCRC-F1, the F2 generation of 2nNCRC was the F1 self-crossed offspring and denoted as 2nNCRC-F2, and the naming procedure was followed for 2nNCRC-F3, 2nNCRC-F4 and 2nNCRC-F5. One specimen of each generation (F1 to F3) was collected, and a total of three mitochondrial genomes of 2nNCRC were sequenced to construct a phylogenetic tree based on two rRNAs and twelve protein-coding genes (ND6 was excluded) (the methods for DNA extraction, amplification, and sequencing are given in Additional file 1 in the Supplementary Information). To perform a comprehensive phylogenetic analysis, another 85 specimens of representative Cyprinidae fish (including 29 specimens of Carassius, 19 of Cyprinus, 14 of Megalobrama and 23 of other cyprinid fish) and 10 Catostomidae fish with complete mitochondrial genomes (both rRNAs and twelve protein-coding genes) were retrieved from GenBank (see Additional file 2 in Supplementary Information). In addition, a total of 831 cytb genes of the specimens (including three specimens in each generation of 2nNCRC-F1 to F5) in the genus Carassius widely distributed across Eurasia were also acquired from GenBank (see Additional file 3 in Supplementary Information) to obtain the phylogeographic structure of Carassius across Eurasia.
As very few nuclear DNA data are shared for Carassius and other Cyprinidae in GenBank, only HoxA2b was used to reconstruct the phylogenetic tree in present study. Two specimens of each generation (2nNCRC-F1 to F3) were sampled with two clones for each individual, and a total of 12 partial cds genes for HoxA2b were amplified (see Additional file 1 in Supplementary Information). In addition, the partial cds of HoxA2b for both parents (one for M. amblycephala and two for C. carpio) of 2nNCRC were also amplified, and 40 other specimens of Cyprinidae representing 21 species (see Additional file 4 in Supplementary Information) were obtained from GenBank in this study.
A previous study showed that the morphometrics of crucian carp-like fish (2nNCRC) were very similar to those of C. auratus in terms of barbel loss and a few lateral scales and even some measurable traits [13]. In this study, the morphological characteristics of other qualitative and quantitative traits were compared among the species of Carassius and 2nNCRC, as well as their parents (see Table S1 in Supplementary Information). To further test the morphological differences among 2nNCRC, C. auratus, C. carpio and M. amblycephala, geometric morphometrics was used to examine shape variation through principal component analysis (PCA) and canonical variance analysis (CVA) in MorphoJ v2.0 [82]. The left side of a specimen was placed on a moist foam mat at the base to maintain its natural configuration and photographed with a Nikon CoolPix 4500. Then, we digitized sixteen landmarks (see Fig. S1 in Supplementary Information) in each fish to capture the variation across the head and body shape, and the X and Y coordinates were recorded according to Zamani-Faradonbe et al. [83] by the software TpsDig2 [84]. For each specimen, the X and Y landmark coordinates were translated to the origin, rotated, scaled and superimposed by GPA (generalized Procrustes analysis) [85] in TpsRelwarp [86] to remove nonshape variations (such as location, orientation and scale). The specimen number of each species was more than 25 in this study. The CVA differentiates mutually exclusive groups by analyzing their relative positions in the morphospace and mapping shape differences onto the two first variates that explain the majority of the morphospace. This process is performed through the construction of a coordinate system called the canonical variates by determining the scores on those axes for all individuals, and they are referred to as eigenvalues [87].
Furthermore, pairwise genetic distances of inter- and intraspecific variation in the Carassius genus (see Table S5 in Supplementary Information) were calculated under the K2P model for the cytb dataset using MEGA 7 [88], as the cytb dataset is the most even of the datasets in GenBank.
Phylogenetic analysis
All sequences were aligned with MAFFT [89] as implemented in Geneious 4.8.3. For the 98 mitochondrial genomes, the species of Catostomidae (see Additional file 2 in Supplementary Information) that shared a more recent common ancestry with Cyprinidae than with any other extant group of Cypriniform based on the pharyngeal feeding structures [90] were used as outgroups, and all alignments (twelve proteins and two rRNAs) were combined. We tested the 14 genes’ data for saturation using DAMBE v. 6.4.41 [91]. The test revealed an ISS value that was significantly lower than the ISS.c in all cases (P < 0.0000), indicating the suitability of the fourteen genes for phylogenetic analysis. The homogeneity of the fourteen genes was tested in *PAUP [92], and the P value was 0.69 (> 0.05). The 57 partial cds of HoxA2b (nuclear DNA) included 12 clones of 2nNCRC and 43 specimens of other Cyprinidae species, as well as the outgroup Colossoma macropomum and Esox Lucius (see Additional file 4 in Supplementary Information). The alignments (the conservative sequence after removing GAP) were also tested for saturation using DAMBE v. 6.4.41, and the ISS-value was significantly lower than the ISS.c value in all cases (P < 0.0000). To test our first hypothesis that 2nNCRC is C. auratus, ML and Bayesian trees using both the HoxA2b and mitochondrial genomes (incorporating the 14-gene data) were generated using RAxML v. 8.0 [93] with 1000 bootstrap replicates and MrBayes v. 3.2 [94], respectively. In the phylogenetic tree analysis, we determined the best-fitting substitution models for each gene fragment using Moldetest 3.7 [95], which are shown in Table S6 in the Supplementary Information. Mixing of the MCMC chains of the two independent runs was monitored with TRACER v. 1.7.1 [96], and the analysis was terminated after the average standard deviation of the split frequencies fell to less than 0.01. The first 25% of the sampled approximately 20 million generations were discarded as burn-in. The final trees were visualized in FIGTREE v. 1.4.4 [97].
To test our second hypothesis that several geographic lineages of Carassius endemic to specific regions of Eurasia exist, especially for C. auratus in China, we generated two haplotype networks using 831 cytb genes for all in Eurasia and 128 cytb for samples only in China and colored each haplotype by the geographic region from which it was collected (see Additional file 3 in Supplementary Information). Only the cytb gene was used in the haplotype network, as it had the most abundant data with population information for the five species of Carassius and is often used for population genetics in fish. The haplotype networks were constructed using network v. 10 (www.fluxus-engineering.com/sharenet.htm) and applying the median-joining and maximum parsimony options.
Divergence time estimation
To test our third hypothesis that C. auratus originated from the hybridization between C. carpio and M. amblycephala in China, the precondition would need to be that TMRCA of both Cyprinus and Megalobrama had a much earlier divergence than that of the C. auratus complex, and all would have had to have a sympatric distribution in history. Furthermore, these lineages of the C. auratus complex distributed in Eurasia should belong to native lineages distributed in China. The divergence time was estimated using a molecular clock approach as implemented in BEAST. We used the combined data (98 specimens) including twelve proteins and two rRNAs in the phylogenetic tree and employed a (uncorrelated lognormal) relaxed clock, as a likelihood-ratio test (LRT) rejected the strict molecular clock hypothesis for the data (P < 0.01). As the lack of credible and exact fossil data can be found for the species in this study, we used a conservative approach by choosing three calibration points with normal distribution priors: the oldest fossil of Plesiomyxocyprinus arratiae, which is similar to Myxocyprinus asiaticus, was constrained from the middle Eocene or earlier, approximately 40–38 Mya [32], which is often used as the calibration point of M. asiaticus [30, 90]; the minimum age of Catostomidae is 60 Ma based on a catostomid fossil from the Paleocene [31]; and the timing of the drastic uplift of the Tibet Plateau occurred between 25 and 17 Mya [33] and was utilized as one calibration point for the Schizothoracinae as the species endemic to Qinghai-Tibet Plateau. The clade corresponding to each calibration point was constrained to be monophyletic. The GTR + I + G model was the best fit for the combined dataset (Table S6 in Supplementary Information). The ‘speciation: Yule process’ tree prior was used to construct the tree. We ran four independent runs for 20 million generations, logging trees every 2000 generations. Convergence was checked with TRACER v. 1.7.1. Posterior trees from the four runs were combined after removing the first 10% as burn-in in LogCombiner v. 2.5.2 (http://beast.bio.ed.ac.uk/logcombiner). The maximum credibility tree was created in TreeAnnotator v. 2.5.2 available in the BEAST package.
Furthermore, another divergence time was estimated using the cytb data. We used a reduced dataset of cytb: specimens for which two or more sequences from the same region were included. This approach resulted in 247 terminals – 237 representatives of the five species of Carassius and 13 outgroup sequences (C. carpio) from GenBank (see Additional file 5 in Supplementary Information). To minimize error, we used a conservative approach by employing calibration points from previous studies [24]. TMRCA of C. carassius in the northern and central-eastern European drainages and the Danubian catchment was constrained to 2.18–2.15 Mya. Another calibration point was the fossil of Carassius in the Pliocene epoch (5.3–2.6 Mya) in northern China (Yushe, Shanxi Province). The GTR + I + G model was also the best fit for the cytb dataset. The ‘speciation: Yule process’ tree prior was used to construct the tree. We ran four independent runs for 50 million generations, logging trees every 5000 generations. The other settings were same as those noted above.
Reconstruction of ancestral areas
We further performed a biogeographic reconstruction of ancestral areas for species of Carassius using BioGeoBEARS [98] in RASP 4.02 [99] to determine whether overlapping distributions existed between C. auratus and other species of Carassius. The analyses were conducted on a fully resolved topology from the BEAST analysis containing 24 cytb sequences and five species of Carassius (see Additional file 6 in Supplementary Information). Seven major geographical areas were defined based on the worldwide distribution of Carassius according to its current distribution and Jeffries et al. [24]: (A) Europe, except for the southern Alpes Mountains and Danube River basin, (B) the southern Alpes Mountains and Danube River basin in Europe, (C) Western Asia, (D) Siberia in Russia and Mongolia, (E) China, (F) Southeast Asia, and (G) East Asia—Japan. To confirm that TMRCA of both Cyprinus and Megalobrama had a sympatric distribution in China, another biogeographic reconstruction of ancestral areas for Cyprinus, Megalobrama and C. auratus complex just distributed in China was conducted using the mitochondrial genome (twelve proteins and two rRNAs) of 26 specimens (the detailed sample location for each please see Additional file 7) representing 12 individuals of Cyprinus, 8 individuals of Megalobrama, and 6 individuals of Carassius. Six major geographical areas were defined based on their current distribution in China: (a) the Yangtze River basin, (b) the Pearl River basin, (c) the Yellow River basin, (d) the river basin of southeastern China, (e) the Amur River basin, and (f) the river basin of southwestern China.
All six models of geographic range evolution were compared in a likelihood framework (DEC, DEC + j, DIVALIKE, DIVALIKE + j, BAYAREALIKE and BAYAREALIKE + j). The best-fit model was assessed by comparing Akaike’s information criterion and likelihood-ratio tests, and DIVALIKE + j was chosen (see Table S7 in Supplementary Information). To account for phylogenetic uncertainty, 4000 post burn-in trees resulting from the BEAST analysis were integrated for inference. The maximum number of ancestral areas was set to four, as C. auratus and C. carpio can be widespread.
Availability of data and materials
Many genetic data were obtained through the GenBank (Additional file 2 and file 3 in Supplementary Information), and the new sequences data for 2nNCRC have been submitted in GenBank. The new datasets generated and analysed during the current study are available in the GenBank (https://www.ncbi.nlm.nih.gov/nuccore/?term=), and the accession number was OM240550-OM240576 (see Additional files 3 and 4 in Supplementary Information).
References
Harini BP, Ramachandra NB. Evolutionary experimentation through hybridization under laboratory condition in Drosophila: evidence for Recombinational speciation. BMC Evol Biol. 2003;3:20.
Comeault AA, Matute DR. Genetic divergence and the number of hybridizing species affect the path to homoploid hybrid speciation. Proc Natl Acad Sci. 2018;115:9761–6.
Liu S, Luo J, Chai J, Ren L, Zhou Y, Huang F, et al. Genomic incompatibilities in the diploid and tetraploid offspring of the goldfish × common carp cross. Proc Natl Acad Sci. 2016;113:1327–32.
Dowling TE, Secor CL. The role of hybridization and introgression in the diversification of animals. Annu Rev Ecol Syst. 1997;28:593–619.
Kwan YS, Ko MH, Jeon YS, Kim HJ, Won YJ. Bidirectional mitochondrial introgression between Korean cobitid fish mediated by hybridogenetic hybrids. Ecol Evol. 2019;9:1244–54.
Pazmino DA, van Herderden L, Simpfendorfer CA, Junge C, Donnellan SC, Hoyos-Padilla EM, et al. Introgressive hybridisation between two widespread sharks in the East Pacific region. Mol Phylogenet Evol. 2019;136:119–27.
Meier JI, Marques DA, Mwaiko S, Wagner CE, Excoffier L, Seehausen O. Ancient hybridization fuels rapid cichlid fish adaptive radiations. Nat Commun. 2017;8:14363.
Irisarri I, Singh P, Koblmüller S, Torres-Dowdall J, Henning F, Franchini P, et al. Phylogenomics uncovers early hybridization and adaptive loci shaping the radiation of Lake Tanganyika cichlid fishes. Nat Commun. 2018;29:3159.
Svardal H, Quah FX, Malinsky M, Ngatunga BP, Miska EA, et al. Ancestral hybridization facilitated species diversification in the Lake Malawi cichlid fish adaptive radiation. Mol Biol Evol. 2019;37:1100–13.
Wang S, Tang C, Tao M, Qin Q, Zhang C, Luo K, et al. Establishment and application of distant hybridization technology in fish. Sci China Life Sci. 2019;62:22–45.
Liu S. Fish distant hybridization. Beijing: China Social Sciences Press; 2014.
Liu S, Duan W, Tao M, Zhang C, Sun Y, Shen J, et al. Establishment of the diploid gynogenetic hybrid clonal line of red crucian carp × common carp. SCI China Ser C. 2007;50:186–93.
Wang S, Ye X, Wang Y, Chen Y, Lin B, Yi Z, et al. A new type of homodiploid fish derived from the interspecific hybridization of female common carp × male blunt snout bream. SCI Rep UK. 2017;7:4189.
Avise JC. Phylogeography: retrospect and prospect. J Biogeogr. 2009;36:3–15.
Hickerson MJ, Carstens BC, Cavender-Bares J, Crandall KA, Graham CH, Johnson JB, et al. Phylogeography's past, present, and future: 10 years after Avise, 2000. Mol Phylogenet Evol. 2010;54:291–301.
Ivory SJ, Blome MW, King JW, McGlue MM, Cole JE, Cohen AS. Environmental change explains cichlid adaptive radiation at Lake Malawi over the past 1.2 million years. Proc Natl Acad Sci. 2016;113:11895–900.
Jensen MP, Fitzsimmons NN, Bourjea J, Hamabata T, Reece J, Dutton PH. The evolutionary history and global phylogeography of the green turtle (Chelonia mydas). J Biogeogr. 2019;46:860–70.
Song K, Gao B, Halvarsson P, Fang Y, Jiang YX, Sun YH, et al. Genomic analysis of demographic history and ecological niche modeling in the endangered Chinese grouse Tetrastes sewerzowi. BMC Genomics. 2020;21:581.
Dussex N, Alberti F, Heino MT, Olsen RA, van der Valk T, Ryman N, et al. Moose genomes reveal past glacial demography and the origin of modern lineages. BMC Genomics. 2020;21:854.
Hewitt GM. The genetic legacy of the quaternary ice ages. Nature. 2000;405:907–13.
Gao Y, Wang SY, Luo J, Murphy RW, Du R, Wu SF, et al. Quaternary palaeoenvironmental oscillations drove the evolution of the Eurasian Carassius auratus complex (Cypriniformes, Cyprinidae). J Biogeogr. 2012;39:2264–78.
Liu XL, Li XY, Jiang FF, Wang ZW, Li Z, Zhang XJ, et al. Numerous mitochondrial DNA haplotypes reveal multiple independent polyploidy origins of hexaploids in Carassius species complex. Ecol Evol. 2017;7:10604–15.
Liu HT, Su TT. Pliocene fishes from Yüshe Basin, Shansi. Vertebrata PalAsiatica. 1962;6:1–125 (in Chinese with English abstract).
Jeffries DL, Copp GH, Handley LL, Olsén KH, Sayer CD, Hänfling B. Comparing RADseq and microsatellites to infer complex phylogeographic patterns, an empirical perspective in the crucian carp, Carassius carassius, L. Mol Ecol. 2016;25:2997–3018.
Liu XL, Jiang FF, Wang ZW, Li XY, Li Z, Zhang XJ, et al. Wider geographic distribution and higher diversity of hexaploids than tetraploids in Carassius species complex reveal recurrent polyploidy effects on adaptive evolution. SCI Rep UK. 2017;7:5395.
Li J, Wang X, Kong X, Zhao K, He S, Mayden RL. Variation patterns of the mitochondrial 16S rRNA gene with secondary structure constraints and their application to phylogeny of cyprinine fishes (Teleostei: Cypriniformes). Mol Phylogenet Evol. 2008;47:472–87.
Xu P, Xu J, Liu G, Chen L, Zhou Z, Peng W, et al. The allotetraploid origin and asymmetrical genome evolution of common carp Cyprinus carpio. Nat Commun. 2019;10:4625.
Huang WL. The geologic distribution of fish fossiles in China. Chin J Geol. 1960;5:249–55 (in Chinese).
Ruber L, Kottelat M, Tan HH, Ng PK, Britz R. Evolution of miniaturization and the phylogenetic position of Paedocypris, comprising the world’s smallest vertebrate. BMC Evol Biol. 2007;7:38.
Wang Y, Shen YJ, Feng CG, Zhao K, Song ZB, Zhang YP, et al. Mitogenomic perspectives on the origin of Tibetan loaches and their adaptation to high altitude. SCI Rep UK. 2016;6:29690.
Cavender TM. Review of the fossil history of north American freshwater fishes. In: Hocutt CH, Wiley EO, editors. The zoogeography of north American freshwater fishes. New York: Wiley; 1986. p. 699–724.
Liu J, Chang MM. A new Eocene catostomid (Teleostei: Cypriniformes) from northeastern China and early divergence of Catostomidae. Sci China Ser D. 2009;52:189–202.
Harrison TM, Copeland P, Kidd W, Yin A. Raising tibet. Science. 1992;255:1663–70.
Soltis PS, Soltis DE. The role of hybridization in plant speciation. Annu Rev Plant Biol. 2009;60:561–88.
Gross BL, Rieseberg LH. The ecological genetics of homoploid hybrid speciation. J Hered. 2005;96:241–52.
Miao W, Yu YH, Shen YF, Zhang XY. Intraspecific phylogeography of Carchesium polypinum (Peritrichia, Ciliophora) from China, inferred from 18S-ITS1-5.8S ribosomal DNA. Science in China Ser. C. Life Sci. 2004;47:11–7.
Gross BL. Genetic and phenotypic divergence of homoploid hybrid species from parental species. Heredity. 2012;108:157–8.
Stock DW. Zebrafish dentition in comparative context. J Exp Zool Part B. 2007;308B:523–49.
Wang X, Gan X, Li J, Mayden RL, He S. Cyprinid phylogeny based on Bayesian and maximum likelihood analyses of partitioned data: implications for Cyprinidae systematics. Sci China Life Sci. 2012;55:761–73.
Kong XH, Wang XZ, Gan XN, Li JB, He SP. Phylogenetic relationships of Cyprinidae (Teleostei: Cypriniformes) inferred from the partial S6K1 gene sequences and implication of indel sites in intron 1. Sci China Ser C Life Sci. 2007;50:780–8.
Takada M, Tachihara K, Kon T, Yamamoto GJ. Biogeography and evolution of the Carassius auratus-complex in East Asia. BMC Evol Biol. 2010;10:7.
Li XY, Zhang XJ, Li Z, Hong W, Liu W, Zhang J, et al. Evolutionary history of two divergent Dmrt1 genes reveals two rounds of polyploidy origins in gibel carp. Mol Phylogenet Evol. 2014;78:96–104.
Luo J, Gao Y, Ma W, Bi XY, Wang SY, Wang J, et al. Tempo and mode of recurrent polyploidization in the Carassius auratus species complex (Cypriniformes, Cyprinidae). Heredity. 2014;112:415–27.
Zhang J, Sun M, Zhou L, Li Z, Liu Z, Li XY, et al. Meiosis completion and various sperm responses lead to unisexual and sexual reproduction modes in one clone of polyploid Carassius gibelio. Sci Rep UK. 2015;5:10898.
Li XY, Zhang QY, Zhang J, Zhou L, Li Z, Zhang XJ, et al. Extra microchromosomes play male determination role in polyploidy gibel carp. Genetics. 2016;203:1415–24.
Xiong J, Li Y, Zheng W, Zhang P, Lei J, Zhong Y, et al. Climatically driven formation of the Tangxian planation surface in North China: an example from northwestern Zhongtiao Shan of the Shanxi Graben system. Lithosphere. 2018;10:530–44.
Xing LS. Magnetostratigraphic age of quaternary glaciations in the Lushan area. Bull Inst Geomechanics Cags. 1989;13:71–9 (in Chinese).
Jing CR, Liu HP. On the glacial and interglacial stages in quaternary of China. J Chengdu Univ Technol. 1999;26:97–100.
Yan F, Zhou WW, Zhao HT, Yuan ZY, Wang YY, Jiang K, et al. Geological events play a larger role than Pleistocene climatic fluctuations in driving the genetic structure of Quasipaa boulengeri (Anura: Dicroglossidae). Mol Ecol. 2013;22:1120–33.
Yap SY. On the distributional patterns of Southeast–East Asian freshwater fish and their history. J Biogeogr. 2002;29:1187–99.
Gu QH, Husemann M, Wu HH, Dong J, Zhou CJ, Wang XF, et al. Phylogeography of Bellamya (Mollusca: Gastropoda: Viviparidae) snails on different continents: contrasting patterns of diversification in China and East Africa. BMC Evol Biol. 2019;19:82.
Cherfas NB. Gynogenesis in fishes. Genetic bases of fish selection (ed. by V.S. Kirichnikov). Berlin: Springer-Verlag; 1981. p. 255–73.
Gui JF. Retrospects and prospects of studies on the mechanism of natural gynogenesis in silver crucian carp (Carassius auratus gibelio). Bull Natl Nat Sci Found China. 1997;1:11–6 (in Chinese with English abstract).
Meng QW, Su JX, Miao XZ. Fish taxonomy. Beijing: China Agriculture Press; 1995.
Luo J, Zhang YP, Zhu CL, Xiao WH, Huang SY. Genetic diversity in crucian carp (Carassius auratus). Biochem Genet. 1999;37:267–79.
Copp GH, Černý J, Kováč V. Growth and morphology of an endangered native freshwater fish, crucian carp Carassius carassius, in an English ornamental pond. Aquat Conserv. 2008;18:32–43.
Kimura M, editor. The formation of the Ryukyu arc and migration of biota to the arc. Naha: Okinawa Times; 2002. (in Japanese)
Chang MM, Chen YY. Late Mesozoic and tertiary ichthyofaunas from China and some puzzling patterns of distribution. Vertebrata PalAsiatica. 2000;38:161–75.
Park YC, Kitade O, Schwarz M, Kim JP, Kimm W. Intraspecific molecular phylogeny, genetic variation and phylogeography of Reticulitermes speratus (Isoptera: Rhinotermitidae). Mol Cells. 2006;21:89–103.
Dufresnes C, Litvinchuk SN, Borzee A, Jang Y, Li JT, Miura I, et al. Phylogeography reveals an ancient cryptic radiation in east-Asian tree frogs (Hyla japonica group) and complex relationships between continental and island lineages. BMC Evol Biol. 2016;16:253.
Jeffries DL, Copp GH, Maes GE, Lawson Handley L, Sayer CD, Hänfling B. Genetic evidence challenges the native status of a threatened freshwater fish (Carassius carassius) in England. Ecol Evol. 2017;7:2871–82.
Copp GH. Typology of aquatic habitats in the great ouse, a small regulated lowland river. Regul River. 1991;6:125–34.
Sun H, Liu XD. Impacts of the uplift of four mountain ranges on the arid climate and dust cycle of inland Asia. Palaeogeogr Palaeocl. 2018;505:167–79.
Lamichhaney S, Han F, Webster MT, Andersson L, Grant BR, Grant PR. Rapid hybrid speciation in Darwin's finches. Science. 2018;359:224–7.
Luo J, Chai J, Wen YL, Tao M, Lin GL, Liu XC, et al. From asymmetrical to balanced genomic diversification during rediploidization: subgenomic evolution in allotetraploid fish. Sci Adv. 2020;6:eaaz7677.
Yuan J, He Z, Yuan X, Jiang X, Sun X, Zou S. Speciation of Polyploid Cyprinidae fish of common carp, Crucian carp, and silver Crucian carp derived from duplicated Hox genes. J Exp Zool Part B. 2010;314B:445–56.
Cook BD, Kennard MJ, Real K, Pusey BJ, Hughes JM. Landscape genetic analysis of the tropical freshwater fish Mogurnda mogurnda (Eleotridae) in a monsoonal river basin: importance of hydrographic factors and population history. Freshw Biol. 2011;56:812–27.
Ferreira M, Aleixo A, Ribas CC, Santos MPD. Biogeography of the Neotropical genus Malacoptila (Aves: Bucconidae): the influence of the Andean orogeny, Amazonian drainage evolution and palaeoclimate. J Biogeogr. 2017;44:748–59.
Fan DD, Li CX. Reviews on researches of timing of the Yangtze draining the Tibetan plateau to the East China Sea. Mar Geol Quaternary Geol. 2007;27:121–31 (in Chinese with English abstract).
Zheng HB, Wang P, He MY, Luo C, Huang XT, Jia JT. Timing of the establishment of the east-flowing Yangtze River and tectonic-geomorphic implications. Quaternary Sci. 2013;33:621–30 (in Chinese with English abstract).
Gu QH, Husemann M, Ding BQ, Luo Z, Xiong BX. Population genetic structure of Bellamya aeruginosa (Mollusca: Gastropoda: Viviparidae) in China: weak divergence across large geographic distances. Ecol Evol. 2015;5:4906–19.
Zhisheng A, Kutzbach JE, Prell WL, Porter SC. Evolution of Asian monsoons and phased uplift of the Himalaya-Tibetan plateau since Late Miocene times. Nature. 2001;2001(411):62–6.
Xin SZ, Shen J, Zhang WF, Sun WW, Xiao XY. East Asian winter monsoon evolution since the late Pliocene based on a pollen record from Lake Xingkai, Northeast Asia. Quat Res. 2020;93:40–59.
Bai DY, Li CA. Status of quaternary geology research of Dongting Basin. Geol Sci Technol Inf. 2010;29:1–12.
Wang MM. A study on developmental mechanism and tectonic evolution of the Hanzhong Basin. Dissertation, Beijing: Institute of Geology, China Earthquake Administration; 2013.
Shen J. Spatiotemporal variations of Chinese lakes and their driving mechanisms since the last glacial maximum: a review and synthesis of lacustrine sediment archives. Chin Sci Bull. 2013;58:17–31.
Chen P. Ecological niche modeling as a predictive tool: Asiatic freshwater fishes in North America. (PhD thesis). Lawrence: University of Kansas; 2008.
Koblmüller S, Egger B, Sturmbauer C, Sefc KM. Rapid radiation, ancient incomplete lineage sorting and ancient hybridization in the endemic Lake Tanganyika cichlid tribe Tropheini. Mol Phylogenet Evol. 2010;55:318–34.
Danley PD, Husemann M, Ding B, DiPietro LM, Beverly EJ, Peppe DJ. The impact of the geologic history and paleoclimate on the diversification of east African cichlids. Int J Evol Biol. 2012;2012:574851.
Gui JF, Zhou L. Genetic basis and breeding application of clonal diversity and dual reproduction modes in polyploid Carassius auratus gibelio. Sci China Life Sci. 2010;53:409–15.
Gui JF, Zhu ZY. Molecular basis and genetic improvement of economically important traits in aquaculture animals. Sci Bull. 2012;57:1751–60.
Klingenberg CP. MorphoJ: an integrated software package for geometric morphometrics. Mol Ecol Resour. 2011;11:353–7.
Zamani-Faradonbe M, Keivany Y, Dorafshan S. Phenotypic diversity in Hari garra, garra rossica (Nikol'skii, 1900) populations (Cyprinidae) in Iranian river systems. J Wildl Biodivers. 2019;3:1–9.
Rohlf FJ. tpsDig, digitize landmarks and outlines, version 2.05. Department of Ecology and Evolution, State University of New York at Stony Brook; 2005.
Rohlf FJ, Slice DE. Extensions of the Procrustes method for the optimal superimposition of landmarks. Syst Zool. 1990;39:40–59.
Rohlf FJ. tpsDig, tpsRegr, tpsRelw. 2004–2007. Distributed by the author at http://life.bio.sunysb.edu/morph/.
Zelditch ML, Swiderski DL, Sheets DH. Geometric morphometrics for biologists: a primer. 2nd ed. Waltham: Academic; 2012.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2006;33:1870–4.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80.
Tao WJ, Yang L, Mayden RL, He SP. Phylogenetic relationships of Cypriniformes and plasticity of pharyngeal teeth in the adaptive radiation of cyprinids. Sci China Life Sci. 2019;62:553–65.
Xia X. DAMBE5: a comprehensive software package for data analysis in molecular biology and evolution. Mol Biol Evol. 2013;30:1720–8.
Swofford DL. PAUP*. Phylogenetic analysis using parsimony (*and other methods). Version 4. Sunderland (Massachusetts): Sinauer Associates; 2002.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3.
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–42.
Posada D, Crandall KA. MODELTEST: testing the model of DNA substitution. Bioinformatics (Oxford, England). 1998;14:817–8.
Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA. Posterior summarization in Bayesian Phylogenetics using Tracer 1.7. Syst Biol. 2018;67:901–4.
Rambaut A. FigTree v1.4.4 2006-2018: Tree Figure Drawing Tool; 2018. Available at: http://tree.bio.ed.ac.uk/software/figtree/.
Matzke N. BioGeoBEARS: BioGeography with Bayesian (and likelihood) evolutionary analysis in R scripts. Berkeley: University of California; 2013.
Yu Y, Harris AJ, Blair C, He XJ. RASP (reconstruct ancestral state in phylogenies): a tool for historical biogeography. Mol Phylogenet Evol. 2015;87:46–9.
Acknowledgements
We thank reviewers for very valuable comments on previous versions of the manuscript. All authors approved the final version of the manuscript.
Funding
This work received funding and design inspiration from Natural Science Foundation of Hunan (Grant No. 2021JJ30442), National Natural Science Foundation of China (Grant No. 31730098, U19A2040, 31802287) and the China Postdoctoral Science Foundation (Grant No. 2018 M632969), the High-Level Talent Agglomeration Program of Hunan, China (Grant No. 2019RS1044), the Key Research and Development Program of Hunan Province (Grants No. 2018NK2072) and the Hunan Provincial Natural Science and Technology Major Project (Grant No.2017NK1031), the earmarked fund for China Agriculture Research System (Grant No. CARS-45), 111 Project (D20007), supply enough funding and equipment for the cultivation of crucian carp-like fish in our lab.
Author information
Authors and Affiliations
Contributions
S.J.L conceived and designed the study, Q.H.G drafted the manuscript, analyzed the data, and co-wrote the manuscript. S.W made fish measurements, and carried out the molecular laboratory work, and co-wrote the manuscript, H.Z. and Y.H. carried out tissue dissection, DNA isolation, sample preparation for sequencing, and co-wrote the manuscript. J.L.Y, C.H.Y., X.X.H, X.W.X., Y.D.W., Z.H.W. and J.W. carried out the molecular laboratory work and collect the mtDNA data from GenBank. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
We confirm that all methods were carried out in accordance with relevant guidelines and regulations. All methods are reported in accordance with ARRIVE guidelines (https://arriveguidelines.org). In this study, many genetic data were obtained through the Genbank (see Additional files 2, 3, 4), while some samples (homoploid 2nNCRC, Cyprinus carpio and Megalobrama amblycephala) used in this study were cultured in ponds at the State Key Laboratory of Developmental Biology of Freshwater Fish, Hunan Normal University, and fed adequate artificial feed. Before taking pterygiophore and muscle tissue, the fish were deeply anaesthetized with 100 mg/L MS-222 (Sigma-Aldrich, St. Louis, MO, USA) prior to dissection. And they were snap-frozen in liquid nitrogen and stored at − 80 °C before use. Fish work was performed in strict accordance with the recommendations in the Guidelines for the Care and Use of Laboratory Animals of the National Advisory Committee for Laboratory Animal Research in China and approved by the Animal Care Committee of Hunan Normal University (Permit Number: 4237).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing of interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
DNA extraction, PCR amplification, cloning and sequencing.
Additional file 2.
Sequence information including the GenBank accession number, species name and ID for reconstructing the phylogenetic tree based on 98 mitochondrial genome.
Additional file 3.
Sequence information including the GenBank accession number, species name, sampling location of 831 cytb for haplotype network.
Additional file 4.
Sequence information including the GenBank accession number, species name and ID for reconstructing the phylogenetic tree based on HoxA2b gene.
Additional file 5.
Sequence information including the GenBank accession number, species name, sampling location of 247 cytb for divergence time estimation.
Additional file 6.
Sequence information including species name, GenBank accession number and sampling localities of 24 cytb for ancestral range reconstruction.
Additional file 7.
Sequence information including species name, GenBank accession number and River system of 26 mitochondrial genome for ancestral range reconstruction of Cyprinus, Megalobrama and C. auratus complex just distributed in China.
Additional file 8: Figure S1.
Positions of 16 landmarks superimposed on a photograph of fish.
Additional file 9: Figure S2.
Ancestral range reconstruction for the Carassius across Eurasia using 24 sequence of cytb. The colors of the charts correspond to the most likely ancestral areas inferred, and the black color means the unknown area. Letters represent the biogeographic regions same with that in Fig. 3a. The blue curves in the distribution map of Carassius mean the river systems.
Additional file 10: Table S1.
Morphological data of qualitative and quantitative traits including barbells, lateral scales, first gill rakers number, Spines and Soft-rays in Dorsal fin and Anal fin, as well as pharyngeal teeth formula of among 2nNCRC (F1-F5 generations), C. auratus, C. carpio, M. amblycephala, C. gibelio, C. cuvieri, and C. langsdorfii, and C. carassius (Kalous et al., 2007; Fishbase: https://www.fishbase.se/Nomenclature/ScientificNameSearchList.php?). Table S2. Principal component analysis on the variation of geometrical morphology among C. auratus, 2nNCRC, C. carpio, M. amblycephala, including the eigenvalues, percentage of variance and cumulative percentage of the first three principle components. Table S3. Canonical variate analysis on the variation of geometrical morphology among C. auratus, 2nNCRC, C. carpio, M. amblycephala, including the eigenvalues, percentage of variance and cumulative percentage of the three canonical variate axes. Table S4. Differences based on geometric morphometrics of fish shape among C. auratus, 2nNCRC, C. carpio, M. amblycephala. Mahalanobis (lower triangular) and Procrustes (upper triangular) distances computed from the Canonical Variate analysis, P-values for the significance of the interspecies distances were computed using permutation tests (10,000 replications); all P < 0.0001. Table S5. Pairwise genetic distance among groups including C. auratus, 2nNCRC, C. cuvieri, C. langsdorfii, C. gibelio, and C. carassius were calculated under the K2P model for the cytb dataset using MEGA v7.0 (Kumar et al., 2016), within group genetic distance are in bold. Table S6. Descriptive statistics for the fourteen genes alignments giving alignment length in base pairs (bp); model of sequence evolution for phylogenetic analysis. Table S7. Comparison of models used for BioGeoBEARS based on 24 cytb and 26 mtDNA; likelihood scores (LnL), number of parameters (numparams), dispersal rate (d), extinction rate (e), free parameter controlling the relative probability of founder-event speciation events at cladogenesis (j), corrected Akaike Information Criterion (AICc), and AICc model weights.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gu, Q., Wang, S., Zhong, H. et al. Phylogeographic relationships and the evolutionary history of the Carassius auratus complex with a newly born homodiploid raw fish (2nNCRC). BMC Genomics 23, 242 (2022). https://doi.org/10.1186/s12864-022-08468-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-022-08468-x