
Abstract
Background
Genetic improvement for disease resilience is anticipated to be a practical method to improve efficiency and profitability of the pig industry, as resilient pigs maintain a relatively undepressed level of performance in the face of infection. However, multiple biological functions are known to be involved in disease resilience and this complexity means that the genetic architecture of disease resilience remains largely unknown. Here, we conducted genome-wide association studies (GWAS) of 465,910 autosomal SNPs for complete blood count (CBC) traits that are important in an animal’s disease response. The aim was to identify the genetic control of disease resilience.
Results
Univariate and multivariate single-step GWAS were performed on 15 CBC traits measured from the blood samples of 2743 crossbred (Landrace × Yorkshire) barrows drawn at 2-weeks before, and at 2 and 6-weeks after exposure to a polymicrobial infectious challenge. Overall, at a genome-wise false discovery rate of 0.05, five genomic regions located on Sus scrofa chromosome (SSC) 2, SSC4, SSC9, SSC10, and SSC12, were significantly associated with white blood cell traits in response to the polymicrobial challenge, and nine genomic regions on multiple chromosomes (SSC1, SSC4, SSC5, SSC6, SSC8, SSC9, SSC11, SSC12, SSC17) were significantly associated with red blood cell and platelet traits collected before and after exposure to the challenge. By functional enrichment analyses using Ingenuity Pathway Analysis (IPA) and literature review of previous CBC studies, candidate genes located nearby significant single-nucleotide polymorphisms were found to be involved in immune response, hematopoiesis, red blood cell morphology, and platelet aggregation.
Conclusions
This study helps to improve our understanding of the genetic basis of CBC traits collected before and after exposure to a polymicrobial infectious challenge and provides a step forward to improve disease resilience.
Similar content being viewed by others

Background
The prevalence of infectious diseases caused by a multitude of pathogens results in high economic losses for the pig industry [1, 2]. Genetic improvement for disease resilience is a practical option to help address the problem of infectious disease as it can ensure production efficiency, because resilient animals are defined as maintaining a relatively undepressed performance in the face of disturbances caused by infection [3, 4]. Disease resilience is a complex trait composed of multiple biological functions, such as growth, health, nutrient status, and other dynamic elements, including the efficiency of immune response and the rate of recovery from infection [5]. This complexity makes disease resilience hard to properly characterize and little is known about the genetic architecture that drives disease resilience. Alternatively, indirect selection of disease resilience based on immune-related traits may be a feasible breeding strategy, because the disease response of an animal largely depends upon its immunity [6, 7].
Blood cells comprise white blood cells, red blood cells, and platelets that are important elements of an animal’s immune status [8]. Complete blood count (CBC) is one of the most common clinical tests performed to evaluate concentrations and relative proportions of these circulating blood cells, which may help to uncover the layers of immune system complexity [9]. Our previous study [10] found that CBC traits collected from blood samples of pigs in both healthy and challenged conditions at 2-weeks before, and 2 and 6-weeks after exposure to a polymicrobial challenge were moderately to highly heritable (0.08 ± 0.04 to 0.53 ± 0.05). Changes of each CBC trait between blood samples collected at different time points (e.g. the change of a CBC level from 2-weeks before to 2-weeks after exposure to the challenge) were also found to be heritable, with estimates ranging from 0.06 ± 0.04 to 0.24 ± 0.04 [10]. These heritability estimates indicate the importance of the genetic component of CBC traits. Moreover, significant genetic correlations (either positive or negative) were found for several CBC traits collected after exposure to the challenge with the economically important production traits of grow-to-finish growth rate (GFGR) and treatment rate (TR) in response to the polymicrobial challenge (− 0.82 ± 0.47 to 0.89 ± 0.26) [10], which may further indicate the potential of developing those CBC traits as indicator traits of disease resilience. In addition to these significant genetic correlations for CBC with GFGR and TR, our previous study [10] also found high genetic correlations (≥ 0.40 ± 0.04) between the CBC traits. Changes in CBC traits between each time point were also found to be genetically correlated, with significant estimates ranging from − 0.42 ± 0.21 to − 0.92 ± 0.11 to 0.44 ± 0.22 to 0.98 ± 0.03 [10]. This allows multivariate models to be used for joint analyses of these genetically correlated traits, which provides the potential to improve statistical power and explore pleiotropy [11,12,13,14].
To date, some quantitative trait loci (QTL) have been identified for some blood cell traits in pigs under either healthy or disease challenged status by linkage and association analyses [15,16,17,18,19,20,21,22]. However, due to the use of a pathogen-specific challenge or a relatively low density of genetic markers, the genetic components of blood cell traits in pigs under typical commercial environments, where multiple disease-causing pathogens are present, remains largely unknown.
In this study, CBC traits were collected from pigs in a natural polymicrobial disease challenge model, as described by Bai et al. [10]. Standard univariate genome-wide association studies (GWAS) and multivariate GWAS based on a relatively high-density panel of 465,910 autosomal single-nucleotide polymorphisms (SNPs) were conducted for these CBC traits. The objectives were: (1) to reveal the genomic regions associated with the CBC traits and with their changes in response to the polymicrobial challenge; and (2) to explore the underlying genetic architecture for disease resilience of pigs in the face of a polymicrobial infectious challenge.
Results
Descriptive statistics and genetic parameters
Descriptive statistics (mean, standard deviation, minimum, maximum, and distribution) for the CBC data after removing the outliers, are shown in Additional file 1: Fig. S1, S2 and S3. Details about genetic parameters for the evaluated CBC traits, including heritabilities and genetic correlations, can be found in our previous study [10], which used the same 2593 genotyped animals. In addition to the genetic correlations with resilience already reported for these data by Bai et al. [10], we also found significant genetic correlations for platelet concentration in Blood 3 collected at 2-weeks after exposure to a polymicrobial infectious challenge with GFGR (0.40 ± 0.22) and TR (− 0.46 ± 0.26), and for the change of monocyte concentration from Blood 1 to Blood 3 (MONOΔ13) collected at 2-weeks before and 2-weeks after exposure to the challenge with GFGR (0.63 ± 0.21).
Population structure
As false positive results can be introduced in GWAS by confounding effects due to population stratification, multidimensional scaling (MDS) plots (Additional file 2: Fig. S4) were generated to provide a visualization of the population structure in the first three dimensions (C1, C2, and C3). Animals tended to cluster by farm of origin, as they shared a similar genetic background when they came from the same farm. Since batches were nested within farms and coded uniquely, population stratification associated with the farm effect was accounted for in the association analysis model by fitting the fixed effect of batch. The genomic inflation factors of single-step (SSGWAS) for the CBC traits ranged from 0.98 to 1.06, suggesting that there was no population stratification that confounded the GWAS results.
Association results and estimates for SNP effects
White blood cell traits
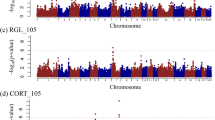
Five genomic regions were found to be significantly associated with white blood cell traits at a genome-wise false discovery rate (FDR) of 0.05 by univariate SSGWAS. Of note, SNPs located on Sus scrofa chromosome (SSC) 4, SSC10, and SSC12 were found to be associated with eosinophil concentration in Blood 3, which was collected 2 weeks after exposure to the challenge (EOSB3). Meanwhile, SNPs on SSC2 and two adjacent floating SNPs (significant SNPs without a group of supportive SNPs) on SSC9 were identified to be associated with MONOΔ13. The Manhattan and quantile-quantile (Q-Q) plots for EOSB3 and MONOΔ13 are shown in Additional file 2: Figs. S5 and S6. Top lead SNPs (the most significant SNP with a group of supportive SNPs) for significant associations (genome-wise FDR < 0.05) with EOSB3 and MONOΔ13 are shown in Table 1. For EOSB3, the additive genetic variances explained by the 1 Mb window of the top lead SNPs (SNP1, SNP2, SNP3) and their adjacent SNPs on SSC4, SSC10, and SSC12 were estimated to be 0.46, 0.35, and 0.53% of the additive genetic variance for EOSB3, respectively. SNP4 was a floating SNP on SSC2 and its 1 Mb window explained 0.12% of the additive genetic variance for MONOΔ13. The 1 Mb window for SNP5, the top lead SNP on SSC9, was estimated to explain about 1.23% of the additive genetic variance for MONOΔ13.
Estimates of additive and dominance effects for the top significant SNPs (genome-wise FDR < 0.05, including both top lead and top floating SNPs) associated with EOSB3 and MONOΔ13 are summarized in Table 1. A significant dominance effect (p < 0.05) was only identified for SNP2, which was associated with EOSB3. Estimates of additive effects were found to be significant (p < 0.05) for all SNPs that were associated with EOSB3 and MONOΔ13. For EOSB3, estimates of additive effects were − 0.05 ± 0.01, 0.14 ± 0.04, and − 0.06 ± 0.01 for SNP1, 2 and 3, respectively. Estimates of additive effects for MONOΔ13 were 0.08 ± 0.02 and − 0.08 ± 0.02 for SNP 4 and 5, respectively.
Due the relatively low genetic correlations and large standard errors between white blood cell traits [10], no genomic region was found to be significantly associated with white blood cell traits at FDR < 0.05 from multivariate SSGWAS.
Red blood cell and platelet traits
Nine genomic regions were found to be significantly associated with red blood cell and platelet traits at the genome-wise FDR of 0.05 by univariate and or multivariate SSGWAS. The Manhattan plots and Q-Q plots are shown in Additional file 2: Figs. S7, S8, S9, S10, S11 and S12. The four top lead SNPs for significant associations (genome-wise FDR < 0.05) and estimates of additive genetic variances explained by these top lead SNPs and their adjacent SNPs in a 1 Mb window are summarized in Table 2. Five floating SNPs (genome-wise FDR < 0.05) that explained small amounts of the additive genetic variance (0.05 to 0.21%) for associated red blood cell and platelet traits were found and are summarized in Table 3. Of note, several pleiotropic SNPs associated with red blood cell or platelet traits were identified by multivariate SSGWAS of CBC traits in Blood 1, 3, and 4 (collected at 2-weeks before, 2- and 6-weeks after the challenge, respectively). High genetic correlations were found between mean corpuscular hemoglobin (MCH), mean corpuscular volume (MCV), and red blood cell concentration (RBC) traits (Additional file 1: Table S1), and also between all three sampling time points for each of these traits (≥ 0.77 ± 0.08) [10]. Therefore, pleiotropic SNP7 on SSC6 was identified as the top lead and pleiotropic SNP for MCH in Blood 1 and for both MCV and RBC traits in all three blood samples (Table 2). The percentage of additive genetic variance explained by the 1 Mb window of SNP7 and its adjacent SNPs ranged from 0.29 to 0.57% for its associated traits. Moreover, SNP8 was the top lead and pleiotropic SNP on SSC8, which was associated with MCH, MCV, and RBC traits in all three blood samples. The percentages of additive genetic variance explained by SNP8 and its adjacent SNPs in a 1 Mb window were estimated to range from 0.28 to 0.35% for its associated traits. SNP9 on SSC17 was the top lead and pleiotropic SNP for mean platelet volume (MPV) in Blood 1 and 4. Together with adjacent SNPs in a 1 Mb window, SNP7 was estimated to explain about 0.49 and 0.40% of the additive genetic variances for MPV in Blood 1 and 4, respectively. Significant associations (genome-wise FDR < 0.05) for SNP7 with MCV in Blood 1 (genome-wise FDR = 0.003) and for SNP8 with MCV in Blood 4 (genome-wise FDR = 0.04) were also found by univariate SSGWAS but at a lower significance level compared to the multivariate SSGWAS. Meanwhile, univariate SSGWAS only indicated suggestive associations (genome-wise FDR of 0.10) for SNP8 with RBC in Blood 1 (genome-wise FDR = 0.09) and with MCV in Blood 1 (genome-wise FDR = 0.08).
For red blood cell and platelet traits, the estimates of additive and dominance effects for the top lead SNPs are summarized in Table 2 and for the top floating SNPs in Table 3. Of note, the additive effects for pleiotropic SNPs showed a tendency of affecting each CBC trait in the three blood samples in the same way, including SNP7 for MCV, SNP8 for MCH and MCV, SNP9 for MPV, SNP10 for MCV, SNP11 for PLT, and SNP12 for MCH. For pleiotropic SNP8, no significant additive effect was found for RBC traits.
Candidate genes and functional enrichment results
Browsing regions for candidate genes located within a maximum distance of 1 Mb on either side of the lead SNPs based on a genome-wise FDR < 0.1 for the associated CBC traits are summarized in Additional file 3: Table S2. Candidate gene functions for each CBC trait were explored with the Ingenuity Pathway Analysis (IPA) database. The top five enriched (p < 0.05) biological functions among diseases, molecular and cellular functions, and physiological system development and function categories for each CBC trait are summarized in Additional file 4. Enriched functions such as inflammatory responses, cell-to-cell signaling and interaction, cellular development, cell morphology, cellular growth and proliferation, and hematological system development and function were commonly identified for the candidate gene lists for white blood cell traits collected after exposure to the challenge, and / or the pleiotropic candidate gene lists for red blood cell and platelet traits collected before and after exposure to the challenge.
Candidate genes that have been reported by previous studies of pigs, human, mice, or rats to be functionally and biologically related to the same category of blood cells, as explored here, are summarized in Table 4. A group of immunity genes on SSC2 has been reported to be functionally and biologically related to monocytes, including TICAM2 (toll-like receptor adaptor molecule 2), TMED7(transmembrane emp24 domain-containing protein 7 precursor), and CDO1 (cysteine dioxygenase type 1), which were located proximal to SNP4, and COMMD10 (COMM domain containing 10), which harbored SNP4 (Table 4). An overview of the location of these candidate genes and the distribution of all the SNPs in this region on SSC2 is shown in the linkage disequilibrium (LD) haplotype map in Additional file 3: Fig. S13. SNP6 on SSC4 is intronic within candidate gene SPTA1 (spectrin alpha, erythrocytic 1) and the LD haplotype map for this region is shown in Additional file 3: Fig. S14. In Table 4, a group of candidate genes, including THAP11 (THAP domain containing protein 11), PSMB10 (proteasome subunit beta type 10), LCAT (lecithin-cholesterol acyltransferase), and SLC12A4 (Potassium/Chloride Cotransporter 1), was reported to be functionally related to red blood cells and were located close to SNP7 in the same haplotype block on SSC6 (Additional file 3: Fig. S15). SNP8 on SSC8 was found to be in LD (r2 > 0.30) with SNPs in the PDGFRA (platelet derived growth factor receptor alpha) gene (Additional file 3: Fig. S16).
Discussion
Potential roles of candidate genes
Functional enrichment analyses for the candidate gene lists for CBC traits indicated multiple enriched functions that can be considered as functionally and biologically relevant to white blood cell traits in response to a polymicrobial infectious challenge, and red blood cell and platelet traits that were collected before and after exposure to the challenge, such as inflammatory response, cell growth and proliferation, cell-to-cell signaling and interaction, and hematological system development and function.
The candidate genes in Table 4 have been reported to be relevant to particular types of CBC traits by studies in pigs, human, mice, and rat, which may help us to further understand the functions of these candidate genes related to CBC traits in response to the polymicrobial challenge. Of note, candidate genes ARHGEF2 (Rho/Rac guanine nucleotide exchange factor), TGFB2 (transforming growth factor beta 2), and MIR21 (microRNA miR-21) were identified to be functionally and biologically relevant to eosinophils. The product of ARHGEF2 regulates the activity of GTPases and has been identified to be highly expressed in eosinophils. GTPases are known to be involved in mediator release from granulocytes, which is a crucial event in the activation of eosinophils and neutrophils during inflammation [23, 24]. TGFB2 has also been found to be expressed mainly in eosinophils, and greater expression of TGFB2 has been identified to be associated with persistent eosinophilic inflammation (severe asthma) in human [25]. However, in the polymicrobial challenge, an increase in the number of eosinophils may be associated with parasitic infection (e.g. Ascaris suum) rather than respiratory disease. Eosinophils play an important role of killing larvae by releasing the toxic content of their granules as part of the immune response [26]. Thus, further investigations are warranted to investigate the functional relationships between the expression of TGFB2 and response to the challenge. Expression of MIR21 has not been identified in eosinophils but in other white blood cells, including lymphocytes, monocytes, macrophages, and dendritic cells, which work collaboratively with eosinophils in the immune response [27,28,29]. Although the mRNA targets for MIR21 are complex and remain an area of active investigation, it has been demonstrated that MIR21 acts as a key signal mediating the balance of the inflammatory reaction to promote healing, resolution, and a return to homeostasis [27].
For the candidate genes on SSC2, the product of COMMD10 has been found to be related to the function of phagosomes in murine macrophages, which promotes phagolysosome maturation and facilitates the timely killing of pathogens [30, 31]. The product of ATG12 (autophagy related 12) is involved in autophagy of circulating monocytes for degradation and recycling of cellular components, which prevents apoptosis (programmed cell death) of monocytes and is essential for monocyte-macrophage differentiation and cytokine production in the innate immune response [32, 33]. The product of CDO1, cysteine dioxygenase type 1, catalyzes taurine synthesis and it is commonly accepted that taurine plays an important role in the immune system as an antioxidant to protect phagocytes, including macrophages, from oxidative stress caused by the generation of reactive oxygen species at the site of inflammation [34,35,36,37]. Both TMED7 and TICAM2 are immunity genes and their products are involved in the function of toll-like receptors (TLRs), which are expressed on macrophages and monocytes and are responsible for the sensing of pathogen-associated-molecular-patterns in the extracellular environment and in endosomes [38,39,40]. Of note, overexpression of TMED7 has been found to be associated with inhibition of MyD88-independent TLR4 signaling and the protein encoded by TICAM2 has been identified as a bridge adaptor recruiting TLRs to mediate innate immune responses [38,39,40]. In addition, NAMPT (nicotinamide phosphoribosyl transferase) on SSC9 has been found to be functionally and biologically related to monocytes, and its gene product has been found to play an important role in governing monocyte recruitment and in monocyte-macrophage differentiation [41, 42].
For red blood cells, the majority of candidate genes reported here have been identified as key components involved in hematopoiesis and erythropoiesis responsible for the differentiation and development of red blood cells, including MNDA (myeloid cell nuclear differentiation antigen) on SSC4, CBFB (core-binding factor subunit beta) and THAP11 on SSC6, PDGFRA and KIT (KIT proto-oncogene, receptor tyrosine kinase) on SSC8, and RARA (retinoic receptor alpha) and THRA (thyroid hormone receptor alpha) on SSC12 [43,44,45,46,47,48,49,50,51,52]. In addition, SPTA1 on SSC4 encodes a protein in the red blood cell membrane, the products of LCAT and SLC12A4 on SSC6 regulate the lipid composition in the red blood cell membrane and cell swelling, respectively, and all these gene products work together to maintain the normal volume and biconcave shape of red blood cells, which helps to ensure the biological and biomechanical functions of the cells [53,54,55,56,57]. ACKR1 (atypical chemokine receptor 1) on SSC4 and PSMB10 on SSC6 are candidate genes that have been shown to be involved in the immune response of red blood cells. The receptor ACKR1 expressed in red blood cells was found to regulate immune responses by interacting with chemokines, and which works as a blood-based chemokine buffer involved with the uptake and degradation of chemokines [58]. Meanwhile, ACKR1 has also been identified as an essential regulator of hematopoiesis and erythropoiesis promoting interactions between nuclear progenitor red blood cells and hematopoietic stem cells in the bone marrow [58, 59]. PSMB10 is found to be responsible for intracellular protein degradation and generation of peptides that bind to class I major histocompatibility complex (MHC) molecules [60]. The MHC molecules display these peptides to cytotoxic CD8+ T cells to support their activity of immune surveillance [61]. Further, through a study of anemia caused by congenital red blood cell aplasia in human, PSMB10 has been suggested to be functional in the MHC class I machinery in mature red blood cells in response to inflammatory signaling [62].
Candidate genes for platelet traits were annotated into two major functions, platelet aggregation and megakaryopoiesis. Platelet aggregation involves platelet-to-platelet adhesion, which is essential for effective hemostasis following injury and bleeding, and megakaryopoiesis is the process of differentiation and development of platelets [63]. Among them, CD9 (CD9 antigen) on SSC5 encodes a major platelet cell surface glycoprotein and plays dual roles in megakarypoiesis and platelet aggregation [64,65,66,67]. The products of VWF (von Willebrand factor), PHB2 (prohibitin 2), and GNB3 (G protein subunit beta 3) on SSC5 and GNAS (guanine nucleotide binding protein) on SSC17 were found to be involved in platelet aggregation [68,69,70,71,72,73]. In addition to megakaryopoiesis, tubulin beta class VI coded by TUBB1 (tubulin beta 1 class VI) on SSC17 has been reported to play a role in maintaining platelet morphology [74,75,76].
Overlap with previously discovered QTL
In addition to the novel QTL for CBC traits identified in this study, some of the QTL identified have been previously reported. QTL on SSC8 located nearby the KIT gene were found to be associated with MCH, MCV, and RBC in this study. In addition, this region has also been identified to show a significant effect on the levels of NEU and HCT in the crossbreds of European Wild Boar × Yorkshire and Landrace × Yorkshire subsequent to stress and disease challenges [15, 22]. For QTL on SSC5 that associated with PLT traits here, Reiner et al. [17] found them to be associated with red blood cell traits in Pietrain × Meishan pigs including HCT, HGB, and RBC traits. These results may be caused by the common myeloid progenitors for all cells mentioned above. Moreover, it may also further indicate the pleiotropic roles of QTL involved in the functions of different blood cells. Apart from studies in pigs, the candidate gene SPTA1 associated with MCHC has also been identified by GWAS for red blood cell traits in human, which also functions in maintaining the shape and deformability of human red blood cells [77].
Potential links with disease resilience
Although the QTL uncovered for blood cell traits have small effects in this study, which has also been found in previous GWAS for blood cell traits of pigs and human [21, 77], the genes involved in these QTL are suggested to be involved in hematopoiesis and immune responses in the face of a polymicrobial infectious challenge. In turn, they may contribute to disease resilience, as hematopoiesis and immune response are collaborative mechanisms that play essential roles in defending against pathogens, maintaining homeostasis, and preventing death from the infection [6, 78, 79]. None of the QTL identified for the CBC traits were pleiotropic with GFGR or TR in response to the challenge. However, some candidate genes are known to have pleiotropic effects among different CBC traits and play roles in both hematopoiesis and immune response. For example, KIT may be a pleiotropic gene for multiple blood cell populations in response to stress and disease challenge, and ACKR1 exhibits pleiotropic effects on hematopoiesis and immune responses, as discussed above [15, 22, 58, 59]. Accordingly, these results highlight the importance of further investigating and validating the function of such pleiotropic genes in disease resilience.
Conclusions
In this study, we identified 14 genomic regions that were significantly associated (genome-wise FDR < 0.05) with CBC traits collected from the natural polymicrobial challenge model, including five for white blood cell traits and nine for red blood cell and platelet traits. Candidate genes or regions located nearby significant SNPs were found to have potential roles in immune response pathways, red blood cell morphology, platelet aggregation, and hematopoiesis, including granulopoiesis and granulocytic differentiation, erythropoiesis, and megakarypoiesis. These results complement previous GWAS for blood cell traits in pigs and contribute to improving our understanding of the genetic basis of blood cell composition before and after exposure to a polymicrobial infectious challenge. This study also advances understanding of the genetic control of disease resilience, as blood cells are key players in an animal’s immune response and are recruited by hematopoiesis. Validation and identification of the candidate genes and causal mutations are necessary to further investigate and develop the use of CBC traits to enhance genetic improvement of disease resilience for the pig industry.
Methods
Natural disease challenge model and phenotypic traits
Details of the natural disease challenge model (NDCM) and the collection of phenotypic traits are described in Bai et al. [10] and Putz et al. [80]. Briefly, the NDCM was established to simulate a polymicrobial infectious challenge and severe disease pressure often found at the commercial level of pig production. A 3-week healthy quarantine nursery after weaning and a test station that consisted of a 4-week second-stage nursery and an approximately 16-week grow-to-finish stage were the two main facilities in the NDCM. A total of 2743 healthy F1 crossbred (Landrace × Yorkshire) barrows owned by company Centre de Développement du Porc du Québec, Inc. were introduced into the NDCM in 42 batches at 3-week intervals after weaning at an average age of 21 days old. Each batch consisted of 60 or 75 weaned crossbred barrows that were provided by one of the seven members of PigGen Canada from healthy multiplier farms. All pigs from each batch were sourced from one multiplier farm, and over time, 13 farms were involved, and each supplied one to six batches of pigs. Pigs that went through the NDCM were first exposed to the polymicrobial infectious challenge in the second-stage nursery. The challenge was established by co-introducing four groups of 12 to 28 commercial seeder pigs from three strategically selected commercial farms with known disease outbreaks together with the first four batches of healthy barrows into the NDCM. Common disease-causing pathogens found in commercial farms were the major target pathogens in the NDCM, including multiple strains of porcine reproductive and respiratory syndrome virus (PRRSV) and swine influenza A virus, various respiratory and enteric bacterial pathogens (such as Mycoplasma hyopneumoniae, Haemophilus parasuis, Brachyspira hampsonii, Salmonella enterica serovar typhimurium, and Streptococcus suis), and two parasites (Cystoisospora suis and Ascaris suum). Subsequently, the challenge model was maintained as a continuous flow system by nose-to-nose direct contact between the new batch of healthy barrows and the preceding challenged group during the first week of challenge nursery period, except during the period of excessively high challenge pressure when indirect contact was used to help maintain the mortality rate below the target level established by the Animal Protection Committee. Serum samples were randomly collected from a subset of individuals for RT-PCR 4 weeks post-challenge and ELISA 6 weeks post-challenge to confirm that every batch had been exposed to PRRSV in the test station and to monitor the pathogens in the test station for each batch. The disease pressure varied by batch; not all pigs were exposed to all the same pathogens because not all pathogens were identified in all batches, as would be the case on a commercial farm. Clinical signs and mortality were monitored in the NDCM for treatment decisions made by herd veterinarians and trained staff, including individual treatments that were given on a case-by-case basis and group medication through water and feed on a batch-level, as necessary, to maintain a balance between disease pressure and animal welfare. Of a total of 2743 pigs, some of the animals died due to infectious diseases after exposure to the challenge (n = 561), a few animals died due to non-infectious and unclear reasons (n = 164) and their phenotypes were set to missing, the other animals (n = 2018) reaching the target slaughter weight at approximately 181 days old were slaughtered commercially and entered the food chain after the study.
In total, four sets of blood samples (Blood 1, Blood 2, Blood 3, and Blood 4) were collected from the jugular vein. Blood 2 was collected immediately before entry into the challenge nursery and polymicrobial infectious challenge at 40 days of age. Whole EDTA-anticoagulated blood was collected for CBC analyses at Blood 1, Blood 3, and Blood 4 (2-weeks before, and at 2- and 6-weeks after exposure to the challenge) at an average age of 26 days, 54 days, and 82 days, respectively, using the ADVIA® 2120i Hematology System (Siemens Healthineer, Erlangen, Germany) within 24 to 48 h of collection. The standard deviation of the bleeding age was 2, 3, and 3 days for Blood 1, Blood 3, and Blood 4, respectively. CBC traits used for this study were described and the outliers were removed previously [10]. There are three categories of CBC traits: (1) six white blood cell traits, including total white blood cell concentration (WBC, 103/μL), neutrophil concentration (NEU, 103/μL), lymphocyte concentration (LYM, 103/μL), monocyte concentration (MONO, 103/μL), eosinophil concentration (EOS, 103/μL), and basophil concentration (BASO, 103/μL); (2) seven red blood cell traits, consisting of red blood cell concentration (RBC, 106/μL), hemoglobin concentration (HGB, g/L), hematocrit (HCT, %), mean corpuscular volume (MCV, fL), mean corpuscular hemoglobin (MCH, pg), mean corpuscular hemoglobin concentration (MCHC, g/L), and red blood cell distribution width (RDW, %); and (3) two platelet traits, including platelet concentration (PLT, 103/μL) and mean platelet volume (MPV, fL). CBC traits were assessed at individual time points (Blood 1, Blood 3, Blood 4) as well as calculating and testing the changes between time points: Blood 1 to Blood 3 (Δ 13, calculated as Blood 3 − Blood 1), Blood 3 to Blood 4 (Δ 34, Blood 4 − Blood 3), and Blood 1 to Blood 4 (Δ 14, Blood 4 − Blood 1). All white blood cell traits in Blood 1, Blood 3, and Blood 4 were log10-transformed to reduce skewness of the distribution.
Body weights and veterinary treatments were recorded on an individual pig basis and used to calculate production traits, grow-to-finish growth rate (GFGR), and treatment rate (TR), which were regarded as economically important traits related to disease resilience. The GFGR for each animal was estimated using linear regression of body weights measured every 3 weeks in the grow-to-finish phase on ages from an average age of 69 days of age to the endpoint, i.e., either mortality due to infectious diseases or reaching the target slaughter weight at approximately 181 days old. The TR for each animal was the number of individual treatment events given on a case-by-case basis standardized by the number of days the animal spent in the NDCM (TR = number of treatment events/days × 100%). The TR for animals that died before receiving any treatment was set to missing.
SNP array genotyping and quality control
The genotyping using the 650 K Affymetrix Axiom® Porcine Genotyping Array was performed at Delta Genomics (Edmonton AB, Canada). Raw Affymetrix SNP data were processed by Delta Genomics with the Axiom Analysis Suite, using all defaults (sample call rate ≥ 97%; SNP call rate ≥ 97%; number of minor alleles observed ≥2). Imputation of sporadic missing genotypes was completed using FImpute [81]. The pedigree was utilized for imputation but only included the dam, since sire was typically unknown due to the use of pooled semen. The preGSf90 software in the BLUPF90 suite of programs was used to remove SNPs with minor allele frequency lower than 0.01 [82]. After quality control, there were 2593 genotyped animals with 465,910 autosomal SNPs remained for the subsequent analyses.
Population stratification and linkage disequilibrium estimation
Population stratification among genotyped animals was investigated using PLINK 1.90 [83] based on pairwise identity-by-state (IBS) distance, which was estimated using SNP genotypes. A multidimensional scaling (MDS) plot based on IBS pairwise distance was drawn by the ‘ggplot2’ package in R [84] to show the first three dimensions of the population structure. The genomic inflation factor and quantile–quantile (Q–Q) plots were applied to assess genomic inflation of the test statistics using the R packages of ‘GenABEL’ and ‘qqman’ [84,85,86]. The linkage disequilibrium (LD) of pairwise SNPs was measured as the squared correlation (r2) of allele counts for the two SNPs and haplotype blocks were built using the Haploview software [87, 88].
Single-step GWAS and models
Univariate and multivariate single-step GWAS (SSGWAS) for CBC traits were implemented in the BLUPF90 suite of programs [82, 89] with the joint pedigree-genomic relationship matrix (H) for single-marker associations, accommodating both genotyped (n = 2593) and non-genotyped (n = 150) animals. Details for algorithms employed for these analyses have been described by Aguilar et al. [89]. Briefly, BLUPF90 combines the algorithms for single-step GBLUP and for back-solving to obtain estimates and p-values for SNP associations from estimates of breeding values. The genomic relationship matrix (G) for genotyped animals was constructed as ZZ′/2 ∑ pi(1 − pi), where the Z matrix contains centered SNP genotype codes and pi is the minor allele frequency for SNP i [90]. The p-values for SNP associations were adjusted for multiple testing by the Benjamini and Hochberg correction (false discovery rate, FDR) [84, 91]. An FDR threshold of 0.05 was used to control false positive results and to declare significant associations. The most significant SNP above the genome-wise FDR of 0.05 in each genomic region were referred to as the top significant SNP, which were further separated into top lead and top floating SNPs, which referred to top significant SNPs in a genomic region with or without a group of supportive SNPs, respectively.
The univariate mixed linear model used for GWAS can be described as follows:
where y is a vector of observations on a CBC trait for all individuals, b is a vector of fixed effects, including the effect of batch and the covariate of bleeding age, X is a design matrix relating observations to the fixed effects, a is a vector of breeding values, Z is a design matrix that relates observations to breeding values, including genotyped and ungenotyped animals, and e is a vector of residual effects. Vector c represents a stack of vectors (cLitter, cPen1, cPen2, and cPen3) of independent and uncorrelated random environmental effects, including litter (cLitter) and pen effects in the quarantine unit (cPen1), in the test station second-stage nursery (cPen2), and in the test station grow-to-finish stage (cPen3). These random environmental effects were tested and fitted in the model for each CBC trait when they were significant (p < 0.05). Matrix W (WLitter, WPen1, WPen2, and WPen3) is a stack of incidence matrices that relate observations to the corresponding random environmental effects. The random effects fitted for each of CBC traits were the same as Bai et al. [10].
Assuming the random effects c and e are uncorrelated and identically distributed, the (co-)variances of random effects for univariate models are:
where H is the joint pedigree-genomic relationship matrix for genotyped and non-genotyped animals as mentioned above, I is the identity matrix, \( {\upsigma}_{\mathrm{a}}^2 \) is the additive genetic variance, \( {\boldsymbol{\upsigma}}_{\mathbf{c}}^{\mathbf{2}} \) represents a stack of random effect variances (e.g. \( {\boldsymbol{\upsigma}}_{\mathbf{c}}^{\mathbf{2}}=\left[\begin{array}{cc}{\upsigma}_{{\mathrm{c}}_{\mathrm{Litter}}}^2& 0\\ {}0& {\upsigma}_{{\mathrm{c}}_{\mathrm{Pen}1}}^2\end{array}\right] \), when the random effects cLitter and cPen1 are significant and fitted in the model for a trait), and \( {\upsigma}_{\mathrm{e}}^2 \) is the residual variance.
The model for multivariate analyses resembles a stack of univariate models for each of the traits that were found to be highly genetic correlated in [10], which can be written as [11, 92]:
For each trait in the multivariate model, the same effects were fitted as in the univariate models. For multivariate models, assuming random effects cn and residual effects en for the nth trait (n = 1, 2, 3) are uncorrelated and identically distributed, the (co-) variances of random effects are:
where \( {\upsigma}_{{\mathrm{a}}_{12}}={\upsigma}_{{\mathrm{a}}_{21}} \), \( {\upsigma}_{{\mathrm{a}}_{13}}={\upsigma}_{{\mathrm{a}}_{31}} \), and \( {\upsigma}_{{\mathrm{a}}_{23}}={\upsigma}_{{\mathrm{a}}_{32}} \) are additive genetic covariances between traits, \( {\boldsymbol{\upsigma}}_{{\mathbf{c}}_{\mathbf{12}}}={\boldsymbol{\upsigma}}_{{\mathbf{c}}_{\mathbf{21}}} \), \( {\boldsymbol{\upsigma}}_{{\mathbf{c}}_{\mathbf{13}}}={\boldsymbol{\upsigma}}_{{\mathbf{c}}_{\mathbf{31}}} \), and \( {\boldsymbol{\upsigma}}_{{\mathbf{c}}_{\mathbf{23}}}={\boldsymbol{\upsigma}}_{{\mathbf{c}}_{\mathbf{32}}} \) are covariances for common random effects between two traits, \( {\upsigma}_{{\mathrm{e}}_{12}}={\upsigma}_{{\mathrm{e}}_{21}} \), \( {\upsigma}_{{\mathrm{e}}_{13}}={\upsigma}_{{\mathrm{e}}_{31}} \), and \( {\upsigma}_{{\mathrm{e}}_{23}}={\upsigma}_{{\mathrm{e}}_{32}} \) are covariances for residual effects between two traits.
Post-GWAS marker effect analyses
The percentage of additive genetic variance explained by a 1 Mb window (with a median of 224 adjacent SNPs) was estimated by conducting window-based inferences for additive genetic variance in the BLUPF90 suit of programs [82]. Each chromosome was evaluated by using a sliding (moving) 1 Mb window by using every SNP on the chromosome as a starting SNP for a window segment [82]. Therefore, a top significant SNP was contained within multiple windows and among them, the largest percentage of additive genetic variance explained by a window that contained that top significant SNP was reported for each trait.
Additive and dominance effects of each top significant SNP were estimated using the BLUPF90 suite of programs [82] based on the following model:
where y, X, b, Z, a, W, c, and e are the same as for the univariate model described above; v is a vector of the top significant SNP genotypes coded as − 1, 0, and 1 for the AA, AB, and BB, respectively; α is the additive effect; d is a vector of dominance coded as 1 for heterozygous genotype (AB) and 0 for homozygous genotypes (AA and BB); δ is the dominance effect. Vectors v and d were fitted as covariates and the top significant SNPs were fitted one by one in the model. The likelihood ratio test was used to test the significance of the additive and dominance effects for each of the top significant SNPs by comparing full models to restricted models that constrained additive or dominance effects to zero using the REMLF90 program of BLUPF90 [82]. When the dominance effect was not significant (p > 0.05), the additive effect for a SNP was re-estimated by removing the dominance effect from the model.
Post-GWAS bioinformatics analyses
Ingenuity Pathway Analysis (IPA) (Ingenuity® Systems, Redwood City, CA; https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/, IPA Spring 2020 release) was used for functional enrichment analyses of candidate genes in significant genomic regions for the CBC trait. A maximum distance of 1 Mb on either side of the lead SNPs based on a genome-wise FDR < 0.10 was used to search for candidate genes for white blood cell traits of EOSB3 and MONOΔ13. The lead pleiotropic SNPs at genome-wise FDR < 0.10 were used to search for common candidate genes located within 1 Mb on either side of the SNPs for red blood cell (MCH, MCV, and RBC) and (MPV and PLT) platelet traits in different time points before and after exposure to the challenge. A relaxed FDR < 0.10 threshold for associated SNPs was used here to increase identification of true positives for the significance of biological and functional relevance of candidate genes [93]. Identification of positional candidate genes was conducted using the UCSC Genome Browser for the Ensembl annotation of the Sscrofa11.1 build of the swine genome (https://genome.ucsc.edu). One collective gene list was created for each trait by combing all candidate genes in associated genomic regions for IPA [94, 95]. Human, mouse, and rat genes in the IPA knowledge base database were used as background for biological function analyses in diseases, molecular and cellular functions, and physiological system development and function categories. A biological function was considered significantly enriched if the p-value for the overlap comparison test between the input list of candidate genes and the IPA database was less than 0.05 [94,95,96].
Availability of data and materials
The data that support the findings of this study were generated on samples from commercially owned animals provided by members of the PigGen Canada consortium. Restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. Data are however available from the corresponding author GP upon non-commercial use and reasonable request and with permission of the PigGen Canada consortium.
Abbreviations
- BASO:
-
Basophil concentration
- CBC:
-
Complete blood count
- DNA:
-
Deoxyribonucleic acid
- EOS:
-
Eosinophil concentration
- FDR:
-
Benjamini and Hochberg correction for false discovery rate
- GFGR:
-
Grow-to-finish growth rate
- GWAS:
-
Genome-wide association studies
- HCT:
-
Hematocrit
- HGB:
-
Hemoglobin concentration
- IBS:
-
Identity-by-state
- IPA:
-
Ingenuity Pathway Analysis
- LD:
-
Linkage disequilibrium
- LYM:
-
Lymphocyte concentration
- LSMs:
-
Least-squares means
- MCH:
-
Mean corpuscular hemoglobin
- MCHC:
-
Mean corpuscular hemoglobin concentration
- MCV:
-
Mean corpuscular volume
- MDS:
-
Multidimensional scaling
- MHC:
-
Major histocompatibility complex
- MONO:
-
Monocyte concentration
- MPV:
-
Mean platelet volume
- NDCM:
-
Natural disease challenge model
- NEU:
-
Neutrophil concentration
- PCA:
-
Principle component analysis
- PLT:
-
Platelet concentration
- PRRSV:
-
Porcine reproductive and respiratory syndrome virus
- Q-Q plot:
-
Quantile-quantile plot
- QTL:
-
Quantitative trait loci
- RBC:
-
Red blood cell concentration
- RDW:
-
Red blood cell distribution width
- SSC:
-
Sus scrofa chromosome
- SSGWAS:
-
Single-step genome-wide association studies
- SNP:
-
Single-nucleotide polymorphisms
- TLRs:
-
Toll-like receptors
- TR:
-
Treatment rate
- WBC:
-
Total white blood cell concentration
References
Davies G, Genini S, Bishop SC, Giuffra E. An assessment of opportunities to dissect host genetic variation in resistance to infectious diseases in livestock. Animal. 2008;3:415–36.
Tomley FM, Shirley MW. Livestock infectious diseases and zoonoses. Philos Trans Royal Soc B Biol Sci. 2009;364(1530):2637–42. https://doi.org/10.1098/rstb.2009.0133.
Albers GA, Gray GD, Piper LR, Barker JS, Le Jambre LF, Barger IA. The genetics of resistance and resilience to Haemonchus contortus infection in young merino sheep. Int J Parasitol. 1987;17(7):1355–63. https://doi.org/10.1016/0020-7519(87)90103-2.
Mulder HA, Rashidi H. Selection on resilience improves disease resistance and tolerance to infections. J Anim Sci. 2017;95(8):3346–58. https://doi.org/10.2527/jas.2017.1479.
Friggens NC, Blanc F, Berry DP, Puillet L. Review: deciphering animal robustness. A synthesis to facilitate its use in livestock breeding and management. Animal. 2017;11(12):2237–51. https://doi.org/10.1017/S175173111700088X.
Calder PC. Feeding the immune system. P Nutr Soc. 2013;72(3):299–309. https://doi.org/10.1017/S0029665113001286.
Knap PW, Bishop SC. Relationships between genetic change and infectious disease in domestic livestock. BSAP Occas Publ. 2000;27:65–80. https://doi.org/10.1017/S1463981500040553.
Abbas AK, Lichtman AH, Pillai S. Basic immunology: functions and disorders of the immune system. Amsterdam: Elsevier; 2015.
George-Gay B, Parker K. Understanding the complete blood count with differential. J Perianesth Nurs. 2003;18(2):96–117. https://doi.org/10.1053/jpan.2003.50013.
Bai X, Putz AM, Wang Z, Fortin F, Harding JCS, Dyck MK, et al. Exploring phenotypes for disease resilience in pigs using complete blood count data from a natural disease challenge model. Front Genet. 2020;11:216. https://doi.org/10.3389/fgene.2020.00216.
Lu Y, Vandehaar MJ, Spurlock DM, Weigel KA, Armentano LE, Connor EE, et al. Genome-wide association analyses based on a multiple-trait approach for modeling feed efficiency. J Dairy Sci. 2018;101(4):3140–54. https://doi.org/10.3168/jds.2017-13364.
Fatumo S, Carstensen T, Nashiru O, Gurdasani D, Sandhu M, Kaleebu P. Complimentary methods for multivariate genome-wide association study identify new susceptibility genes for blood cell traits. Front Genet. 2019;10:334. https://doi.org/10.3389/fgene.2019.00334.
Park SH, Lee JY, Kim S. A methodology for multivariate phenotype-based genome-wide association studies to mine pleiotropic genes. BMC Syst Biol. 2011;5(Suppl 2):S13.
Shriner D. Moving toward system genetics through multiple trait analysis in genome-wide association studies. Front Genet. 2012;3:1.
Edfors-Lilja I, Wattrang E, Andersson L, Fossum C. Mapping quantitative trait loci for stress induced alterations in porcine leukocyte numbers and functions. Anim Genet. 2000;31(3):186–93. https://doi.org/10.1046/j.1365-2052.2000.00628.x.
Edfors-Lilja I, Wattrang E, Marklund L, Moller M, Andersson-Eklund L, Andersson L, et al. Mapping quantitative trait loci for immune capacity in the pig. J Immunol Baltim. 1998;161:829–35.
Reiner G, Fischer R, Hepp S, Berge T, Köhler F, Willems H. Quantitative trait loci for red blood cell traits in swine. Anim Genet. 2007;38(5):447–52. https://doi.org/10.1111/j.1365-2052.2007.01629.x.
Reiner G, Fischer R, Hepp S, Berge T, Köhler F, Willems H. Quantitative trait loci for white blood cell numbers in swine. Anim Genet. 2008;39(2):163–8. https://doi.org/10.1111/j.1365-2052.2008.01700.x.
Ponsuksili S, Reyer H, Trakooljul N, Murani E, Wimmers K. Single- and Bayesian multi-marker genome-wide association for haematological parameters in pigs. PLoS One. 2016;11(7):e0159212. https://doi.org/10.1371/journal.pone.0159212.
Gong Y-F, Lu X, Wang Z-P, Hu F, Luo Y-R, Cai S-Q, et al. Detection of quantitative trait loci affecting haematological traits in swine via genome scanning. BMC Genet. 2010;11(1):56. https://doi.org/10.1186/1471-2156-11-56.
Zou Z, Ren J, Yan X, Huang X, Yang S, Zhang Z, et al. Quantitative trait loci for porcine baseline erythroid traits at three growth ages in a white Duroc × Erhualian F2 resource population. Mamm Genome. 2008;19(9):640–6. https://doi.org/10.1007/s00335-008-9142-9.
Wattrang E, Almqvist M, Johansson A, Fossum C, Wallgren P, Pielberg G, et al. Confirmation of QTL on porcine chromosomes 1 and 8 influencing leukocyte numbers, haematological parameters and leukocyte function. Anim Genet. 2005;36(4):337–45. https://doi.org/10.1111/j.1365-2052.2005.01315.x.
Lacy P. The role of rho GTPases and SNAREs in mediator release from granulocytes. Pharmacol Ther. 2005;107(3):358–76. https://doi.org/10.1016/j.pharmthera.2005.03.008.
Turton, KB. Case studies of cryptic proteins contributing to shape change in eosinophils. 2018. https://search.proquest.com/docview/2089435247?pq-origsite=gscholar&fromopenview=true. Accessed 5 May 2020.
Balzar S, Chu HW, Silkoff P, Cundall M, Trudeau JB, Strand M, et al. Increased TGF-β2 in severe asthma with eosinophilia. J Allergy Clin Immunol. 2005;115(1):110–7. https://doi.org/10.1016/j.jaci.2004.09.034.
Masure D, Vlaminck J, Wang T, Chiers K, Van den Broeck W, Vercruysse J, et al. A role for eosinophils in the intestinal immunity against infective Ascaris suum larvae. PLoS Negl Trop Dis. 2013;7(3):e2138. https://doi.org/10.1371/journal.pntd.0002138.
Sheedy FJ. Turning 21: induction of miR-21 as a key switch in the inflammatory response. Front Immunol. 2015;6:19.
Cobb BS, Hertweck A, Smith J, O’Connor E, Graf D, Cook T, et al. A role for dicer in immune regulation. J Exp Med. 2006;203(11):2519–27. https://doi.org/10.1084/jem.20061692.
Wu H, Neilson JR, Kumar P, Manocha M, Shankar P, Sharp PA, et al. miRNA profiling of naïve, effector and memory CD8 T cells. PLoS One. 2007;2:e1020.
Dill BD, Gierlinski M, Härtlova A, Arandilla AG, Guo M, Clarke RG, et al. Quantitative proteome analysis of temporally resolved phagosomes following uptake via key phagocytic receptors. Mol Cell Proteomics. 2015;14(5):1334–49. https://doi.org/10.1074/mcp.M114.044594.
Shlomo SB, Mouhadeb O, Cohen K, Varol C, Gluck N. COMMD10-guided phagolysosomal maturation promotes clearance of Staphylococcus aureus in macrophages. Iscience. 2019;14:147–63. https://doi.org/10.1016/j.isci.2019.03.024.
Zhang Y, Morgan MJ, Chen K, Choksi S, Liu Z. Induction of autophagy is essential for monocyte-macrophage differentiation. Blood. 2012;119(12):2895–905. https://doi.org/10.1182/blood-2011-08-372383.
Geng J, Klionsky DJ. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. EMBO Rep. 2008;9(9):859–64. https://doi.org/10.1038/embor.2008.163.
Wang L, Zhao N, Zhang F, Yue W, Liang M. Effect of taurine on leucocyte function. Eur J Pharmacol. 2009;616(1-3):275–80. https://doi.org/10.1016/j.ejphar.2009.05.027.
Schaffer SW, Azuma J, Mozaffari M. Role of antioxidant activity of taurine in diabetes. Can J Physiol Pharmacol. 2009;87(2):91–9. https://doi.org/10.1139/Y08-110.
Booken N, Gratchev A, Utikal J, Weiß C, Yu X, Qadoumi M, et al. Sézary syndrome is a unique cutaneous T-cell lymphoma as identified by an expanded gene signature including diagnostic marker molecules CDO1 and DNM3. Leukemia. 2008;22(2):393–9. https://doi.org/10.1038/sj.leu.2405044.
Marcinkiewicz J, Kontny E. Taurine and inflammatory diseases. Amino Acids. 2012;46:7–20.
Oshiumi H, Sasai M, Shida K, Fujita T, Matsumoto M, Seya T. TIR-containing adapter molecule (TICAM)-2, a bridging adapter recruiting to toll-like receptor 4 TICAM-1 that induces interferon-β. J Biol Chem. 2003;278(50):49751–62. https://doi.org/10.1074/jbc.M305820200.
Mekonnen E, Bekele E, Stein CM. Novel polymorphisms in TICAM2 and NOD1 associated with tuberculosis progression phenotypes in Ethiopian populations. Global Heal Epidemiol Genom. 2018;3:e1. https://doi.org/10.1017/gheg.2017.17.
Doyle SL, Husebye H, Connolly DJ, Espevik T, O’Neill LAJ, McGettrick AF. The GOLD domain-containing protein TMED7 inhibits TLR4 signalling from the endosome upon LPS stimulation. Nat Commun. 2012;3(1):707. https://doi.org/10.1038/ncomms1706.
Schilling E, Wehrhahn J, Klein C, Raulien N, Ceglarek U, Hauschildt S. Inhibition of nicotinamide phosphoribosyltransferase modifies LPS-induced inflammatory responses of human monocytes. Innate Immun. 2011;18:518–30.
Travelli C, Colombo G, Mola S, Genazzani AA, Porta C. NAMPT: a pleiotropic modulator of monocytes and macrophages. Pharmacol Res. 2018;135:25–36. https://doi.org/10.1016/j.phrs.2018.06.022.
Xie J, Briggs JA, Briggs RC. Human hematopoietic cell specific nuclear protein MNDA interacts with the multifunctional transcription factor YY1 and stimulates YY1 DNA binding. J Cell Biochem. 1998;70(4):489–506. https://doi.org/10.1002/(SICI)1097-4644(19980915)70:4<489::AID-JCB6>3.0.CO;2-F.
Kong X-Z, Yin R-H, Ning H-M, Zheng W-W, Dong X-M, Yang Y, et al. Effects of THAP11 on erythroid differentiation and megakaryocytic differentiation of K562 cells. PLoS One. 2014;9(3):e91557. https://doi.org/10.1371/journal.pone.0091557.
Cools J, DeAngelo DJ, Gotlib J, Stover EH, Legare RD, Cortes J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. New Engl J Medicine. 2003;348(13):1201–14. https://doi.org/10.1056/NEJMoa025217.
Zhai P-F, Wang F, Su R, Lin H-S, Jiang C-L, Yang G-H, et al. The regulatory roles of microRNA-146b-5p and its target platelet-derived growth factor receptor α (PDGFRA) in erythropoiesis and megakaryocytopoiesis. J Biol Chem. 2014;289(33):22600–13. https://doi.org/10.1074/jbc.M114.547380.
Moller MJ, Chaudhary R, Hellmén E, Höyheim B, Chowdhary B, Andersson L. Pigs with the dominant white coat color phenotype carry a duplication of the KIT gene encoding the mast/stem cell growth factor receptor. Mamm Genome. 1996;7(11):822–30. https://doi.org/10.1007/s003359900244.
Thorén LA, Liuba K, Bryder D, Nygren JM, Jensen CT, Qian H, et al. Kit regulates maintenance of quiescent hematopoietic stem cells. J Immunol. 2008;180(4):2045–53. https://doi.org/10.4049/jimmunol.180.4.2045.
Kastner P, Chan S. Function of RARα during the maturation of neutrophils. Oncogene. 2001;20(49):7178–85. https://doi.org/10.1038/sj.onc.1204757.
Kastner P, Lawrence HJ, Waltzinger C, Ghyselinck NB, Chambon P, Chan S. Positive and negative regulation of granulopoiesis by endogenous RARα. Blood. 2001;97(5):1314–20. https://doi.org/10.1182/blood.V97.5.1314.
Kendrick TS, Payne CJ, Epis MR, Schneider JR, Leedman PJ, Klinken SP, et al. Erythroid defects in TRα−/− mice. Blood. 2008;111(6):3245–8. https://doi.org/10.1182/blood-2007-07-101105.
Zhu J, Heyworth CM, Glasow A, Huang Q-H, Petrie K, Lanotte M, et al. Lineage restriction of the RARα gene expression in myeloid differentiation. Blood. 2001;98(8):2563–7. https://doi.org/10.1182/blood.V98.8.2563.
Diez-Silva M, Dao M, Han J, Lim C-T, Suresh S. Shape and biomechanical characteristics of human red blood cells in health and disease. MRS Bull. 2010;35(5):382–8. https://doi.org/10.1557/mrs2010.571.
Godin DV, Gray GR, Frohlich J. Erythrocyte membrane alterations in lecithin:cholesterol acyltransferase deficiency. Scand J Clin Lab Invest Suppl. 1978;150:162–7.
Karai I, Fukumoto K, Horiguchi S. Alterations of lipids of the erythrocyte membranes in workers exposed to lead. Int Arch Occup Environ Health. 1982;50(1):11–6. https://doi.org/10.1007/BF00432489.
Bize I, Güvenç B, Buchbinder G, Brugnara C. Stimulation of human erythrocyte K-cl cotransport and protein phosphatase type 2A by n-ethylmaleimide: role of intracellular mg++. J Membr Biol. 2000;177(2):159–68. https://doi.org/10.1007/s002320001109.
Rust MB, Alper SL, Rudhard Y, Shmukler BE, Vicente R, Brugnara C, et al. Disruption of erythroid K-cl cotransporters alters erythrocyte volume and partially rescues erythrocyte dehydration in SAD mice. J Clin Invest. 2007;117(6):1708–17. https://doi.org/10.1172/JCI30630.
Permanyer M, Bošnjak B, Förster R. Dual role for atypical chemokine receptor 1 in myeloid cell hematopoiesis and distribution. Cell Mol Immunol. 2018;15(4):399–401. https://doi.org/10.1038/cmi.2017.79.
Bonavita O, Poeta VM, Massara M, Mantovani A, Bonecchi R. Regulation of hematopoiesis by the chemokine system. Cytokine. 2018;109:76–80. https://doi.org/10.1016/j.cyto.2018.01.021.
Wu X, Yang SL, Yerle M, Zhu ZM, Wang HL, Wang H, et al. Genomic organization, localization and polymorphism of porcine PSMB10, a gene encoding the third beta-type proteasome subunit of 26S proteasome complex. J Anim Breed Genet. 2006;123(5):331–6. https://doi.org/10.1111/j.1439-0388.2006.00592.x.
Leone P, Shin E-C, Perosa F, Vacca A, Dammacco F, Racanelli V. MHC class I antigen processing and presenting machinery: organization, function, and defects in tumor cells. Jnci J Natl Cancer Inst. 2013;105(16):1172–87. https://doi.org/10.1093/jnci/djt184.
Pesciotta EN, Lam H-S, Kossenkov A, Ge J, Showe LC, Mason PJ, et al. In-depth, label-free analysis of the erythrocyte cytoplasmic proteome in diamond blackfan anemia identifies a unique inflammatory signature. PLoS One. 2015;10(10):e0140036. https://doi.org/10.1371/journal.pone.0140036.
Rumbaut RE, Thiagarajan P. Platelet-Vessel Wall interactions in hemostasis and thrombosis. Colloq Ser Integ Syst Physiol Mol Funct. 2010;2(1):1–75. https://doi.org/10.4199/c00007ed1v01y201002isp004.
Kaprielian Z, Cho K, Hadjiargyrou M, Patterson P. CD9, a major platelet cell surface glycoprotein, is a ROCA antigen and is expressed in the nervous system. J Neurosci. 1995;15(1):562–73. https://doi.org/10.1523/JNEUROSCI.15-01-00562.1995.
Boucheix C, Benoit P, Frachet P, Billard M, Worthington RE, Gagnon J, et al. Molecular cloning of the CD9 antigen. A new family of cell surface proteins. J Biol Chem. 1991;266(1):117–22. https://doi.org/10.1016/S0021-9258(18)52410-8.
Clay D, Rubinstein E, Mishal Z, Anjo A, Prenant M, Jasmin C, et al. CD9 and megakaryocyte differentiation. Blood. 2001;97(7):1982–9. https://doi.org/10.1182/blood.V97.7.1982.
Worthington RE, Carroll RC, Boucheix C. Platelet activation by CD9 monoclonal antibodies is mediated by the FCγII receptor. Brit J Haematol. 1990;74(2):216–22. https://doi.org/10.1111/j.1365-2141.1990.tb02568.x.
Kanaji S, Fahs SA, Shi Q, Haberichter SL, Montgomery RR. Contribution of platelet vs. endothelial VWF to platelet adhesion and hemostasis. J Thromb Haemost. 2012;10(8):1646–52. https://doi.org/10.1111/j.1538-7836.2012.04797.x.
Zhang Y, Wang Y, Xiang Y, Lee W, Zhang Y. Prohibitins are involved in protease-activated receptor 1-mediated platelet aggregation. J Thromb Haemost. 2012;10(3):411–8. https://doi.org/10.1111/j.1538-7836.2011.04607.x.
Frey UH, Aral N, Müller N, Siffert W. Cooperative effect of GNB3 825C>T and GPIIIa PI(a) polymorphisms in enhanced platelet aggregation. Thromb Res. 2003;109(5-6):279–86. https://doi.org/10.1016/S0049-3848(03)00253-6.
Dusse F, Frey UH, Bilalic A, Dirkmann D, Görlinger K, Siffert W, et al. The GNB3 C825T polymorphism influences platelet aggregation in human whole blood. Pharmacogenet Genomics. 2012;22(1):43–9. https://doi.org/10.1097/FPC.0b013e32834e1674.
Freson K, Izzi B, Geet CV. From genetics to epigenetics in platelet research. Thromb Res. 2012;129(3):325–9. https://doi.org/10.1016/j.thromres.2011.11.050.
Freson K, Izzi B, Labarque V, Helvoirt MV, Thys C, Wittevrongel C, et al. GNAS defects identified by stimulatory G protein α-subunit signalling studies in platelets. J Clin Endocrinol Metab. 2008;93(12):4851–9. https://doi.org/10.1210/jc.2008-0883.
Kunishima S, Nishimura S, Suzuki H, Imaizumi M, Saito H. TUBB1 mutation disrupting microtubule assembly impairs proplatelet formation and results in congenital macrothrombocytopenia. Eur J Haematol. 2014;92(4):276–82. https://doi.org/10.1111/ejh.12252.
Schwer HD, Lecine P, Tiwari S, Italiano JE, Hartwig JH, Shivdasani RA. A lineage-restricted and divergent β-tubulin isoform is essential for the biogenesis, structure and function of blood platelets. Curr Biol. 2001;11(8):579–86. https://doi.org/10.1016/S0960-9822(01)00153-1.
Burley K, Westbury SK, Mumford AD. TUBB1 variants and human platelet traits. Platelets. 2018;29(2):209–11. https://doi.org/10.1080/09537104.2017.1411587.
Ganesh SK, Zakai NA, van Rooij FJA, Soranzo N, Smith AV, Nalls MA, et al. Multiple loci influence erythrocyte phenotypes in the CHARGE consortium. Nat Genet. 2009;41(11):1191–8. https://doi.org/10.1038/ng.466.
Boettcher S, Manz MG. Regulation of inflammation- and infection-driven hematopoiesis. Trends Immunol. 2017;38(5):345–57. https://doi.org/10.1016/j.it.2017.01.004.
Baldridge MT, King KY, Goodell MA. Inflammatory signals regulate hematopoietic stem cells. Trends Immunol. 2011;32(2):57–65. https://doi.org/10.1016/j.it.2010.12.003.
Putz AM, Harding JCS, Dyck MK, Fortin F, Plastow GS, Dekkers JCM, et al. Novel resilience phenotypes using feed intake data from a natural disease challenge model in wean-to-finish pigs. Front Genet. 2019;9:660. https://doi.org/10.3389/fgene.2018.00660.
Sargolzaei M, Chesnais JP, Schenkel FS. A new approach for efficient genotype imputation using information from relatives. BMC Genomics. 2014;15(1):478. https://doi.org/10.1186/1471-2164-15-478.
Misztal I, Tsuruta S, Strabel T, Auvray B, Druet T, Lee DH. BLUPF90 and related programs (BGF90). In: Proceedings of 7th World Congress of Genetics Applied to Livestock Production, (Montpellier); 2002. Available on line at: http://www.wcgalp.org/system/files/proceedings/2002/blupf90-and-related-programs-bgf90.pdf. Accessed 5 May 2020.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75. https://doi.org/10.1086/519795.
R core team. R: A Language and Environment for Statistical Computing. Vienna: R foundation for statistical computing. Available online at: https://www.R-project.org/. Accessed 1 Jan 2019
Aulchenko YS, Ripke S, Isaacs A, van Duijn CM. GenABEL: an R library for genome-wide association analysis. Bioinformatics. 2007;23(10):1294–6. https://doi.org/10.1093/bioinformatics/btm108.
Turner SD. qqman: an R package for visualizing GWAS results using Q-Q and manhattan plots; 2014. https://doi.org/10.1101/005165.
VanLiere JM, Rosenberg NA. Mathematical properties of the r2 measure of linkage disequilibrium. Theor Popul Biol. 2008;74(1):130–7. https://doi.org/10.1016/j.tpb.2008.05.006.
Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21(2):263–5. https://doi.org/10.1093/bioinformatics/bth457.
Aguilar I, Legarra A, Cardoso F, Masuda Y, Lourenco D, Misztal I. Frequentist p-values for large-scale-single step genome-wide association, with an application to birth weight in American Angus cattle. Genet Sel Evol. 2019;51(1):28. https://doi.org/10.1186/s12711-019-0469-3.
VanRaden P. Efficient methods to compute genomic prediction. J Dairy Sci. 2008;91(11):4414–23. https://doi.org/10.3168/jds.2007-0980.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Royal Stat Soc Ser B Methodol. 1995;57:289–300.
Mrode RA. Linear models for the prediction of animal breeding values. Boston: CABI; 2013.
Waide EH, Tuggle CK, Serão NVL, Schroyen M, Hess A, Rowland RRR, et al. Genomewide association of piglet responses to infection with one of two porcine reproductive and respiratory syndrome virus isolates1. J Anim Sci. 2017;95(1):16–38. https://doi.org/10.2527/jas.2016.0874.
Zhang F, Wang Y, Mukiibi R, Chen L, Vinsky M, Plastow G, et al. Genetic architecture of quantitative traits in beef cattle revealed by genome wide association studies of imputed whole genome sequence variants: I: feed efficiency and component traits. BMC Genomics. 2020;21(1):36. https://doi.org/10.1186/s12864-019-6362-1.
Wang Y, Zhang F, Mukiibi R, Chen L, Vinsky M, Plastow G, et al. Genetic architecture of quantitative traits in beef cattle revealed by genome wide association studies of imputed whole genome sequence variants: II: carcass merit traits. BMC Genomics. 2020;21(1):38. https://doi.org/10.1186/s12864-019-6273-1.
Wysocki M, Chen H, Steibel JP, Kuhar D, Petry D, Bates J, et al. Identifying putative candidate genes and pathways involved in immune responses to porcine reproductive and respiratory syndrome virus (PRRSV) infection. Anim Genet. 2012;43(3):328–32. https://doi.org/10.1111/j.1365-2052.2011.02251.x.
Acknowledgements
The following individuals served as collaborators and representatives for member companies of the PigGen Canada Consortium: Mr. D. Vandenbroek and Mr. B. DeVries, Alliance Genetics Canada, St. Thomas, ON, Canada; Dr. N. Dion and Ms. S. Blanchette, AlphaGene, St.-Hyacinthe, QU, Canada; Dr. T. Rathje, DNA Genetics, Columbus, NE, United States; Mr. M. Duggan, FastGenetics, Saskatoon, SK, Canada; Dr. R. Kemp, Genesus, Oakville, MB, Canada; Dr. P. Charagu, Hypor, Regina, SK, Canada; Dr. P. Mathur, Topigs Norsvin, Helvoirt, Netherlands; JH, University of Saskatchewan, Saskatoon, SK, Canada. They participated in the development and implementation of the project, coordinated the sources of piglets and the collection of associated data, and discussed with the research team regularly during the execution of the natural challenge protocol to contribute to the project. Thanks to Mr. Michael Lowing at Delta Genomics (Edmonton, AB, Canada) for processing the raw genotypes from the Affymetrix SNP chips. Thanks to Dr. Jason Grant at Livestock Gentec, University of Alberta for managing the project database and helping with questions concerning genotyping and SNP map file questions. Thanks to Ms. Yan Meng, Ms. Janelle Jiminez, Ms. Ziqi Yang, and Mr. Jiehan Lim at the University of Alberta for helping with the processing of blood samples and managing the database of samples and CBC measures. Thanks to Ms. Susan Goruk and Ms. Marnie Newell at the University of Alberta for helping with the processing of blood samples and problem solving of technical problems with the CBC instrument.
Funding
This project was funded by the Genome Canada, Genome Alberta, PigGen Canada, Swine Innovation Porc, and Alberta Agriculture and Forestry. PigGen Canada also intimately involved in the design of the project and the development of protocols for the natural disease challenge model. This research is also part of the AMR – One Health Consortium, funded by the Major Innovation Fund program of the Alberta Ministry of Economic Development, Trade and Tourism.
Author information
Authors and Affiliations
Consortia
Contributions
XB analyzed the data and wrote the manuscript with GP and help from TY, ZW and CL. FF, JH, MD, PC, JD, and GP designed the project and developed protocols for the natural disease challenge model. FF oversaw the sample collection and scheduling. JH was in charge of veterinary oversight on the project. CF provided support on CBC data measurement and interpretation. GP was in charge of the database and genotyping for the project. AP and JD further processed the genotype data and provided the genomic relationship matrix for the project. All authors helped with the interpretation of results and reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The animal study was reviewed and approved by the Animal Protection Committee of the Centre de Recherche en Sciences Animales de Deschambault (15PO283) and the Animal Care and Use Committee at the University of Alberta (AUP00002227).
Consent for publication
Not applicable.
Competing interests
FF is employed by company Centre de Développement du Porc du Québec, Inc. who manage the challenge model developed by the team. We have also included the PC consortium as co-authors as they are intimately involved in the research studies as identified in the manuscript and this is custom and practice in our publication to date. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
Violin plots for descriptive statistics for white blood cell traits in Blood 1, Blood 3, and Blood 4 collected at 2-weeks before, and at 2- and 6-weeks after the challenge, respectively. Figure S2. Violin plots for descriptive statistics for red blood cell traits in Blood 1, Blood 3, and Blood 4 collected at 2-weeks before, and at 2- and 6-weeks after the challenge, respectively. Figure S3. Violin plots for descriptive statistics for platelet traits in Blood 1, Blood 3, and Blood 4 collected at 2-weeks before, and at 2- and 6-weeks after the challenge, respectively. Table S1. Genetic correlations between red blood cell traits of MCH, MCV, and RBC traits in Blood 1, Blood 3, and Blood 4.
Additional file 2: Figure S4.
Multidimensional scaling (MDS) plots showing the first three dimensions (C1, C2, and C3) of the population structure for genotyped animals based on pairwise identity-by-state distance. Figure S5. (A) Manhattan plot for EOSB3. (B) Quantile-Quantile plot for EOSB3; Figure S6. (A) Manhattan plot for MONO ∆ 13. (B) Quantile-Quantile plot for MONO ∆ 13; Figure S7. Manhattan plots (A, C, E) and Quantile-Quantile plots (B, D, F) for MCH in Blood 1, Blood 3, and Blood 4, respectively; Figure S8. Manhattan plots (A, C) and Quantile-Quantile plots (B, D) for MCHC in Blood 3 and for the change of MCHC from Blood 1 to Blood 4, respectively; Figure S9. Manhattan plots (A, C, E) and Quantile-Quantile plots (B, D, F) for MCV in Blood 1, Blood 3, and Blood 4, respectively; Figure S10. Manhattan plots (A, C, E) and Quantile-Quantile plots (B, D, F) for RBC in Blood 1, Blood 3, and Blood 4, respectively; Figure S11. Manhattan plots (A, C) and Quantile-Quantile plots (B, D) for MPV in Blood 1 and Blood 4, respectively; Figure S12. Manhattan plots (A, C, E) and Quantile-Quantile plots (B, D, F) for PLT in Blood 1, Blood 3, and Blood 4, respectively.
Additional file 3: Table S2.
Browsing regions for candidate genes located within 1-Mb on either side of lead SNPs (FDR < 0.10) associated with complete blood cell count traits; Figure S13. Haplotype block pattern (r2-scheme) for the region of candidate genes on SSC2 located within the maximum distance of 1 Mb on either side of the top lead SNP4 (SSC2, 120,341,201 bp); Figure S14. Haplotype block pattern (r2-scheme) for the region of candidate genes on SSC4 located within the maximum distance of 1 Mb on either side of the top lead SNP6 (SSC4, 91,591,493 bp); Figure S15. Haplotype block pattern (r2-scheme) for the region of candidate genes on SSC6 located within the maximum distance of 1 Mb on either side of the top lead SNP7 (SSC6, 28,511,423 bp); Figure S16. Haplotype block pattern (r2-scheme) for SNPs (40,946,144 bp to 41,198,574 bp) on SSC8 located within the maximum distance of 1 Mb on either side of the top lead SNP8 (SSC8, 41,156,538 bp).
Additional file 4.
This file contains a list of top five enriched biological functions (p-value < 0.05) for the candidate gene lists for EOSB3, MONO ∆ 13, and for MCH, MCV, RBC, MPV, and PLT in Blood 1, Blood 3, and Blood 4.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bai, X., Yang, T., Putz, A.M. et al. Investigating the genetic architecture of disease resilience in pigs by genome-wide association studies of complete blood count traits collected from a natural disease challenge model. BMC Genomics 22, 535 (2021). https://doi.org/10.1186/s12864-021-07835-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-021-07835-4