
Abstract
Background
Broad-spectrum fluoroquinolone antibiotics are central in modern health care and are used to treat and prevent a wide range of bacterial infections. The recently discovered qnr genes provide a mechanism of resistance with the potential to rapidly spread between bacteria using horizontal gene transfer. As for many antibiotic resistance genes present in pathogens today, qnr genes are hypothesized to originate from environmental bacteria. The vast amount of data generated by shotgun metagenomics can therefore be used to explore the diversity of qnr genes in more detail.
Results
In this paper we describe a new method to identify qnr genes in nucleotide sequence data. We show, using cross-validation, that the method has a high statistical power of correctly classifying sequences from novel classes of qnr genes, even for fragments as short as 100 nucleotides. Based on sequences from public repositories, the method was able to identify all previously reported plasmid-mediated qnr genes. In addition, several fragments from novel putative qnr genes were identified in metagenomes. The method was also able to annotate 39 chromosomal variants of which 11 have previously not been reported in literature.
Conclusions
The method described in this paper significantly improves the sensitivity and specificity of identification and annotation of qnr genes in nucleotide sequence data. The predicted novel putative qnr genes in the metagenomic data support the hypothesis of a large and uncharacterized diversity within this family of resistance genes in environmental bacterial communities. An implementation of the method is freely available at http://bioinformatics.math.chalmers.se/qnr/.
Similar content being viewed by others


Background
Antibiotics are one of our most powerful tools for treating and preventing bacterial infections and have since their introduction vastly improved human health and drastically reduced mortality rates. The high use of antibiotics in human and veterinary medicine has however resulted in an accelerated development of multiresistant bacteria [1, 2]. Bacteria can adapt to an antibiotic selection pressure by altering their genome, either by mutations in pre-existing DNA or through the acquisition of resistance genes [3]. Since resistance genes can be horizontally transferred between bacterial cells, antibiotic resistance can rapidly spread within and between bacterial communities [4–6]. Many types of antibiotics are derived from compounds that are naturally found in the environment and bacteria have developed resistance genes as a protection mechanism. Environmental bacterial communities have therefore been hypothesized to contain a large and unexplored collection of antibiotic resistance genes [7–10]. Antibiotic resistance genes were present in environmental bacterial communities long before they emerged in human pathogens [11]. As a consequence, many of the antibiotic resistance genes found in clinical settings have been horizontally transferred from environmental bacteria [12, 13].
The broad-spectrum fluoroquinolone antibiotics were introduced in the early 1960’s and are today extensively used in human and veterinary medicine. Fluoroquinolones interacts with the essential bacterial type II topoisomerases (topoisomerase IV and DNA gyrase) and thereby inhibits DNA replication. The most effective fluoroquinolone resistance mechanism is chromosomal mutations in the antibiotic target proteins which confers high levels of resistance in several bacterial species [14, 15]. Recently, a family of mobile fluoroquinolone antibiotic resistance genes called qnr was discovered [16, 17]. These mobile plasmid-mediated quinolone resistance genes (sometimes labeled PMQR) have been grouped into five recognized classes; qnrA, qnrB, qnrC, qnrD, and qnrS and it is currently unknown whether more classes exist. The qnr genes encode proteins that prevent fluoroquinolones from interacting with DNA/type-II-topoisomerase complexes formed during DNA replication, thus preventing fluoroquinolone inhibition. The levels of resistance conferred by qnr genes are generally lower than chromosomal mutations but can reach up to 1 mg/L (minimum inhibitory concentration) depending on the organism and specific antibiotic compound [18].
The qnr genes belong to the larger family of pentapeptide repeat proteins (PRP), which are ubiquitously present with more than 500 variants described in all forms of life [19]. All PRPs are characterized by a sequence feature consisting of repeating subunits of five amino acid residues following the form A(D/N)LXX. This repetitive pattern makes PRPs fold into a β-helix that performs a wide range of cellular functions and they are found both membrane bound and in the cytoplasm [20]. For qnr genes the β-helix resembles the structure of the DNA spiral and interacts with type II topoisomerases and thereby prevent fluoroquinolone antibiotics to inhibit the function of the complex [21, 22]. Despite the strong similarity in the repeating amino acid pattern between qnr sequences and other PRPs it is unclear exactly why qnr genes provide resistance to fluoroquinolones.
Further characterization of qnr genes is necessary to fully understand their function and estimate their diversity. Assuming the presence of antibiotic resistance genes in clinical settings is the result of transfer of mobile genetic elements from the environment, it is natural to search environmental microbial communities to find previously unidentified qnr genes. Recent culture-independent methods such as metagenomics enables unprecedented exploratory analysis of the genetic basis in microbial communities [23, 24]. This is especially true considering that more than 99% of environmental bacterial communities do not submit easily to cultivation and would consequently be missed with sampling and analysis of individual strains [25, 26]. In combination with next-generation DNA sequencing technologies metagenomics provide means for culture-independent studies of bacterial communities at a very high resolution. However, high-throughput sequencing equipment can currently only produce short DNA fragments (typically 75-400 nucleotides long) which substantially limits the sensitivity and specificity of identifying genes such as qnr[27].
In-silico approaches have previously been used to identify novel variants of qnr genes. For example, Fonseca et al. identified qnr VC1 and qnr VC2 in Vibrio cholerae using sequence comparison to existing plasmid-mediated qnr genes [28]. A similar approach was used by Sanches et al. to identify several chromosomal qnr variants, including multiple members of the class Smqnr from Stenotrophomonas maltophilia[29], and by Velasco et al. to discover Smaqnr in Serratia marcescens[30]. However, all of these studies used sequence alignment tools such as BLAST which do not explicitly make use of the repetitive structure of the qnr genes. Furthermore, none of the previous suggested methods were adapted to short sequence lengths and high volumes of data which makes them inapplicable to sequences from shotgun metagenomics.
In this paper, we describe a novel method to identify fluoroquinolone antibiotic resistance genes in DNA sequence data. By using hidden Markov models combined with a length-dependent classification rule, the method is able to discriminate between qnr and other pentapeptide repeat proteins not associated with a resistance phenotype. Cross-validation estimated that the method had a high statistical power of detecting fragments of qnr genes in metagenomic data, even at fragment lengths as short as 100 nucleotides. The method was applied to sequence data from various databases and both known and novel putative qnr genes were identified. An implementation of the method is freely available at http://bioinformatics.math.chalmers.se/qnr/.
Results
A hidden Markov model (HMM) was constructed from a multiple sequence alignment of all currently known and experimentally verified plasmid-mediated qnr resistance gene amino acid sequences [31]. Using the database search software HMMER3, we analyzed the empirical bit score distributions produced by applying the HMM to two sources of protein sequence data; a) true qnr fragments, created from randomly fragmented qnr sequences and b) non-qnr fragments, created from pentapeptide repeat protein (PRP) sequences not associated with a fluoroquinolone resistance phenotype (see Methods). To visualize the bit score distributions of fragmented sequences, random fragments of qnr and non-qnr sequences were created for each fragment length between 10 and 210 amino acid residues (i.e. full length qnr sequences) and their scores against the HMM were plotted as a function of fragment length. As indicated by Figure 1 true qnr fragments had bit scores that were approximately linear in relation to their fragment length while the bit score distribution of the non-qnr fragments was centered around 33. A two-part linear classification function was therefore introduced to discriminate between true qnr and non-qnr fragments. For fragments up to a length threshold (D), the classification function was linear with an intercept M and slope K. For fragments longer than D, the function used a fixed cutoff C = K × D + M (Figure 1A, Additional files 1, 2, 3, 4, 5).

Fragment bit scores and classification rule. A) The figure shows the distribution of the fragment bit scores at different fragment lengths. The separation between the qnr fragments (light blue) and non-qnr fragments (light red) increase for longer fragment lengths. The solid blue and red lines show the average bit scores for qnr and non-qnr fragments, with their 99th and 1st percentiles in grey dashed lines above and below, respectively. The thick dashed line in black shows the classification function with the optimized parameters K=0.778, M=-7.89, D=150.64 [see Additional file 1: Figure S1, Additional file 2: Figure S2, Additional file 3: Figure S3, Additional file 4: Figure S4, Additional file 5: Figure S5 for plots corresponding to each separate class of qnr]. B) The bit scores when compared to the hidden Markov model for 33 amino acid long fragments, corresponding to the approximately 100 nucleotides long sequence reads common in next-generation sequencing technologies. At this fragment length, the qnr fragments (blue) are clearly separated from the non-qnr (red) with only a small overlap.
Cross-validation was used to optimize the parameters M, K and D of the classification function for identification of novel classes of qnr genes. The optimization was performed for five different models where each model was created by excluding one class of plasmid-mediated qnr proteins (i.e. QnrA, QnrB, QnrC, QnrD and QnrS). The cross-validation was then performed using disjoint set of fragments, one for parameter estimation (training) and one for evaluation of the corresponding performance (validation). The training and validation data sets were created from fragments of both qnr genes and non-qnr PRP genes without any associated resistance phenotype. For each of the five models, the excluded qnr class was also removed from the training dataset. The corresponding performance was, on the other hand, evaluated only using the excluded qnr class and a set of non-qnr genes. Thus, the ability to classify novel qnr genes was evaluated on fragments of gene classes not included in the model. The cross-validation was performed with random fragments ranging from 10 to 209 amino acid residues, each length repeated 2500 times. The parameters of the classification function were estimated to M = -7.89 (1.37), K = 0.778 (0.084), D = 150.64 (27.05) (average over all five models, standard deviation in brackets). The corresponding fixed cutoff C was calculated to C = 109.64 (16.40).
The optimized classification function parameters were then used to validate the statistical power to detect novel putative fragments. At a fragment length as short as 33 amino acids the average power for correctly classifying fragments from novel putative qnr gene classes was 94% (Figure 2A). The results differed between the five models (Figure 2B): for a 33 amino acid long sequence, the power to identify a QnrD fragment (given a model built from QnrA, B, C and S) was highest (99.04%) while the power of identifying a QnrB fragment (given QnrA, C, D and S) was the lowest (88.02%). The specificity was estimated to be above 99.27% for all models and all fragment lengths [Additional file 6: Figure S6]. See Methods for full details.

Estimated power. A) The figure shows the estimated power of detecting fragments from novel classes of qnr as a function of fragment length in nucleotides (averaged over the five different models used in the cross-validation). At a fragment length of 33 amino acids (approximately 100 nucleotides), the power to detect fragments from novel classes of qnr genes was estimated to 94% which increased to 100% for 100 amino acid long fragments. B) A magnification of the upper left region showing the power of detecting each class of qnr genes: QnrA (black), QnrB (red), QnrC (green), QnrD (dark blue) and QnrS (cyan). Corresponding plots for the specificity are available as [Additional file 6: Figure S6].
To search for novel putative qnr gene variants a model based on all five classes of plasmid-mediated qnr genes together with the classifier with the optimized parameter values was applied to protein sequences from various databases and metagenomic sequencing projects; GenBank [32], CAMERA [33], MG-RAST [34], contigs from Meta-HIT [35], and several data sets from SRA [36] (see Table 1 and Methods). A smaller metagenomic dataset from a recent study where a high abundance of qnr genes was detected was also included [37]. The total number of fragments available in all datasets was 478,025,600 comprising 214,168,682,742 nucleotides. In total, 1733 (3.6 × 10-4%) sequence fragments classified as qnr by the method. For the metagenomes the proportion of qnr fragments was estimated to 2.8 × 10-4% (1275 out of 463,364,852 metagenomic fragments), reflecting the low abundance of qnr genes in the environment. All fragments that classified as qnr were stringently clustered into 475 groups (where 165 contained more than one fragments) and annotated against GenBank and the list of known qnr genes [31] [Additional file 7: Table S1]. Among these clusters, all five classes of plasmid-mediated qnr were represented as well as 28 previously described chromosomally located variants [28, 29, 38–41]. In addition, one contig in group #1, which consisted entirely of metagenomic fragments, represented a full length sequence of a novel putative qnr gene with 93% identity (97% similarity) to QnrB1. During the course of this project this sequence was accepted as a novel QnrB variant, QnrB35, and submitted to GenBank [GenBank:AEL00456] [39]. The method was hence capable of reconstructing complete qnr sequences directly from fragmented metagenomic data. This was particularly evident since the complete sequence of QnrB35 was not available in any of the datasets at the time of their retrieval in this project.
The method discovered 732 fragments of metagenomic origin that clustered in 440 groups which did not contain any of the previously described plasmid-mediated or chromosomal qnr genes. An additional 11 sequences of novel putative qnr genes in the genomes of 9 sequenced bacteria were also discovered [Additional file 7: Table S1]. Table 2 shows five examples of groups containing sequences classified as novel putative qnr genes by the method. Sequence #1 was constructed from fragments originating from baby stool metagenomes [43] [SRA accession SRX032366] and shared 79% sequence identity with QnrB37. Sequence #2 was discovered in an environmental samples from coastal sea water outside the North American coast [MG-RAST accession 4441580] as a part of the Gene Ocean Sampling Expedition [44]. This sequence is a 218 amino acid long fragment that shares 33% sequence identity with QnrC. The next three sequences were discovered in bacterial genomes in GenBank. Sequence #3 was discovered in the chromosome of Dickey dadantii 3937 [GenBank:NC_014500.1] and was a 213 amino acid long sequence with 68% identity to QnrB28. Sequence #4 was found in the chromosome of Xenorhabdus bovienii [GenBank:NC_013892.1] and was a 211 amino acid long sequence with 66% identity to QnrB19. Sequence #5 came from the chromosome of Vibrio furnissii [GenBank:CP002378.1] and was a 218 amino acid long sequence sharing 72% identity with QnrC. Full results, including all 475 groups and their annotation, are available in [Additional file 7: Table S1].
Discussion
Qnr genes provide resistance to broad-spectrum fluoroquinolone antibiotics and can move between bacteria using horizontal gene transfer. However, the total number of qnr classes and their diversity in environmental bacterial communities is not clear. We therefore developed a novel method to identify new classes of qnr genes in fragmented metagenomic data. The method uses a hidden Markov model (HMM) to identify candidate qnr fragments which are then further classified based on their model score and sequence length. Cross-validation confirmed that the method had a high sensitivity and specificity to detect fragments from novel classes of known qnr genes, even at fragments as short as 33 amino acid residues. This makes the method applicable to many forms of nucleotide data, including sequences generated by next-generation DNA sequencers. From public sequence repositories the method classified 1733 sequence fragments (3.6 × 10-4%) as qnr, which were further clustered into 475 groups. The method also identified 39 chromosomal qnr variants in 33 bacterial species.
Several of the novel putative qnr genes identified in this study have to the authors’ best knowledge previously not been described in literature. Experimental verification, including phenotypic profiling in multiple bacterial hosts, is therefore necessary to fully evaluate the resistance potential of our predictions. However, the cross-validation demonstrated that the proposed method had a high sensitivity and could discriminate between fragments from classes of known qnr and pentapeptide repeat proteins without a resistance phenotype (Figure 2A). The method was also able to identify all previously reported classes of qnr genes, including the variant qnr B35 which was at the time for this analysis not submitted to the database and thus not included in the hidden Markov model. This shows that the method has a high predictive power and it is therefore possible that several of the predictions indeed represent previously unidentified novel classes or variants of qnr genes.
Many of the identified putative qnr gene fragments were discovered in metagenomes sampled from different types of environments, e.g. human gut [43], seawater [44] and river sediment [37] [see Additional file 7: Table S1]. This indicates that there is an unexplored diversity of qnr genes within environmental bacterial communities and that these can be identified by metagenomic sequencing. However, the amount of nucleotide data currently represented in the sequence repositories merely reflects a tiny fraction of the total microbial diversity on earth [45–47]. In addition, the estimated relative abundance of unknown fragments from putative qnr genes was 2.8 × 10-4% (1275 out of 463,364,852 metagenomic fragments) underlining the vast amounts of sequence data needed to identify and assemble qnr genes from environmental data. It is therefore possible, and even likely, that there are additional variants of qnr genes present in the environmental bacterial communities currently not represented in the sequence repositories due to the heavy undersampling. The data that is currently being generated by large-scale reference metagenome projects, such as the Earth Microbiome Project [48] and the Gene Ocean Sampling [44], will offer a substantially higher sequencing depth and may therefore reveal additional classes and variants of qnr genes.
Our results show that hidden Markov models are highly suitable for identifying sequence fragments from qnr genes. The model used in this study was derived from a multiple alignment of qnr genes and can thereby infer information on the degree of variability at different amino acid positions in the sequence [49]. This is especially useful for pentapeptide repeat proteins which generally have a low sequence similarity except in the conserved residues of the distinctive repetitive A(D/N)LXX motif. In contrast, traditional sequence alignment tools such as BLAST cannot distinguish between important variation in the repeating pattern and variation in the intermediate regions. Previous methods to identify novel qnr genes from DNA sequence data have used BLAST and may therefore have limited sensitivity and specificity [49]. The proposed method has, on the other hand, demonstrated a high power of detecting new classes of qnr genes (Figure 2) and is hence a more suitable approach for identification and annotation of qnr genes.
Controlling the number of false predictions is vital for large-scale data analysis. A low specificity can generate a massive amount of false positives and thereby decrease the quality of analysis and the biological interpretation of the downstream results (in this case the sequence groups). Based on the distribution of bit scores for the sequence fragments (Figure 1A) it is clear that a traditional cut-off would not be sufficient to discriminate between qnr and non-qnr PRPs with both a high sensitivity and specificity. Indeed, a single bit score cut-off would have to be set to 75 to minimize false positives across all fragment lengths, effectively removing the ability to classify fragments shorter than 100 amino acid residues (300 nucleotides). Instead, a linear classification function dependent on fragment length for short fragments enabled correct identification while maintaining a high specificity [Additional file 6: Figure S6]. This makes the method suitable for analysis of large datasets consisting of short sequence fragments and the method is therefore directly applicable to data from next-generation sequencing technologies such as Illumina’s sequencing by synthesis, Life Technologies’ sequencing by ligation (SOLiD) or Roche’s 454 pyrosequencing [50].
The hidden Markov model used by the method was created from all known plasmid-mediated qnr genes with experimentally validated resistance phenotype. However, several recent studies have described chromosomally located qnr genes in wide range of species (e.g. Vibrio spp. alginolyticus, Vibrio harveyi and Aeromonas hydrophilia) [28, 29, 38, 40, 41]. These chromosomal qnr genes show a relatively high sequence similarity to their plasmid-mediated relatives and some have been shown to confer resistance towards fluoroquinolones when expressed in E. coli (e.g. SmaQnr and SmQnr) [29, 41]. Their potential to transfer horizontally between bacteria is however not clear. Even though the hidden Markov model was based on plasmid-mediated gene variants, the method demonstrated a high sensitivity to detect qnr genes in bacterial chromosomes. In fact, the method identified 28 previously reported chromosomally located qnr genes in 24 species. In addition, 11 potentially novel chromosomal qnr genes in 9 different species were also identified [Additional file 7: Table S1]. Interestingly, four previously suggested chromosomal qnr genes were not classified as such by the method. These genes, which are located in Alkaliphilus metalliredigens, Bacteroides thetaiotaomicron, Bacillus weihenstephanensis and Anabaena variabilis have previously been identified as putative qnr genes using BLAST [38]. However, all these genes share low sequence similarity to other qnr genes and their resistance phenotype has so far not been validated. The four genes received very low scores by our model, which may indicate that these are false predictions and hence not qnr genes. All other previously described qnr genes received high scores by the model and were thus classified as qnr.
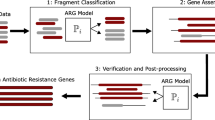
The method described in this study has been implemented as a freely available application in Python. The application searches any specified sequence dataset, classifies the matching sequences as qnr or non-qnr and clusters the results into groups of putative qnr genes [see Additional file 8: Figure S7 for an overview]. The implementation is straightforward to use, has been optimized to handle data sizes of the order of terabytes, and is suitable for use on standard desktop computers. The package is documented with internal functions thoroughly commented in the distributed source code, making it possible to interface them directly from related applications. The application can be installed and run on any modern GNU/Linux system and it is available from http://bioinformatics.math.chalmers.se/qnr/.
Conclusions
In this study we proposed a new method to detect and annotate novel classes of qnr antibiotic resistance genes in nucleotide sequence data. The method uses a hidden Markov model with a fragment length-dependent classification rule and has a high sensitivity and specificity, even for sequences as short at 100 nucleotides. This makes the method directly applicable to the immense amount of data generated by the next-generation DNA sequencing techniques. Based on sequence data currently available in the repositories, the method was able to identify all previously reported plasmid-mediated qnr genes as well as the vast majority of the previously reported chromosomal variants. In addition, the method predicted several novel putative qnr genes and some of these were discovered in shotgun metagenomes, which may indicate a large and unknown diversity of qnr genes in uncultured environmental bacteria.
Methods
A hidden Markov model was based on a multiple sequence alignment of sequences from the reference list of acknowledged and experimentally verified plasmid-mediated qnr genes [31]. Peptide qnr sequences were aligned using MAFFT [51] with default settings. The alignment quality was manually assessed and then used as input for the construction of the hidden Markov model using HMMER3 [49] with default settings.
Investigation of the empirical bit score distribution of the HMM was performed by drawing random fragments of both qnr and non-qnr genes (Figure 1). This led to the creation of a classifier consisting of a two-part linear discrimination function using information of fragment length (Lf) and fragment bit score (Sf) from the hidden Markov model from HMMER. The classifier was defined by three parameters; linear intercept (M), linear slope (K), and long fragment definition (D). A fragment with length, Lf, and domain bit score, Sf, was classified as qnr if Lf < D and Sf ≥ K × Lf + M, or if Lf ≥ D and Sf ≥ K × D + M.
Cross-validation was used to estimate the parameters and to evaluate the performance of the model. Five different models were created and for each model one class of plasmid-mediated qnr genes was excluded. Two different kinds of sequences were used in the cross-validation: true qnr genes and non-qnr pentapeptide repeat protein sequences. The source of true qnr sequences was the reference list of qnr sequences [31] and the source of non-qnr sequences was sequences from GenBank annotated as pentapeptide repeat proteins (PRP) with the COG1357 annotation, but without a known resistance phenotype.
Two sets of data were created for each model in the cross-validation; a training and a validation set. The training sets consisted of a combination of true qnr sequences excluding the class which was left out from the model in question and a set of 90 random non-qnr genes. The validation sets contained all known variants of the previously excluded qnr class plus a different set of 421 non-qnr genes. For example, the first model was based on all known plasmid-mediated qnr sequences excluding the sequences from the class qnrA. This model was then applied to training data consisting of true qnr sequences excluding qnrA and a set of non-qnr genes. The classification function was then applied to validation data consisting exclusively of qnrA and a different set of non-qnr sequences where the performance of the model to identify unknown (i.e. novel) classes of plasmid-mediated qnr was estimated. The fragments used in the cross-validation were randomly generated from the training and validation data sets for each model by randomly drawing a qnr/non-qnr fragments with equal probability. For each dataset, 2500 random fragments were created for each fragment length between 10-210 amino acids. A relatively high mutation rate on amino acid sequence was added by randomly substituting each residue for another with the probability of 5% to introduce a substantial amount of noise.
Parameter values for the classification function were optimized using particle swarm optimization where the parameter spaces for the three parameters were explored (ranges in brackets): M [-20, 30], K [0, 2], and D [30, 210]. Optimization was performed six times using a swarm size of 30 particles with 50 iterations in each run with randomized starting points in parameter space. The objective was to achieve a high true positive rate (TPR) without letting the false positive rate (FPR) becoming too high. The objective function was therefore set to TPR-FPR. The statistical power of the model for identifying novel plasmid-mediated qnr gene variants was computed by using the average parameter values from the six optimization runs when applying the model to the validation data sets (Figure 2).
The nucleotide datasets used in this project (Table 1) were public sequence data sets downloaded in April 2011 (GenBank version 183). Data from the NCBI Sequence Read Archive (SRA) was selected using the search string “metagenom* AND (454 AND (flx OR titanium)) NOT 16S NOT V6 NOT V9” which generated 1756 hits at the time data was sourced for this project. The sequence data was first translated into all six reading frames using bacterial translation table 11 in EMBOSS transeq[52]. The translated sequences were fed into HMMER3 program hmmsearch to find hits against the model. The only non-default settings used were --notextw and --cpu 8, with no change from default settings for inclusion or reporting thresholds. All hits discovered by HMMER3 were instead subjected to the classification function and hits that classified as qnr were clustered using Blastclust [53]. Clustering parameters used were fragment similarity threshold 90% and minimum length coverage 25%. Cluster groups (containing hits/sequence fragments) were aligned using MAFFT to produce overlapping multiple alignments. The aligned groups were then manually adjusted to identify overlapping fragments that formed longer contigs and complete qnr gene contigs. Finally, such contig sequences were annotated using a combination of the reference qnr compilation [31] and the GenBank data displayed in Table 1.
References
Davies J, Davies D: Origins and evolution of antibiotic resistance. Microbiol Mol Biol Rev. 2010, 74: 417-433. 10.1128/MMBR.00016-10.
Neu HC: The crisis in antibiotic resistance. Science (New York, N.Y.). 1992, 257: 837-842.
Martinez J, Baquero F: Mutation frequencies and antibiotic resistance. Antimicrobial agents. 2000, 44: 1771-1777. 10.1128/AAC.44.7.1771-1777.2000.
Stokes HW, Gillings MR: Gene flow, mobile genetic elements and the recruitment of antibiotic resistance genes into Gram-negative pathogens. FEMS Microbiol Rev. 2011, 35: 790-819. 10.1111/j.1574-6976.2011.00273.x.
Boucher Y, Labbate M, Koenig JE, Stokes HW: Integrons: mobilizable platforms that promote genetic diversity in bacteria. Trends Microbiol. 2007, 15: 301-309. 10.1016/j.tim.2007.05.004.
Gillings M, Boucher Y, Labbate M, Holmes A, Krishnan S, Holley M, Stokes HW: The evolution of class 1 integrons and the rise of antibiotic resistance. J Bacteriol. 2008, 190: 5095-5100. 10.1128/JB.00152-08.
Wright GD: The antibiotic resistome: the nexus of chemical and genetic diversity. Nat Rev Microbiol. 2007, 5: 175-186. 10.1038/nrmicro1614.
Cantón R: Antibiotic resistance genes from the environment: a perspective through newly identified antibiotic resistance mechanisms in the clinical setting. Clin Microbiol Infect. 2009, 15 (Suppl 1): 20-25.
D’Costa VM, Griffiths E, Wright GD: Expanding the soil antibiotic resistome: exploring environmental diversity. Curr Opin Microbiol. 2007, 10: 481-489. 10.1016/j.mib.2007.08.009.
D’Costa VM, McGrann KM, Hughes DW, Wright GD: Sampling the antibiotic resistome. Science (New York, N.Y.). 2006, 311: 374-377. 10.1126/science.1120800.
D’Costa VM, King CE, Kalan L, Morar M, Sung WWL, Schwarz C, Froese D, Zazula G, Calmels F, Debruyne R, Golding GB, Poinar HN, Wright GD: Antibiotic resistance is ancient. Nature. 2011, 477: 457-461. 10.1038/nature10388.
Martínez JL: Antibiotics and antibiotic resistance genes in natural environments. Science (New York, N.Y.). 2008, 321: 365-367. 10.1126/science.1159483.
Allen HK, Donato J, Wang HH, Cloud-Hansen KA, Davies J, Handelsman J: Call of the wild: antibiotic resistance genes in natural environments. Nat Rev Microbiol. 2010, 8: 251-259. 10.1038/nrmicro2312.
Von Groll A, Martin A, Jureen P, Hoffner S, Vandamme P, Portaels F, Palomino JC, da Silva PA: Fluoroquinolone resistance in Mycobacterium tuberculosis and mutations in gyrA and gyrB. Antimicrob Agents Chemother. 2009, 53: 4498-4500. 10.1128/AAC.00287-09.
Shah SQA, Nilsen H, Bottolfsen K, Colquhoun DJ, Sørum H: DNA Gyrase and Topoisomerase IV Mutations in Quinolone-Resistant Flavobacterium psychrophilum Isolated from Diseased Salmonids in Norway. Microb Drug Resist (Larchmont, N.Y.). 2012, 207-214.
Martínez-Martínez L, Pascual A, Jacoby GA: Quinolone resistance from a transferable plasmid. Lancet. 1998, 351: 797-799. 10.1016/S0140-6736(97)07322-4.
Robicsek A, Jacoby GA, Hooper DC: The worldwide emergence of plasmid-mediated quinolone resistance. Lancet Infect Dis. 2006, 6: 629-640. 10.1016/S1473-3099(06)70599-0.
Strahilevitz J, Jacoby GA, Hooper DC, Robicsek A: Plasmid-mediated quinolone resistance: a multifaceted threat. Clin Microbiol Rev. 2009, 22: 664-689. 10.1128/CMR.00016-09.
Vetting MW, Hegde SS, Fajardo JE, Fiser A, Roderick SL, Takiff HE, Blanchard JS: Pentapeptide repeat proteins. Biochemistry. 2006, 45: 1-10. 10.1021/bi052130w.
Bateman A, Murzin AG, Teichmann SA: Structure and distribution of pentapeptide repeats in bacteria. Protein Sci. 1998, 7: 1477-1480. 10.1002/pro.5560070625.
Xiong X, Bromley EHC, Oelschlaeger P, Woolfson DN, Spencer J: Structural insights into quinolone antibiotic resistance mediated by pentapeptide repeat proteins: conserved surface loops direct the activity of a Qnr protein from a gram-negative bacterium. Nucleic Acids Res. 2011, 39: 3917-3927. 10.1093/nar/gkq1296.
Vetting MW, Hegde SS, Wang M, Jacoby GA, Hooper DC, Blanchard JS: Structure of QnrB1, a plasmid-mediated fluoroquinolone resistance factor. J Biol Chem. 2011, 286: 25265-25273. 10.1074/jbc.M111.226936.
Handelsman J: Metagenomics: application of genomics to uncultured microorganisms. Microbiol Mol Biol Rev. 2004, 68: 669-10.1128/MMBR.68.4.669-685.2004.
Kristiansson E, Hugenholtz P, Dalevi D: ShotgunFunctionalizeR: an R-package for functional comparison of metagenomes. Bioinformatics (Oxford, England). 2009, 25: 2737-2738. 10.1093/bioinformatics/btp508.
Hugenholtz P, Goebel BM, Pace NR: Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol. 1998, 180: 4765-4774.
Streit WR, Schmitz RA: Metagenomics–the key to the uncultured microbes. Curr Opin Microbiol. 2004, 7: 492-498. 10.1016/j.mib.2004.08.002.
Wommack KE, Bhavsar J, Ravel J: Metagenomics: read length matters. Appl Environ Microbiol. 2008, 74: 1453-1463. 10.1128/AEM.02181-07.
Fonseca ÉL, Santos Freitas F, Vicente ACP: New qnr Gene Cassettes Associated with Superintegron Repeats in Vibrio cholerae O1. Emerg Infect Dis. 2008, 14: 1129-1131. 10.3201/eid1407.080132.
Sánchez MB, Hernández A, Rodríguez-Martínez JM, Martínez-Martínez L, Martínez JL: Predictive analysis of transmissible quinolone resistance indicates Stenotrophomonas maltophilia as a potential source of a novel family of Qnr determinants. BMC Microbiol. 2008, 8: 148-10.1186/1471-2180-8-148.
Rodríguez-Martínez JM, Velasco C, Briales A, García I, Conejo MC, Pascual A: Qnr-like pentapeptide repeat proteins in gram-positive bacteria. J Antimicrob Chemother. 2008, 61: 1240-1243. 10.1093/jac/dkn115.
qnr Numbering and Sequence.http://www.lahey.org/qnrStudies,
Benson DA, Boguski MS, Lipman DJ, Ostell J, Ouellette BF, Rapp BA, Wheeler DL: GenBank. Nucleic Acids Res. 1999, 27: 12-17. 10.1093/nar/27.1.12.
Seshadri R, Kravitz SA, Smarr L, Gilna P, Frazier M: CAMERA: a community resource for metagenomics. PLoS Biol. 2007, 5: e75-10.1371/journal.pbio.0050075.
Meyer F, Paarmann D, D’Souza M, Olson R, Glass EM, Kubal M, Paczian T, Rodriguez A, Stevens R, Wilke A, Wilkening J, Edwards RA: The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinforma. 2008, 9: 386-10.1186/1471-2105-9-386.
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li SSS, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto J, Hansen T, Le D, Linneberg A, Nielsen HB, Pelletier E, Renault P, Sicheritz-Ponten T, Turner K, Zhu H, Yu C, Jian M, Zhou Y, Li Y, Zhang X, Guarner F, Qin N, Yang H, Wang JJ, Brunak S, Dore J, Le Paslier D, Doré J, Kristiansen K, Pedersen O, Parkhill J, Weissenbach J, Bork P, Ehrlich SD: A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010, 464: 59-65. 10.1038/nature08821.
Leinonen R, Sugawara H, Shumway M: The sequence read archive. Nucleic Acids Res. 2011, 39: D19-D21. 10.1093/nar/gkq1019.
Kristiansson E, Fick J, Janzon A, Grabic R, Rutgersson C, Weijdegård B, Söderström H, Larsson DGJ: Pyrosequencing of antibiotic-contaminated river sediments reveals high levels of resistance and gene transfer elements. PLoS One. 2011, 6: e17038-10.1371/journal.pone.0017038.
Baquirin MHC, Barlow M: Evolution and recombination of the plasmidic qnr alleles. J Mol Evol. 2008, 67: 103-110. 10.1007/s00239-008-9131-3.
Jacoby GA, Griffin CM, Hooper DC: Citrobacter spp. as a source of qnrB Alleles. Antimicrob Agents Chemother. 2011, 55: 4979-4984. 10.1128/AAC.05187-11.
Wang MM, Guo Q, Xu X, Wang X, Ye X, Wu S, Hooper DC: New plasmid-mediated quinolone resistance gene, qnrC, found in a clinical isolate of Proteus mirabilis. Antimicrob Agents Chemother. 2009, 53: 1892-1897. 10.1128/AAC.01400-08.
Velasco C, Rodríguez-Martínez JM, Briales A, de Díaz Alba P, Calvo A, Pascual A: Smaqnr, a new chromosome-encoded quinolone resistance determinant in Serratia marcescens. J Antimicrob Chemother. 2010, 65: 239-242. 10.1093/jac/dkp424.
Overbeek R, Begley T, Butler RM, Choudhuri JV, Chuang H-Y, Cohoon M, de Crécy-Lagard V, Diaz N, Disz T, Edwards R, Fonstein M, Frank ED, Gerdes S, Glass EM, Goesmann A, Hanson A, Iwata-Reuyl D, Jensen R, Jamshidi N, Krause L, Kubal M, Larsen N, Linke B, McHardy AC, Meyer F, Neuweger H, Olsen G, Olson R, Osterman A, Portnoy V, et al: The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005, 33: 5691-5702. 10.1093/nar/gki866.
Morowitz MJ, Denef VJ, Costello EK, Thomas BC, Poroyko V: Strain-resolved community genomic analysis of gut microbial colonization in a premature infant. Proc Natl Acad Sci. 2010, 108: 1128-1133.
Yooseph S, Sutton G, Rusch DB, Halpern AL, Williamson SJ, Remington K, Eisen JA, Heidelberg KB, Manning G, Li W, Jaroszewski L, Cieplak P, Miller CS, Li H, Mashiyama ST, Joachimiak MP, van Belle C, Chandonia J-M, Soergel DA, Zhai Y, Natarajan K, Lee S, Raphael BJ, Bafna V, Friedman R, Brenner SE, Godzik A, Eisenberg D, Dixon JE, Taylor SS, Strausberg RL, Frazier M, Venter JC: The Sorcerer II Global Ocean Sampling expedition: expanding the universe of protein families. PLoS Biol. 2007, 5: e16-10.1371/journal.pbio.0050016.
Hooper SD, Dalevi D, Pati A, Mavromatis K, Ivanova NN, Kyrpides NC: Estimating DNA coverage and abundance in metagenomes using a gamma approximation. Bioinformatics (Oxford, England). 2010, 26: 295-301. 10.1093/bioinformatics/btp687.
Gilbert JA, Dupont CL: Microbial Metagenomics: Beyond the Genome. Ann Rev Mar Sci. 2011, 3: 347-371. 10.1146/annurev-marine-120709-142811.
Gilbert JA, Meyer F, Jansson J, Gordon J, Pace N, Ley R, Fierer N, Field D, Kyrpides N, Glöckner F: The Earth Microbiome Project: Meeting report of the “ 1st EMP meeting on sample selection and acquisition ” at Argonne National Laboratory October 6th 2010. 2010, 249-253.
Gilbert JA, Bailey M, Field D, Fierer N, Fuhrman JA, Hu B, Jansson J, Knight R, Kowalchuk GA, Kyrpides NC, Meyer F, Stevens R: The Earth Microbiome Project: The Meeting Report for the 1st International Earth Microbiome Project Conference, Shenzhen, China, June 13th-15th 2011. Stand Genomic Sci. 2011, 5: 243-247. 10.4056/sigs.2134923.
Eddy SR: Accelerated Profile HMM Searches. PLoS Comput Biol. 2011, 7: e1002195-10.1371/journal.pcbi.1002195.
Metzker ML: Sequencing technologies - the next generation. Nat Rev Genet. 2010, 11: 31-46. 10.1038/nrg2626.
Katoh K, Misawa K, Kuma K, Miyata T: MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30: 3059-3066. 10.1093/nar/gkf436.
Rice P, Longden I, Bleasby A: The European Molecular Biology Open Software Suite EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16: 276-277. 10.1016/S0168-9525(00)02024-2.
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25: 3389-3402. 10.1093/nar/25.17.3389.
Acknowledgements
Thanks to Dr Carl-Fredrik Flach for valuable discussions on the topic of mobile fluoroquinolone resistance. This research was supported by the Life Science Area of Advance at Chalmers University of Technology, Sweden, the Swedish Research Council (VR), the Swedish Research Council for Environment, Agricultural Sciences and Spatial Planning (FORMAS) and the Swedish Society for Medical Research (SSMF). Support from the Gothenburg Bioinformatics Network (GOTBIN) is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare no competing interests.
Authors’ contributions
FB, EK and AJ planned the project. FB developed and implemented the method, performed the cross-validation, interpreted the clustering results and annotated the hits. MBP assisted with the implementation of the method. FB and EK drafted the manuscript. The work was supervised by EK, AJ and DGJL. All authors read and approved the final manuscript.
Electronic supplementary material
12864_2012_4583_MOESM1_ESM.pdf
Additional file 1: Figure S1. Fragment bit scores with HMM constructed without QnrA. Bit scores of fragments against the hidden Markov model where all sequences from QnrA were excluded. (PDF 1 MB)
12864_2012_4583_MOESM2_ESM.pdf
Additional file 2: Figure S2. Fragment bit scores with HMM constructed without QnrB. Bit scores of fragments against the hidden Markov model where all sequences from QnrB were excluded. (PDF 1 MB)
12864_2012_4583_MOESM3_ESM.pdf
Additional file 3: Figure S3. Fragment bit scores with HMM constructed without QnrC. Bit scores of fragments against the hidden Markov model where all sequences from QnrC were excluded. (PDF 1 MB)
12864_2012_4583_MOESM4_ESM.pdf
Additional file 4: Figure S4. Fragment bit scores with HMM constructed without QnrD. Bit scores of fragments against the hidden Markov model where all sequences from QnrD were excluded. (PDF 1 MB)
12864_2012_4583_MOESM5_ESM.pdf
Additional file 5: Figure S5. Fragment bit scores with HMM constructed without QnrS. Bit scores of fragments against the hidden Markov model where all sequences from QnrS were excluded. (PDF 1 MB)
12864_2012_4583_MOESM6_ESM.pdf
Additional file 6: Figure S6. Specificity. The specificity in classification of fragments of novel qnr genes for each of the five models. The line QnrA denotes the specificity of the model constructed without QnrA to accurately classify fragments from QnrA. The same for QnrB, C, D and S. (PDF 125 KB)
12864_2012_4583_MOESM8_ESM.pdf
Additional file 8: Figure S7. Overview of the pipeline implementation. A flowchart describing the major parts of the pipeline implemented in Python. (PDF 46 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Boulund, F., Johnning, A., Pereira, M.B. et al. A novel method to discover fluoroquinolone antibiotic resistance (qnr) genes in fragmented nucleotide sequences. BMC Genomics 13, 695 (2012). https://doi.org/10.1186/1471-2164-13-695
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-13-695