
Abstract
Background
Fluoroquinolones are broad-spectrum antibiotics used to prevent and treat a wide range of bacterial infections. Plasmid-mediated qnr genes provide resistance to fluoroquinolones in many bacterial species and are increasingly encountered in clinical settings. Over the last decade, several families of qnr genes have been discovered and characterized, but their true prevalence and diversity still remain unclear. In particular, environmental and host-associated bacterial communities have been hypothesized to maintain a large and unknown collection of qnr genes that could be mobilized into pathogens.
Results
In this study we used computational methods to screen genomes and metagenomes for novel qnr genes. In contrast to previous studies, we analyzed an almost 20-fold larger dataset comprising almost 13 terabases of sequence data. In total, 362,843 potential qnr gene fragments were identified, from which 611 putative qnr genes were reconstructed. These gene sequences included all previously described plasmid-mediated qnr gene families. Fifty-two of the 611 identified qnr genes were reconstructed from metagenomes, and 20 of these were previously undescribed. All of the novel qnr genes were assembled from metagenomes associated with aquatic environments. Nine of the novel genes were selected for validation, and six of the tested genes conferred consistently decreased susceptibility to ciprofloxacin when expressed in Escherichia coli.
Conclusions
The results presented in this study provide additional evidence for the ubiquitous presence of qnr genes in environmental microbial communities, expand the number of known qnr gene variants and further elucidate the diversity of this class of resistance genes. This study also strengthens the hypothesis that environmental bacterial communities act as sources of previously uncharacterized qnr genes.
Similar content being viewed by others
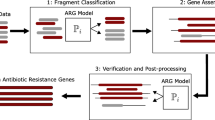


Background
Fluoroquinolones are widely used synthetic broad-spectrum antibiotics that inhibit the type II topoisomerase complexes essential for bacterial DNA replication. There are three main mechanisms of resistance to fluoroquinolones: 1) mutations in the target enzyme reducing the binding affinity of the antibiotic, 2) efflux pumps facilitating export of the antibiotic, and 3) qnr genes encoding proteins thought to sterically prevent the antibiotic to interact with the topoisomerase enzymes [1]. In contrast to fluoroquinolone resistance mechanisms such as chromosomal mutations and efflux pumps, qnr genes are often located on mobile plasmids and can be shared between bacterial cells through the process of horizontal gene transfer. The first plasmid-mediated fluoroquinolone resistance gene family, qnrA, was discovered in 1998 [2], and since then, five additional families have been described: qnrS [3], qnrB [4], qnrC [5], qnrD [6], and most recently qnrVC [7]. Novel variants within the established families have been reported over the last years, bringing the current total number of mobile qnr gene variants across all families to 115 [8]. In addition, sequence homology searches have revealed qnr genes on chromosomes from a wide range of bacterial species [9, 10]. Similarly to many other resistance genes, qnr genes are hypothesized to have originated from harmless environmental bacteria and subsequently mobilized into human pathogens [11,12,13]. Recent reports suggest that qnr genes are ubiquitously present in microbial communities from many environments, including marine and lake water, soil, river sediment, and the human microbiome [11, 14,15,16,17]. It is therefore possible that these communities also maintain a large collection of novel qnr genes that have not yet been transferred to, or discovered in, pathogenic bacteria. Describing this unknown diversity of qnr genes is important to fully understand their biological and ecological roles. It also has the potential to uncover currently undescribed resistance genes that might appear in clinical settings in the future [18,19,20].
Modern high-throughput DNA sequencing technologies have enabled direct analysis of the DNA from bacterial communities without the need for cultivation of individual isolates. The resulting metagenomic data provides information on the combined set of genes maintained by the microorganisms present in the sample [21, 22]. In Boulund et al. (2012) we used a computational method to screen nucleotide sequences from metagenomes for novel qnr genes. By analyzing approximately 700 gigabytes of sequence data, evidence of several novel qnr genes could be identified. Two of the identified genes selected for experimental validation were confirmed to confer decreased fluoroquinolone susceptibility in an E. coli model system [23]. Over the last years, the body of metagenomic data has grown, much thanks to the continuously increasing throughput of shotgun sequencing technologies [24, 25]. In particular, the widely used Illumina sequencing platforms have provided the ability to characterize bacterial communities at an unprecedented sequencing depth [26, 27]. However, the generated metagenomic sequencing data is extensive and highly fragmented, and the identification of novel qnr genes therefore requires computationally efficient methods with high sensitivity and specificity.
In this report, we expand upon the work presented in Boulund et al. (2012). Here, we modified and improved the approach and also analyzed a substantially larger data set containing more than 12 terabases of sequence data from bacterial genomes and metagenomes. We identified 362,843 qnr gene fragments from which 611 qnr gene sequences could be reconstructed. In total 52 qnr genes were reconstructed from metagenomic data, and 20 of these were predictions of novel and previously undescribed qnr variants. Nine novel qnr genes were selected for experimental validation, and six of these (88.8%) conferred consistently reduced susceptibility against fluoroquinolone in E. coli.
Methods
In this study, we applied an extended and improved version of the method for identification of novel resistance genes described in Boulund et al. (2012). The method has been extensively validated on short nucleotide fragments produced by next generation sequencing platforms [10]. In particular, the sensitivity to identify 100 nucleotide long fragments from novel qnr genes is 94% with a specificity of 99.8% against other non-qnr pentapeptide repeat proteins. Details for the performance for individual qnr classes are available in Boulund et al. 2012 [10]. The original method operated by translating each nucleotide sequence into amino acids in all six reading frames. Then, a profile hidden Markov model (HMM), based on a multiple alignment of 66 well-characterized plasmid-mediated qnr genes, was used to score each fragment based on its sequence [10]. Discrimination between fragments from qnr genes and fragments of other origins (including fragments from pentapeptide proteins without fluoroquinolone resistance phenotype) was done by a classifier based on alignment bit score and fragment length. Finally, a clustering step was used to group the qnr fragments into clusters representing full-length genes. Complete details are available in Boulund et al. (2012). In this work, we have extended the previous method to make it applicable to larger volumes of data than was feasible before. In particular, the management of the input sequence data has been optimized by minimizing the storage of translated amino acid sequence data and other intermediate files. The last step, where the previous approach clustered all identified putative qnr fragments, has also been modified. The reads classified as containing qnr fragments are now assembled using metagenome assembly methods, instead of merely clustering them. This makes the method applicable to larger sets of fragmented DNA sequence data, such as those commonly produced in recent metagenomic studies, characterized by short-read high-throughput sequencing techniques (e.g. Illumina sequencing).
In this study, the HMM-based method was used to search for the presence of new qnr genes in a large collection of nucleotide sequence data sets, including metagenomes (from human-associated microbial communities, polluted environments, and waste water treatment plants), fully assembled bacterial genomes, and assorted nucleotide sequences from public repositories (e.g. NCBI Genbank). All sequence data (Table 1) were downloaded between May and December 2015 and processed as follows. Sequence files in FASTQ format were quality filtered and converted to FASTA using the FASTX-toolkit version 0.13.2 [28]. Quality filtering was performed with fastq_quality_filter; option: “-q 30”, and FASTQ to FASTA conversion was done with fastq_to_fasta (default settings). All nucleotide data was translated into peptide sequences in all six reading frames using EMBOSS transeq version 6.3.1 [29]; options: “-frame 6 -Table 11”. After translation, HMMER3 (hmmsearch) version 3.1b1 [30] was used together with the hidden Markov model constructed from experimentally verified plasmid-mediated qnr genes [10] to search the data sets for the presence of qnr fragments; options: “--domtblout -E 1000 --domE 1000”. All fragments matching the HMM were filtered using the fragment length dependent filtering criteria from Boulund et al. (2012). Both read pairs were retrieved for each fragment passing the filtering (even if only one of them matched the HMM), and then used in gene assembly for each individual data set. The paired-end reads were trimmed using Trim Galore! [31] version 0.4.1, using default settings. Assembly of the filtered retrieved paired-end reads was done using the SPAdes assembler version 3.7.0, option: “--meta”. The contigs produced in the assembly were then scored and evaluated against the HMM, using the same length dependent filtering criteria as above, to further validate each assembled sequence.
Nine assembled full-length sequences were selected for experimental validation, based on their novelty (no identical sequences in publicly available databases such as NCBI GenBank) and similarity to previously described qnr variants (priority given to sequences with high similarity to plasmid-mediated qnr). Predicted genes were considered novel based on the criteria presented in Jacoby et al. (2008) [32]. The qnr gene candidates were synthesized with KpnI and BamHI restriction sites attached to the 5′ and 3′ ends, respectively. The genes were subcloned into the expression vector pZE21 (EXPRESSYS, Ruelzheim, Germany) under the control of the inducible promoter PLtetO-1 by utilizing the attached restriction sites [33]. The generated plasmids were electroporated into E. coli C600Z1 (EXPRESSYS). Previously described C600Z1 strains harboring pZE21 plasmids without any gene insert or with either qnrA1, qnrB1, or qnrVC1 inserted, were included as controls [23]. The strains carrying the different pZE21 plasmids were cultured overnight on Mueller-Hinton agar (MHA) (Oxoid, Basingstoke, UK) supplemented with kanamycin (50 μg/mL) and subjected to ciprofloxacin susceptibility testing with Etest® gradient strips (BioMérieux SA, Marcy l’Etoile, France) according to the manufacturer’s instructions. The tests were performed on MHA containing the inducer anhydrotetracycline (250 ng/mL) as well as on unsupplemented MHA. E. coli ATCC 25922 was used as quality control strain.
Results
In total, 12.7 terabases of nucleotide data distributed over 130 billion sequences were screened for novel qnr genes. The data consisted of metagenomes (12.7 terabases; 99.41%), NCBI RefSeq bacterial genomes (9.63 gigabases; 0.08%) and the NCBI nt database (62 gigabases; 0.41%) (Table 1). In total, 362,843 sequences were classified as fragments from qnr genes, which corresponded to an overall relative abundance of 2.79 × 10−6. The abundance of qnr gene fragments in metagenomes ranged from 5.99 × 10−7 to 8.59 × 10−4, where the highest levels were found in samples from river and lake sediments contaminated with fluoroquinolones (Table 1). The method identified 611 different qnr gene sequences longer than 200 amino acids across all data sets. As expected, most of the sequences (559; 91.5%) were previously characterized qnr sequences present in NCBI GenBank and NCBI RefSeq bacterial. Of these, 78 sequences were not previously annotated as qnr genes (Additional file 1: Table S1). Of the 5232 analyzed genomes, 49 (0.93%) carried qnr genes. In particular, three genes were found in the chromosomes of Vibrio nigripulchritudo str SFn1 (NC_022543.1), Vibrio campbellii ATCC BAA 1116 (NC_022270.1) and Serratia liquefaciens ATC 27592 (NC_021741.1). These species have to the authors’ best knowledge not been previously reported in the literature to carry qnr genes. A total of 52 (8.51%) full-length sequences were assembled from metagenomic data, out of which 20 were putatively novel qnr genes (either not present or not annotated as a fluoroquinolone resistance gene in Genbank). Full-length qnr genes (>200 aa) were assembled from all data sets but two: one with human gut metagenomes [34] and one with soil metagenomes [35]. All predicted qnr sequences are presented in (Additional file 2).
The 20 novel qnr-like sequences identified in this work had a length between 206 and 223 amino acids and showed, on average, 57.12% amino acid sequence identity to previously described plasmid-mediated qnr genes (Table 2). Out of the 20 novel qnr sequences identified, nine assembled full-length sequences were selected for experimental validation by gene synthesis and transformation into an E. coli host. Induced expression of six of the selected sequences resulted in an up to 11-fold increase of the ciprofloxacin minimum inhibitory concentration (MIC) (0.016 to 0.19 mg/L) (Fig. 1). No increase in MIC could be seen for Wadden 4, while Wadden 5 and Oil spill 2 showed inconsistent results between the replicated experiments. Phylogenetic analysis showed that all of the validated genes formed their own clades within the span of the established plasmid-mediated qnr families. Sequences Wadden 3 and Wadden 4 formed one small subclade, as did Oil spill 2 and Oil spill 3. All other validated genes formed their own individual clades. A complete tree containing: 1) all sequences discovered in this work, 2) all plasmid-mediated qnr, and 3) mfpa, is available as an additional figure (Additional file 3: Figure S1).

Gene tree showing the relationships between previously known plasmid-mediated qnr sequences and the sequences validated in this work. The gene tree was produced with ETE 3 using the “standard_fasttree” workflow [50,51,52]. The non-qnr peptapeptide repeat protein MfpA was used as an outlier to root the tree. The bar plot shows fold change of ciprofloxacin minimum inhibitory concentrations for all the tested sequences from three separate measurements each. MIC values for QnrD1, QnrS1, and QnrC for the first time point were taken from our previous study [23]. Wadden 4 did not display an average increase in MIC. Refer to Table 2 for a detailed listing of all sequences discovered in this work, and to Additional file 5: Table S3 for the raw MIC measurement data
Discussion
In this report, we developed an improved computational approach, appropriate also for deeply sequenced short-read metagenomes, and applied it to screen for new qnr genes in collections of DNA sequence data almost totaling 13 terabases. In contrast to what we previously achieved in [10], this study screened an almost 20 times larger dataset, this time dominated by short read metagenomic data produced by next generation DNA sequencing. This also makes this study, to the authors’ best knowledge, the largest and most exhaustive screening of novel qnr genes in publicly available nucleotide sequence datasets to date. Our study shows that qnr-like genes are widespread in different environments and thus, supports the notion that qnr genes could have originated from harmless environmental bacteria. We identified 20 potentially new, full-length gene sequences, all from metagenomic datasets, which have previously not been described or associated with fluoroquinolone resistance. Validation experiments, where the identified putative novel qnr genes were expressed in an E. coli host, were used to confirm the fluoroquinolone resistance phenotype for six out of nine selected genes, reinforcing confidence in the performance of the predictive model.
The general method applied in this work has previously been shown to reliably predict novel qnr genes in sequence data. In particular, extensive cross-validation experiments have demonstrated high true positive rates while maintaining low false positive rates, even for nucleotide fragments as short as 100 bases [10]. Previous predictions based on this method have been experimentally shown to provide increased fluoroquinolone resistance, yielding minimum inhibitory concentration in the same range as other qnr genes, when expressed in E. coli [23]. Moreover, the method was able to identify all known plasmid-mediated and chromosomally located qnr genes in NCBI Genbank and RefSeq, further underlining the validity of the predictions. In addition, three families of qnr genes (qnrD, qnrS and qnrVC) were identified and fully assembled from the metagenomes sampled from fluoroquinolone polluted river sediments collected up and downstream from an industrial waste water treatment plant in Patancheru, near Hyderabad, India [15, 36, 37]. These specific gene families have previously been shown to be the most abundant in these samples using other molecular techniques, such as 454 pyrosequencing and quantitative PCR [15, 35]. The accuracy of the method was further emphasized by the fact that six out of nine tested genes consistently conferred the predicted resistance phenotype in E. coli. Thus, taken together, our results show that the method is reliable and can correctly identify known and unknown qnr genes present in fragmented sequence data. This also implies that the great majority of the genes found in this study that were not validated are also likely to be correctly predicted functional qnr genes.
Our results show that qnr genes are present in the bacterial chromosomes of 49 (0.93%) of the 5242 analyzed genomes from RefSeq (Additional file 2; Additional file 4: Table S2). This agrees well with reports from previous studies showing that qnr genes are carried by a wide range of different bacterial species [9, 10, 14, 38, 39]. Chromosomal and plasmid-mediated qnr genes have also been shown to be almost ubiquitously present in e.g. soil, sediment, aquatic, and human microbiomes [9, 10, 14, 38, 39], which is reflected in our analysis, where fragments from potential qnr genes were identified in all examined metagenomes. Metagenomes from river and lake sediments polluted by fluoroquinolones had the highest abundance of qnr fragments, suggesting that a continuous selection pressure effectively enriches for resistance genes, increasing their relative abundance in the sequenced communities. Interestingly, qnr gene abundances were especially low in the metagenomes from the human microbiome (Table 1). This is in line with previous studies that have demonstrated low abundances of the six known plasmid-mediated qnr gene families in the human gut microflora [17, 40, 41]. Of the 80,000 potential fragments identified in the Human Microbiome Project (764 samples from 300 individuals), only the previously characterized qnrB62 was fully reconstructed. Instead, the majority of the fragments assembled into contigs that, based on their full length sequences, were not classified as qnr and thus disregarded in the analysis. This indicates that a large proportion of the fragments identified in the human microbiome are likely to be false positives and thus not from true qnr genes. One possible explanation for the lack of fully reconstructed qnr genes from the human microbiome data could be that the abundance of qnr genes is very low. This, possibly combined with a high species diversity between individuals [34, 42,43,44], may negatively influence our ability to completely assemble any qnr genes from these data. Our results indicate, however, that no known or novel qnr genes are ubiquitously spread in high abundance across the metagenomes of the individuals included in this study, suggesting that horizontally transferrable qnr fluoroquinolone resistance genes are rare in the human microbiome. Studies using qPCR, however, indicate that qnr genes can be more common in the fecal flora of people from certain regions, e.g. India [35].
Out of the sequences validated in our E. coli model, all but three caused an increased MIC of ciprofloxacin in E. coli when overexpressed. The strain carrying the sequence Wadden 4 showed no increase in MIC. For Wadden 5 and Oil spill 2, an increase in MIC was only detectable in one and two of the three experiments, respectively. This suggest that these three genes may lack the ability to induce a resistance phenotype to ciprofloxacin. However, the phylogenetic analysis showed that all these three genes were not more distant to the known mobile qnr genes than other genes predicted in this study. For example, Wadden 4 formed a small subclade together with Wadden 3, which provided a minor (2-fold) increase in MIC. This may suggest some of these genes were not expressed properly in our assay. However, all other validated sequences exhibited average MIC fold changes between 2 to 10.6, which is in line with representatives of the plasmid-mediated qnr families that were also included in the validation for reference [45]. Indeed, several of the novel qnr genes identified in this study resulted in an increase of the MIC close, or in some cases, above previously identified mobile qnr genes. For example, Wadden 1 had an average fold change in MIC of 10.6 (as high as 15.8 in one experiment), which was larger than QnrA1, QnrC, QnrD1, QnrS1 and QnrVC1. In addition, Tara 2 demonstrated an increase in MIC (2.9) that was higher than QnrA1. Thus, our results suggest that several of the novel qnr genes identified in this study may, if transferred into pathogens, become future clinical problems and contribute to the growing number of bacteria resisting antibiotic treatment.
The evolutionary distance between the established plasmid-mediated qnr gene families is known to be large and the sequence identity between different variants can be as low as 37.61%, as observed between QnrB24 and QnrS5 [46]. Identification of novel qnr gene families therefore requires alignment strategies with high sensitivity and specificity. The computational approach used in this study is based on profile HMMs implemented in HMMER3 [30]. Profile HMMs incorporate sequence variability by describing position-specific amino acid distributions and their short-range dependencies, where the parameters are typically estimated from a multiple alignment of a set of representative sequences. It should, however, be pointed out that qnr genes exhibits a wide range of other properties that are not specifically modelled by the profile HMM used in this study. One such example is the recently discovered external surface loops, i.e. short regions of the amino acid residue chain that protrude out of the side of the otherwise regular β-helix structure, observed in the majority of the available mobile qnr gene structures [47, 48]. Incorporating information about the fold of the protein requires modelling of long-range dependencies that are hard to accurately capture in an HMM. It might therefore be possible to develop a more sophisticated model that could increase the sensitivity and specificity even further. However, increasing model complexity often comes at the cost of a higher computational burden. The profile HMM implemented in HMMER3 provides both satisfactory sensitivity and specificity in combination with high computational performance, making it well suited for application to large amounts of metagenomic sequencing data.
The amount of nucleotide data screened in this work presents substantial computational challenges. In particular, a large proportion of the data (99.44%) consists of metagenomic DNA sequences that are highly fragmented. A common approach when analyzing this type of data is to assemble each metagenome prior to analysis. However, de novo assembly of large data sets is a daunting task that quickly becomes unfeasible because of the computational and memory complexity, in the worst case, exhibiting quadratic growth with increasing number of fragments [49]. Assembly of all the data included in this study would therefore be highly impractical. Instead, our method essentially applies a data reducing filtering approach where fragments unlikely to represent qnr genes (based on similarity to the HMM) are excluded prior to assembly. This provided an almost million-fold decrease of the sequence data (Table 1), which made analysis of these large data sets possible while still maintaining a very high sensitivity, granting us the ability to apply sensitive assembly algorithms to stitch together the identified fragments. Because our method allows studies of all individual metagenomic sequence fragments without prior assembly, it is in a better position to identify fragments from low abundance sequences. Normally, fragments from lowly abundant sequences potentially risk being discarded as assembly chaff when all sequence data is assembled simultaneously. Our approach is therefore well-suited also for screening of future metagenomes with even higher sequencing depth.
Conclusions
We screened more than 12 terabases of DNA for novel qnr fluoroquinolone resistance genes and were able to identify 20 putatively novel qnr genes. Of the nine selected for experimental validation, six consistently showed an increased MIC to ciprofloxacin at levels comparable to previously established qnr variants when expressed in E. coli. Our results thus expand the number of qnr gene variants and further elucidates the diversity of this important class of resistance genes. The results also reinforce the hypothesis that environmental bacterial communities act as sources of previously uncharacterized qnr genes. Identification of novel antibiotic resistance genes before they are mobilized and transferred to human pathogens enables surveillance at an early stage, facilitating implementation of preventive actions to counter the spread of new variants of multiresistant bacteria in clinical environments.
Abbreviations
- DNA:
-
Deoxyribonucleic acid
- HMM:
-
Hidden Markov model
- MHA:
-
Mueller-Hinton agar
- MIC:
-
Minimum inhibitory concentration
- NCBI:
-
National Center for Biotechnology Information
- PCR:
-
Polymerase chain reaction
- qPCR:
-
Quantitative polymerase chain reaction
References
Redgrave LS, Sutton SB, Webber M. A., Piddock LJ V. Fluoroquinolone resistance: mechanisms, impact on bacteria, and role in evolutionary success. Trends Microbiol. Elsevier Ltd. 2014;22:438–45.
Martínez-Martínez L, Pascual A, Jacoby GA. Quinolone resistance from a transferable plasmid. Lancet. 1998;351:797–9.
Hata M, Suzuki M, Matsumoto M. Cloning of a novel gene for quinolone resistance from a transferable plasmid in Shigella flexneri 2b. Antimicrob agents. 2005;
Jacoby G, Walsh K, Mills D. qnrB, another plasmid-mediated gene for quinolone resistance. Antimicrob. agents. 2006;50.
Wang MM, Guo Q, Xu X, Wang X, Ye X, Wu S, et al. New plasmid-mediated quinolone resistance gene, qnrC, found in a clinical isolate of Proteus Mirabilis. Antimicrob Agents Chemother. 2009;53:1892–7.
Cavaco LM, Hasman H, Xia S, Aarestrup FM. qnrD, a novel gene conferring transferable quinolone resistance in salmonella enterica serovar Kentucky and Bovismorbificans strains of human origin. Antimicrob. Agents Chemother. 2009;53:603–8.
Xia R, Guo X, Zhang Y, Xu H. qnrVC-like gene located in a novel complex class 1 integron harboring the ISCR1 element in an Aeromonas Punctata strain from an aquatic environment in Shandong Province, China. Antimicrob. Agents Chemother. 2010;54:3471–4.
Jacoby GA. qnr Numbering and Sequence [Internet]. Lahey Clin. 2016 [cited 2016 Oct 6]. p. 1. Available from: http://www.lahey.org/qnrStudies/
Velasco C, Rodríguez-Martínez JM, Briales A, Díaz de Alba P, Calvo J, Pascual A. Smaqnr, a new chromosome-encoded quinolone resistance determinant in Serratia marcescens. J Antimicrob Chemother. 2010;65:239–42.
Boulund F, Johnning A, Pereira MB, Larsson DGJ, Kristiansson E. A novel method to discover fluoroquinolone antibiotic resistance (qnr) genes in fragmented nucleotide sequences. BMC Genomics. 2012;13:695.
Poirel L, Liard A, Rodriguez-Martinez J-M, Nordmann P. Vibrionaceae as a possible source of Qnr-like quinolone resistance determinants. J Antimicrob Chemother. 2005;56:1118–21.
Allen HK, Donato J, Wang HH, Cloud-Hansen KA, Davies J, Handelsman J. Call of the wild: antibiotic resistance genes in natural environments. Nat. Rev. Microbiol. Nature Publishing Group. 2010;8:251–9.
Finley R, Collignon P, Larsson DGJ, A. McEwen S, Li X-Z, Ried-Smith R, et al. The scourge of antibiotic resistance: the important role of the environment. Clin. Infect. …. 2013;1–24.
Sánchez MB, Hernández A, Rodríguez-Martínez JM, Martínez-Martínez L, Martínez JL. Predictive analysis of transmissible quinolone resistance indicates Stenotrophomonas maltophilia as a potential source of a novel family of Qnr determinants. BMC Microbiol. 2008;8:148.
Kristiansson E, Fick J, Janzon A, Grabic R, Rutgersson C, Weijdegård B, et al. Pyrosequencing of antibiotic-contaminated river sediments reveals high levels of resistance and gene transfer elements. PLoS One. 2011;6:e17038.
Flach C-F, Johnning A, Nilsson I, Smalla K, Kristiansson E, Larsson DGJ. Isolation of novel IncA/C and IncN fluoroquinolone resistance plasmids from an antibiotic-polluted lake. J Antimicrob Chemother. 2015;70(10):1–9.
Bengtsson-Palme J, Angelin M, Huss M, Kjellqvist S, Kristiansson E, Palmgren H, et al. The human gut microbiome as a transporter of antibiotic resistance genes between continents. Antimicrob. Agents Chemother. 2015;AAC.00933-15.
Perry JA, Wright GD. The antibiotic resistance “mobilome”: searching for the link between environment and clinic. Front Microbiol. 2013;4:1–7.
Vaz-Moreira I, Nunes OC, Manaia CM. Bacterial diversity and antibiotic resistance in water habitats: searching the links with the human microbiome. FEMS Microbiol Rev. 2014;38:761–78.
Bengtsson-Palme J, Larsson DGJ. Antibiotic resistance genes in the environment: prioritizing risks. Nat. Rev. Microbiol. Nature Publishing Group. 2015;260:6–7.
Tringe SG. Von Mering C, Kobayashi a, Salamov a a, Chen K, Chang HW, et al. comparative metagenomics of microbial communities. Science. 2005;308:554–7.
Zhu A, Sunagawa S, Mende DR, Bork P. Inter-individual differences in the gene content of human gut bacterial species. Genome Biol. 2015;16:1–13.
Flach C-F, Boulund F, Kristiansson E, Larsson DGJ. Functional verification of computationally predicted qnr genes. Ann Clin Microbiol Antimicrob. 2013;12:34.
Loman NJ, Constantinidou C, Chan JZM, Halachev M, Sergeant M, Penn CW, et al. High-throughput bacterial genome sequencing: an embarrassment of choice, a world of opportunity. Nat Rev Microbiol Nature Publishing Group. 2012;10:599–606.
Cochrane G, Alako B, Amid C, Bower L, Cerdeño-Tárraga A, Cleland I, et al. Facing growth in the European nucleotide archive. Nucleic Acids Res. 2013;41:D30–5.
Gilbert JA, Dupont CL. Microbial Metagenomics: beyond the genome. Ann Rev Mar Sci Annual Reviews. 2011;3:347–71.
Schmieder R, Edwards R. Insights into antibiotic resistance through metagenomic approaches. Future Microbiol. 2012;7:73–89.
Gordon A, Hannon GJ. Fastx-toolkit. FASTQ/A short reads pre-processing tools (unpublished) 2010.
Rice P, Longden I, Bleasby A. The European Molecular Biology Open Software Suite EMBOSS : The European Molecular Biology Open Software Suite 2000;16:2–3.
Eddy SR. Accelerated profile HMM searches. PLoS Comput Biol. 2011;7:e1002195.
Krueger F. Trim galore!: a wrapper tool around Cutadapt and FastQC to consistently apply quality and adapter trimming to FastQ files. 2015.
Jacoby G, Cattoir V, Hooper D, Martínez-Martínez L, Nordmann P, Pascual A, et al. qnr Gene nomenclature. Antimicrob. Agents Chemother. 2008;52:2297–9.
Lutz R, Bujard H. Independent and tight regulation of transcriptional units in escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 1997;25:1203–10.
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65.
Rutgersson C, Fick J, Marathe N, Kristiansson E, Janzon A, Angelin M, et al. Fluoroquinolones and qnr genes in sediment, water, soil, and human fecal flora in an environment polluted by manufacturing discharges. Environ Sci Technol. 2014;48:7825–32.
Larsson DGJ, de Pedro C, Paxeus N. Effluent from drug manufactures contains extremely high levels of pharmaceuticals. J Hazard Mater. 2007;148:751–5.
Fick J, Soderstrom H, Lindberg RH, Phan C, Tysklind M, Larsson DGJ. Contamination of surface, ground, and drinking water from pharmaceutical production. Environ Toxicol Chem. 2009;28:2522–7.
Jacoby GA, Hooper DC. Phylogenetic analysis of chromosomally determined qnr and related proteins. Antimicrob Agents Chemother. 2013;57:1930–4.
Arsène S, Leclercq R. Role of a qnr-like gene in the intrinsic resistance of Enterococcus faecalis to fluoroquinolones. Antimicrob Agents Chemother. 2007;51:3254–8.
Forslund K, Sunagawa S, Roat Kultima J, Mende D, Arumugam M, Typas A, et al. Country-specific antibiotic use practices impact the human gut resistome. Genome Res. 2013;23(7):1163–9.
Nesme J, Cécillon S, Delmont TO, Monier JM, Vogel TM, Simonet P. Large-scale metagenomic-based study of antibiotic resistance in the environment. Curr Biol. 2014;24:1096–100.
Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174–80.
Huttenhower C, Gevers D, Knight R, Abubucker S, Badger JH, Chinwalla AT, et al. Structure, function and diversity of the healthy human microbiome. Nature Publishing Group. 2012;486:207–14.
Penders J, Stobberingh EE, Savelkoul PHM, Wolffs PFG. The human microbiome as a reservoir of antimicrobial resistance. Front Microbiol. 2013;4:87.
Hooper DC, Jacoby GA. Mechanisms of drug resistance: quinolone resistance. Ann N Y Acad Sci. 2015:1–20.
Strahilevitz J, Jacoby GA, Hooper DC, Robicsek A. Plasmid-mediated quinolone resistance: a multifaceted threat. Clin Microbiol Rev. 2009;22:664–89.
Xiong X, Bromley EHC, Oelschlaeger P, Woolfson DN, Spencer J. Structural insights into quinolone antibiotic resistance mediated by pentapeptide repeat proteins: conserved surface loops direct the activity of a Qnr protein from a gram-negative bacterium. Nucleic Acids Res. 2011;39:3917–27.
Vetting MW, Hegde SS, Wang M, Jacoby GA, Hooper DC, Blanchard JS. Structure of QnrB1, a plasmid-mediated fluoroquinolone resistance factor. J Biol Chem. 2011;286:25265–73.
Miller JR, Koren S, Sutton G. Assembly algorithm for next-Ganeration sequencing data. Genomics. 2010;95:315–27.
Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal omega. Mol Syst Biol. 2011;7:539.
Huerta-Cepas J, Serra F, Bork P. ETE 3: reconstruction, analysis, and visualization of Phylogenomic data. Mol Biol Evol. 2016;33:1635–8.
Price MN, Dehal PS, Arkin AP. FastTree 2 - approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5
Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature Nature Publishing Group. 2012;490:55–60.
Bengtsson-Palme J, Hammarén R, Pal C, Östman M, Björlenius B, Flach C-F, et al. Elucidating selection processes for antibiotic resistance in sewage treatment plants using metagenomics. Sci Total Environ Elsevier BV. 2016;572:697–712.
Bengtsson-Palme J, Boulund F, Fick J, Kristiansson E, Larsson DGJ. Shotgun metagenomics reveals a wide array of antibiotic resistance genes and mobile elements in a polluted lake in India. Front Microbiol. 2014;5:1–14.
Pal C, Bengtsson-palme J, Kristiansson E, Larsson DGJ. The structure and diversity of human, animal and environmental resistomes. Microbiome Microbiome. 2016;4:1–15.
Mason OU, Hazen TC, Borglin S, Chain PSG, Dubinsky EA, Fortney JL, et al. Metagenome, metatranscriptome and single-cell sequencing reveal microbial response to Deepwater Horizon oil spill. ISME J. 2012;6(9):1715–27.
Marathe NP, Pal C, Gaikwad SS, Jonsson V, Kristiansson E, Larsson DGJ. Untreated urban waste contaminates Indian river sediments with resistance genes to last resort antibiotics. Submitted. 2017;
Strous M, Kraft B, Bisdorf R, Tegetmeyer HE. The binning of metagenomic contigs for microbial physiology of mixed cultures. Front Microbiol. 2012;3:1–11.
Pesant S, Not F, Picheral M, Kandels-Lewis S, Le Bescot N, Gorsky G, et al. Open science resources for the discovery and analysis of Tara oceans data. Sci data. 2015;2:150023.
Acknowledgements
Not applicable.
Funding
This research was supported by the Swedish Research Council (VR), Life Science Area of Advance at Chalmers University of Technology, and the Wallenberg Foundation. Funding sources took no part in the design, analysis, or interpretation of the results.
Availability of data and materials
All data generated or analyzed during this study are included in this published article (and its additional information files).
Author information
Authors and Affiliations
Contributions
FB1, FB2, DGJL, and EK designed the study. FB1, FB2, JBP, NM collected, and performed pre- and post-processing of the data. FB2 engineered and ran the data analysis pipeline and optimized the clustering parameters. FB1 analyzed the results, constructed the phylogenetic trees, and drafted the manuscript. CFF performed the validation experiments. All authors contributed to and approved of the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethics approval does not apply. This study has not used humans, animals, or plants directly.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1: Table S1.
Annotations for matches in NCBI nt and bacterial genomes. Best hit annotations for sequences matched by the hidden Markov model in NCBI nt and bacterial genome sequences. (XLSX 15 kb)
Additional file 2:
Assembled putative qnr sequences. Description: Assembled putative qnr sequences (DOCX 18 kb)
Additional file 3: Figure S1.
Complete gene tree. Description: A gene tree containing all assembled putative qnr gene sequences identified in metagenomic data sets, including all established qnr gene families. Gene tree created from amino acid sequences with ETE 3 using the “standard_fasttree” workflow. Tree visualized with Dendroscope. Sequences discovered in this work highlighted in bold. (PDF 22 kb)
Additional file 4: Table S2.
RefSeq genomes with qnr genes. A list of RefSeq genomes where our model detected putative qnr genes. (DOCX 12 kb)
Additional file 5: Table S3.
MIC measurements. Raw data for Fig. 1. MIC measurements of assembled, putatively novel, qnr gene sequences. Measurements were taken on separate days. (XLSX 53 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Boulund, F., Berglund, F., Flach, CF. et al. Computational discovery and functional validation of novel fluoroquinolone resistance genes in public metagenomic data sets. BMC Genomics 18, 682 (2017). https://doi.org/10.1186/s12864-017-4064-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-017-4064-0