
Abstract
Background
The relative abundance of five dominant fatty acids (FAs) (palmitic, stearic, oleic, linoleic and linolenic acids) is a major factor determining seed quality in soybean.
Methods
To clarify the currently poorly understood genetic architecture of FAs in soybean, targeted association analysis was conducted in 421 diverse accessions phenotyped in three environments and genotyped using 1536 pre-selected SNPs.
Results
The population of 421 soybean accessions displayed significant genetic variation for each FA. Analysis of the molecular data revealed three subpopulations, which reflected a trend depending on latitude of cultivation. A total of 37 significant (p < 0.01) associations with FAs were identified by association mapping analysis. These associations were represented by 33 SNPs (occurring in 32 annotated genes); another four SNPs had a significant association with two different FAs due to pleiotropic interactions. The most significant associations were cross-verified by known genes/QTL or consistency across cultivation year and subpopulations.
Conclusion
The detected marker-trait associations represent a first important step towards the implementation of molecular-marker-based selection of FA composition with the potential to substantially improve the seed quality of soybean with benefits for human health and for food processing.
Similar content being viewed by others


Background
Cultivated soybean (Glycine max L. Merr) produces seeds with 15 to 25 % oil and is primarily grown as a major source of plant edible oil [1, 2]. The nutritional value, flavor and stability of soybean oil is determined by its five dominant fatty acids (FAs), including saturated palmitic (16:0) and stearic (18:0), monounsaturated oleic (18:1), and polyunsaturated linoleic (18:2) and alpha-linolenic (18:3). Reduction of saturated palmitic acid and increase of unsaturated FA concentrations in soybean oil is desirable to improve human cardiovascular health. ω-6 linoleic and ω-3 linolenic acids are essential to humans but cannot be produced by human metabolism and therefore must be obtained from the diet. However, the presence of high levels of polyunsaturated fatty acids (PUFAs), especially linolenic acid, increases autoxidation which causes off-flavor, and reduces the shelf life of soybean oil.
The inheritance of the five dominant FAs in soybean is controlled by major and minor genes [3]. Identifying molecular marker or quantitative trait loci (QTL) associated with FAs using marker-assisted selection (MAS) would facilitate the development of improved varieties to meet the widespread demand for healthier soybean oil. Linkage mapping is the traditional strategy for the identification of QTL using bi-parental mapping populations and has relatively high power and a low false positive rate. Several QTLs related to FAs have been reported [3–9] and a number of molecular markers associated with unique FAs were developed subsequently [10–12]. However, the utilization in breeding programs of QTL/molecular markers in the development of MAS or backcrossing for altering FAs has been limited due to low consistency across different genetic backgrounds resulting from the small fraction of the possible alleles sampled. Effectiveness is further restricted by the limited resolution and accuracy of these QTLs resulting from the low number of recombination events within bi-parental mapping populations, especially in genomic regions with high levels of linkage disequilibrium (LD). Therefore, it is necessary to clarify the molecular basis of natural variation and identify molecular markers associate with unique FAs in unrelated soybean germplasm with broad genetic diversity.
LD-based association mapping enables the identification of putative nucleotide polymorphisms responsible for phenotypic differences denoted as quantitative trait nucleotide(s), QTN, by searching for marker-trait associations. Association mapping has four main advantages: high mapping resolution, rich allele number, a reduction in time spent establishing mapping populations and greater utilization in MAS. Therefore, association mapping is increasingly used to dissect the genetic architecture of complex quantitative traits in soybean using universal SNP chips (i.e. Universal Soy Linkage Panel 1.0 with 1536 SNPs, SoySNP6k BeadChip with 5361 SNPs or SoySNP50K iSelect BeadChip with 52,041 SNPs) [13–18], genotyping by sequencing (GBS) [19–22] or re-sequencing [23], as complementary approaches for linkage mapping. Based on these analysis, a set of QTNs had been obtained which are significantly associated with maturity, plant height, seed weight, oil content, protein content, and resistance to soybean cyst nematode, sclerotinia stem rot, or white mold. The genetic basis of FA production, however, has not been fully elucidated using the association mapping approach in soybean.
To identify maximum genetic and phenotypic diversity of FAs, extant genetic resources from representative Chinese soybean core and applied collections [24], were genotyped using a 1536 SNP (mainly non-synonymous) chip and phenotyped in this study over three years. Subsequently, a genome-wide scan for significant markers was performed for further understanding of the genetic basis of differences in FAs and to enable the effective use of FA genetic resources. The results suggest that the association mapping approach is valid for detecting favorable alleles for FAs in soybean.
Methods
Plant materials
A worldwide set of 421 soybean accessions was selected (Additional file 1) comprising 248 genotypes from the Chinese mini-core collection, 142 lines from the applied core collection of the Chinese National Soybean GeneBank (CNSGB) and 31 accessions from other countries worldwide (Fig. 1a). The lines from the Chinese mini-core and applied core collections have been described elsewhere [24, 25]. Each accession used in this study has been examined for phenotypic and/or genotypic homogeneity.

The geographic location, distributions and population structure across the range of cultivated soybean. a The geographic distributions of 421 soybean accessions analyzed in this study. The accessions assigned into three inferred genetic clusters (Northeast, central and South China) are indicated by solid blue, red and green circles respectively. Accessions with mixed genomes are indicated by the solid gray circle. b Inferred population structure of the soybean panel partitioned into three segments (K = 3). Each color represents one cluster, and the length of the colored segment shows the accession’s estimated proportion of membership in that cluster as calculated by STRUCTURE. The accessions are shown in order of latitude (°N) of their origin
Phenotyping data collection and analysis
The 421 accessions were evaluated in field trials in 2010, 2011 and 2012 in Sanya (SY10, SY11 and SY12, 18.2°N, 109.5°E), Hainan Province, China. The experiments were conducted following sowing in early January to early March each year. In all years, three biological replicates (rows) were planted following a complete random design. A row was 0.55 m wide and 1.5 m long with a spacing between plants of 0.10 m.
Seed was harvested after the accessions were grown to final maturity. The seeds from three replicate samples were pooled by accession and analyzed for FA components at the Agricultural Experiment Station chemical laboratories of the Chinese Agricultural University (CAU) in 2010, 2011 and 2012, respectively. The FA components were analyzed using an HP6890 gas chromatogram (GC) (Agilent Technologies, Palo Alto, CA, USA). Individual FA contents were calculated as percentage (%) of total free FA. All of the data were normalized by using both the seed weight and an internal reference. FA data used in the association mapping was the average of three replicates for a genotype. Variance component and heritability of target traits were analyzed using the R software lme4 package assuming random genotype and environment effects (http://www.r-project.org/). Pearson moment correlations between the five FAs pairwise were calculated by PASW statistics [26].
SNP genotyping data collection
Twenty-four metabolic pathways and more than 600 annotated genes have been reported to be associated with the biosynthesis and degradation of acyl-lipids in Arabidopsis [27]. To dissect the genetic basis of FA production in soybean, using a targeted association mapping approach, 1794 putative homologous genes were annotated in the palaeopolyploid soybean by sequence comparison of known gene families in Arabidopsis [27, 28]. A total of 1536 single-nucleotide polymorphism (SNP) were selected from the group of SNPs produced by comparison of 55 whole-genome re-sequenced soybean genomes [29, 30] and the first soybean transcript map [31], to design a SNP genotyping array, which originated from putative homologous genes [28] and known QTL regions [5, 7, 8, 32, 33], randomly selected along chromosomes. Four hundred and twenty-one soybean accessions were assayed using the Illumina BeadArray platform (Illumina Inc., San Diego, CA, USA) following the manufacturer’s protocol. DNA was extracted from a bulk of young leaf tissue of 20–30 plants per accession as previous described [34]. A group of 242 SNPs with a GenCall score of < 80 % and GenTrain < 0.6 were excluded from further analysis since it was difficult to separate homozygote and heterozygote clusters, as described in our previous study [25]. A further 69 SNPs were removed due to either an excessive failure rate (10 % or more of samples) or to apparent heterozygosity (>20 % of samples). Finally, 20 polymorphic loci with minor allele frequency (MAF) < 5 % were removed from association mapping analysis. In the end, the final data set reflected the allelic state at 1205 SNP loci (Additional file 2)
Analysis of population structure
Summary statistics, including the number of alleles, the frequency of major allele, gene diversity, the proportion of heterozygous individuals in the population and heterozygosity for each SNP locus were calculated using PowerMarker 3.25 software [35]. A subset of 756 SNPs which were evenly distributed across all 20 soybean chromosomes was selected to determine the population structure (Additional file 2), using Bayesian Markov Chain Monte Carlo approaches incorporated in the software package STRUCTURE 2.1 [36]. The admixture and independent allele frequency model was employed, using cluster number (K) ranging from 1 to 10. Twenty runs were performed for each value of K, without using previous population information, with a 100,000 burn-in length and 100,000 iterations. The estimated log likelihood values increased as the values of K increased, which indicated no clear genetic structure so the derivative of the log likelihood (∆K) was used to determine the most likely number of sub-clusters as previously reported [37].
Association mapping
Marker/trait associations were tested for 1205 SNP loci with each of five averaged FAs using TASSEL 4.0 standalone software (http://www.maizegenetics.net). Since genome wide association studies can be susceptible to false positive associations from population stratification, a mixed linear model (MLM) method [38] with subpopulation membership percentage (Q matrix) and Kinship (K matrix) were performed. Q matrix and K matrix were inferred from the STRUCTURE and TASSEL 4.0 standalone programs, respectively, using allelic data from 756 evenly distributed SNP markers (Additional file 2). The threshold p-values for significant marker-traits associations were set at 0.001 by considering the scale of SNPs used in this study.
Results and discussion
Genotyping results
Four hundred and twenty-one soybean accessions were fingerprinted by a genotyping array designed with 1536 SNPs known to be present in 55 representative soybean accessions [29]. After exclusion of SNPs with a high failure rate or heterozygosity, 1205 markers met the threshold of quality control in this panel. Since an annotated gene approach was adopted to design the genotyping array, the majority (93 %) of the 1205 SNP were located in coding regions (CDS), the untranslated region (UTR) and the introns of 1074 annotated genes (Glyma v1.1). Of the 1077 SNPs in coding regions, 345 (32 %) were synonymous whereas 702 (65.2 %) were non-synonymous. In addition, 16 SNPs created a stop codon and 14 were found to have caused a change of open reading frame. The detailed information for each SNP can be found in Additional file 2.
Population structure
For inferring population structure in the 421 soybean accessions, 756 SNPs evenly distributed across 20 soybean chromosomes were selected from 1205 SNPs. STRUCTURE analysis showed that the logarithm of the data likelihood (Ln P(D)) continued to increase with increasing numbers of assumed subpopulations (K) from 1 to 10. The ad-hoc quantity based on the second order rate of change in the log probability (ΔK) revealed that the uppermost model value of K was at K = 3 suggesting three genetically distinct subpopulations with limited evidence of admixture among them (Fig. 1b, c). The three subpopulations illustrated a trend related to latitude of original cultivation (Fig. 1a, c), and were subsequently denoted as NER (North East region of China), NR (North region of China) and SR (South region of China). The NER subpopulation consisted of 117 accessions mainly from high latitude areas (>40° N, 112 accessions); the NR subpopulation comprised 48 accessions mainly from areas between latitudes of 35–40° N (33 accessions); and SR subpopulation contained 180 accessions mainly from low latitude areas (<35° N, 172 accessions).
Phenotyping and statistical analysis for five FA components
Four hundred and twenty-one diverse soybean accessions were evaluated in field trials across three years in Sanya, the southernmost city in China. Five dominant FAs of the seeds, i.e. linoleic, linolenic, oleic, palmitic and stearic acids, were determined using gas chromatography. Abundant variations were observed which ranged from a 1.2-fold difference in palmitic acid component to a 1.8-fold difference in linolenic acid (Table 1). Correspondingly, broad-sense heritability values for all of five FA components obtained from three years of phenotypic characterization were moderate to high with a range from 0.5 to 0.7.
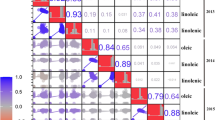
These five FAs showed significant differences among the three subpopulations (p at 0.05 level) (Fig. 2). NER had the lowest proportions of linolenic and linoleic acids, and the highest amounts of oleic and stearic acids. NR had the lowest oleic acid level and the highest linoleic and palmitic acid components. SR had the lowest levels for both palmitic and stearic acids. These differences may be attributed to natural selection due to environment factors such as temperature and planting days as well as artificial selection during domestication and genetic improvement. For example, the presence of a high component of linolenic acid is responsible for autoxidative instability and off-flavors associated with the oils, and thus farmers or breeders have always selected or developed soybean accessions with inherently low levels of linolenic acid. It has been reported that on average cultivated soybeans contain only around two-thirds of the linolenic acid found in wild soybean (Glycine soja Sieb. & Zucc., the progenitor of cultivated soybean) [39]. Wild soybean accessions produced up to 23 % linolenic acid [40, 41]. This study detected a significant difference in linolenic acid contents between the three subpopulations. Accessions from NER, the main high-oil soybean producing area in China, contained the lowest proportion of linolenic acid (8.7 %), followed by SR (9.3 %) and NR (9.5 %). These results agree with the suggestion that linolenic acid levels underwent selection to meet the demands of oil industry during domestication and the expansion of domesticated soybean production [39, 42]. The patterns of correlation coefficients (r) calculated across all five traits (Fig. 3) coincided with the previous observations [43]. Oleic acid levels were significantly negatively correlated (p < 0.01) with the other four traits. The closest relationship was detected with linoleic acid (r = −0.89) and the least significant relationship with stearic acid (r = −0.14). Most of the relationships among the other four traits were significantly (p < 0.01) positive. Among them, linolenic acid showed the closest relationship with linoleic acid (r = 0.45), followed by palmitic acid (r = 0.36). The relationship between linolenic and stearic acids was essentially random, with an r value of 0.01.

Variation in five fatty acids among different subpopulations of the soybean diversity panel. The error bar represents the standard error. NER, NR and SR contained 117, 48 and 180 soybean accessions, respectively

Pearson’s co-efficient of correlation for pairwise comparison of five fatty acids in soybean accessions. Differences significant at p < 0.05 are marked *; those significant at p < 0.01 are marked **
Marker-trait association analysis
Following the distinct population structure detected within this panel by STRUCTURE, the MLM model, which takes account of both kinship matrix and genetic structure (K + Q), was used to reveal QTNs associated with the five FAs. In total, 37 significant (p < 0.01) marker-trait associations were identified for these FA (Table 2). Around one-third of these QTNs were located within or near known QTLs (Table 2). The number of QTNs in the current study associated with a single FA ranged from five for oleic acid and stearic acid to 12 for linolenic acid (Fig. 4). A high consistency of associations (30 of 37 QTNs) was found between significant SNPs and alleles with the target trait in at least two of three environments. These QTNs were represented by 33 unique SNPs and lay in 32 annotated genes. Four SNPs (Map-3670, −6135, −6325, and −6520) exhibited significant (p < 0.01) associations with more than one trait. Of the 32 SNPs with significant (p < 0.01) associations, 28 were located in coding regions (87.5 %). The effects of 18 of these were determined: non-synonymous polymorphisms (15), stop codons (2) and frameshift (1) (Table 2). In the following we discuss in detail the marker-trait associations found for each of the five FAs:

Manhattan plots of p values indicating SNP markers associated with five fatty acids. The x- axis shows SNP markers along each soybean chromosome; y- axis is the –log10 (p value), horizontal lines designate 1E-02 thresholds for significant associations. The association of SNP markers with highly significant associations (p < 0.001) are shown by red dots
Linoleic acid
A total of eight significant marker-trait associations were detected each located in a distinct candidate gene associated with linoleic acid biosynthesis (Fig. 4, Table 2). Three SNPs were non-synonymous (Map-6283, −6325 and −6520). Individually, each of the eight putative QTNs explained a small proportion of the phenotypic variance, with effects ranging from 1.7 % (Map-6135 and −6325) to 2.7 % (Map-6146). Of eight QTNs, two (Map-6325 and −6326) were neighboring with a 41.4-kbp genomic distance. Map-6326 is a synonymous polymorphism in Glyma08g17190, which was predicted to be involved in fatty acid elongation and the wax biosynthesis pathway by comparison with homologs in Arabidopsis thaliana [28]. We identified a strong pairwise LD between Map-6325 and −6326 with r2 of 0.854.
Oleic acid
Five significant (p < 0.01) marker-trait associations were observed, which explained 1.7-2.5 % of oleic acid component variation (Table 2, Fig. 4). These SNPs lay in five annotated genes and two of them (Map-6325 and −3670) were deduced to be non-synonymous polymorphisms. Map-6056 was a synonymous polymorphism in Glyma03g41850 which, by comparison with homologs in Arabidopsis thaliana, is likely to be involved in the phospholipid signaling pathway.
Linolenic acid
Twelve Significant (p < 0.01) marker-trait associations were detected located on Gm01 (3), Gm02 (1), Gm05 (1), Gm08 (1), Gm15 (1), and Gm18 (5). Map-6017, a synonymous polymorphism within Glyma02g15600, had the most significant association (p = 2.9E-04) and explained 3.2 % of the variation in proportion of linolenic acid. Glyma02g15600 encoded one of three isoforms of △9-stearoyl-acyl carrier protein-desaturase (SACPD-B or GmFAB2B) [44, 45] and was predicted to play a role in fatty acid synthesis and fatty acid elongation [28]. It has been reported that a 1-bp insertion in exon 3 of SACPD-B which caused 28 amino acids changes compared to Williams 82 SACPD-B, was associated with linolenic acid production in soybean [45]. Map-6017 was 46-bp away from this 1-bp insertion. For each year, the accessions with allele T had significantly (p < 0.01) higher linolenic acid than those with allele C (Fig. 5a). Allele T in the SNP Map-6017 is a minor allele with a frequency of 9.5 % in this representative cultivated soybean panel, suggesting that this locus potentially underwent selection for low linolenic acid levels during soybean domestication. Further evidence that selection against allele T took place in genetic improvement arises from the decrease in frequency from landrace (13 %) to modern cultivars (2.2 %). Geographical distribution of bi-alleles in Map-6017 was uneven across the three subpopulations. The allele T occurred in 25 % of accessions from NR (the predicted domestication site of cultivated soybean [34]), its frequency decreased to 10.5 % in SR and to 0.9 % in NER subpopulations (Fig. 5b). These findings clearly suggest that this locus has been strongly selected in NER for production of linolenic acid levels.

Diagrams depicting the genetic effects on associated fatty acids across three years, and allele frequencies in three subpopulations inferred from STRUCTURE analysis at three most significant SNP (p < 0.001) loci. a Linolenic acid bi-alleles at Map-6017. b Bi-allele frequency at Map-6017. c Palmitic acid bi-alleles at Map-6064. d Bi-allele frequency at Map-6064. e Stearic acid bi-alleles at Map-6506. f Bi-allele frequency at Map-6506. SY10, SY 11 and SY12 respectively indicate 2010, 2011 and 2012 growing seasons in Sanya
Palmitic acid
Of seven SNPs associated with palmitic acid, Map-0751 and 6064 were the two most significant loci. They cover a 55-kbp genomic region. The most significant locus Map-6064 (p = 2.4E-08) explained 7.7 % of the phenotype variation and is a synonymous SNP within Glyma05g07730, a homolog of a potentially lipid trafficking gene in Arabidopsis thaliana (AT3G25610) [46]. In addition, Map-6064 is located 288-kbp upstream of Glyma05g08060 (GmFATB1a). A nonsense mutation within exon 1 of GmFATB1a was previously found to be responsible for the reduction of palmitic acid in soybean [47]. Non-synonymous Map-0751 in Glyma05g07630 was found to have significant LD (p =3.6E-44) with Map-6064 (r2 = 0.53, D’ = 0.98).
Since saturated palmitic acid give rise to negative health effects in humans associated with a diverse lipoprotein profile arising from consumption of this fatty acid [48], soybean accessions with reduced palmitic acid are desirable. However, for each year the bi-alleles of Map-6064 locus were observed not to be correlated with palmitic acid variation in our 421 worldwide soybean accessions (p > 0.05) (Fig. 5c). Nevertheless, considering the subpopulations individually, significant differences for palmitic acid (p < 0.05) were detected between the bi-alleles of Map-6064 locus (Additional file 3), suggesting that this locus was affected by population structure. Soybean accessions with Map-6064-C allele tended to contain higher levels of palmitic acid than those with Map-6064-T. Similar frequencies of favored allele were detected in landraces (25.2 %) and modern varieties (19.7 %) suggesting that no significant selection occurred in the Map-6064 locus during soybean genetic improvement. The allele frequencies of favorable Map-6064-T were further estimated for 117 NER, 48 NR and 180 SR accessions (Fig. 5d) and a large change was detected across subpopulations: the majority of NR accessions (77.1 %) possessed the Map-6064-T allele, whereas only a small proportion of NER and SR accessions (25.4 % and 8.3 % respectively) exhibited this favorable allele. Of 48 NR accessions examined, all modern varieties (3) and 34 of 45 landraces with Map-6064-T provide potentially important genetic resources for reducing palmitic acid in soybean seeds.
Stearic acid
Four of five loci significantly associated with stearic acid were near or overlapped with previously identified QTLs. The exception was Map-6568 on Gm15 (Table 2). Notably, the two most significant associations (Map-6506, p = 4.0E-08 and BARC-013927-01275, p = 1.4E-07) on Gm14 overlapped with the fas locus with respect to Satt474, Sat_189 and Satt556 [32, 49]. Map-6506 and BARC-013927-01275 explained 8.5 % and 7.8 % of stearic acid, respectively. Map-6506 is a synonymous mutation within exon 1 of Glyma14g27990 (SACPD-C or GmFAB2C), a gene predicted to be involved in converting stearic acid into oleic acid. Besides Map-6506, two SNPs within SACPD-C - one non-synonymous and one nonsense mutation - had previously been found to cause variation of stearic acid levels in soybean [47, 50]. The second most significant SNP, non-synonymous BARC-013927-01275 in Glyma14g27380 is within 752-kbp upstream of Map-6064, and exhibited a strong LD relationship with r2 of 0.91 and D’ of 0.99.
Unlike palmitic acid, a high level of stearic acid is desirable since it is not related to negative effects on human health and also offers potential for improving soybean oil quality in food-processing applications [51]. Significant differences in stearic acid were detected between bi-alleles at the Map-6506 locus across all three years studied (Fig. 5e). Average stearic acid of the accessions with favorable Map-6506-A allele were 3.86 %, 0.16 % higher than that of accessions with Map-6506-G (3.70 %). Desirable Map-6506-A represented only 27.3 % of accessions in the current diversity panel, and also in its three subpopulations (Fig. 5f).
Estimation of the function of genes exhibiting significant marker-trait associations
In the current study, 32 annotated genes were identified to be significantly associated with one or two fatty acid components. Their function were estimated using comprehensive gene expression profiles of the RNA Seq-Atlas [52] from Soybase database (http://www.soybase.org/soyseq/). The expression level of all of 32 genes were compared among 14 tissues, including six tissues without seeds (root, nodule, young leaf, flower, pod shell-10 days after flowering DAF) and eight tissues with seeds (small pod, seeds-10DAF, −14DAF, −21DAF, −25DAF, −28DAF, −35DAF, and -42DAF) (Additional file 4). Five genes (including Glyma01g24850, Glyma08g13060, Glyma15g17090, Glyma18g05980, and Glyma18g34290) were not expressed in any tissue analyzed. Of the remaining 27 genes which were expressed in more than one tissue, three genes, Glyma05g07630 with Map-0751, Glyma05g34030 with Map-6077, and Glyma14g27990 with Map-6506, exhibited preferential gene expression in tissues with seeds and the gene expression level of the later two increased with the seed development (Additional file 4). Seeds-specific Glyma05g07630 was located nearby GmFATB1a [47] and identified to be significantly (p < 0.001) related with palmitic acid inferred from the association mapping. In Arabidopsis thaliana, FatB encodes a palmitoyl thioesterase, which is primarily involved in regulating the production of palmitic acid by catalyzing the conversion of 16:0-acyl carrier protein (ACP) (palmitic acid with ACP) to 18:0-ACP (stearic acid with ACP) [27]. In addition, it was reported that Glyma05g34030 (GmMFT) may be a negative regulator of seed germination [53]. In this study, GmMFT maybe play another role on regulating linolenic acid in soybean seeds. Therefore, the functions of Glyma05g34030 still need to be further analyzed through functional studies such as genetic transformation.
Glyma14g27990 (SACPD-C) was one of four soybean isoforms of SACPD (also referred to as FAB2), a soluble desaturase which determines the relative proportions of saturated stearic acid and three unsaturated FAs [54]. The other three isoforms in soybean identified by comparative analyses were GmSACPD-A (Glyma07g32850) and SACPD-B (Glyma02g15600) and a pseudogene SACPD-D (Glyma13g08990) [45, 55]. It has been reported that both of SACPD-B and SACPD-C are responsible for the variation of seed stearic acid content in soybean [45, 47, 55]. However, in this study Map-6506 in GmSACPD-C was significantly (p < 0.001) associated with stearic acid, whereas Map-6017 in GmSACPD-B was significantly (p < 0.001) associated with linolenic acid. This suggests that the roles of the two orthologs of GmSACPD (GmSACPD-B and GmSACPD-C) are different (Fig. 3).
Pleiotropic effects
Starting from saturated stearic acid, three unsaturated fatty acids, including oleic, linoleic and linolenic are synthesized as a result of the fatty acid desaturation pathway. Consequently, strong phenotypic correlations due to pleiotropy are expected for these four FA traits [5]. In accordance with this expectation we observed in our study that five out of the six pair-wise comparisons among these four FA traits were significantly (p < 0.01) correlated, except linolenic vs stearic acid. Moreover, association mapping analysis identified four SNPs (Map-6135, −6325, −6520 and −3670) exhibiting pleiotropic effects on stearic, oleic, linoleic or linolenic acid (Table 2). For example, as the precursor of linoleic acid, oleic acid exhibited a significant negative correlation with linoleic acid (Fig. 3). Map-6135 and Map-6325 were found to be significantly associated with both oleic and linoleic acid, but showed significant opposite effects, a decrease of oleic acid and an increase of linoleic acid with the Map-6135-A and Map-6325-C. These findings indicate that the QTNs exhibit strong pleiotropic effects.
Conclusions
Due to predominant self-pollination, intensive selection during domestication and following genetic improvement, and genetic drift, extensive LD was observed for FA traits in soybean, especially in cultivated soybean (i.e. landrace and modern accessions) [23, 29, 56, 57]. Therefore, although resolution was limited, the association mapping approach employing diverse soybean cultivars allowed the identification of QTNs using a relatively small density of markers. In the present study, marker-trait association analysis detected 33 SNPs associated with at least one FA trait. Although further validations in independent panels or bi-parental populations are desirable, the co-localization of a group of associated loci with known genes/QTLs (as evidenced for Map-6017 in GmSACPD-B, Map-6506 in GmSACPD-C, and Map-6064 near GmFATB1a related with linolenic acid, stearic acid, and palmitic acid, respectively) suggests that the LD-based association mapping approach is suitable for detecting reliable associations with FA traits. These functional SNPs are essential tools for molecular soybean breeding programmes aimed at improving the FA quality of seeds and processed oil. In the future, labor/time-saving and cost effective SNP assays, such as Kompetitive Allele Specific PCR (KASP) and Cleaved Amplified Polymorphic Sequence (CAPS) assays, might be exploited based on these functional SNPs for assisted selection of specific desirable FA compositions, such as soybean varieties with increased stearic acid (>20 %) for food and industrial products [58]. It is important to note that some associations may have failed to be detected in this study owing to the limited marker density. A high density marker or sequencing-based analysis (i.e. re-sequencing, Genotyping by Sequencing etc.) should be conducted to further deepen our understanding of the genetic architecture of FAs in soybean using LD-based association mapping approach.
Availability of data and materials
Four additional files in machine-readable format were uploaded in BMC Genomic website for supporting the results and findings found in this study, including
References
Nichols DM, Glover KD, Carlson SR, Specht JE, Diers BW. Fine mapping of a seed protein QTL on soybean linkage group I and its correlated effects on agronomic traits. Crop Sci. 2006;46(2):834–9.
Hurburgh CR. Long-term soybean composition patterns and their effect on processing. J Am Oil Chem Soc. 1994;71(12):1425–7.
Wang X, Jiang G-L, Green M, Scott RA, Hyten DL, Cregan PB. Quantitative trait locus analysis of unsaturated fatty acids in a recombinant inbred population of soybean. Mol Breeding. 2014;33(2):281–96.
Hyten DL, Pantalone VR, Sams CE, Saxton AM, Landau-Ellis D, Stefaniak TR, et al. Seed quality QTL in a prominent soybean population. Theor Appl Genet. 2004;109(3):552–61.
Panthee DR, Pantalone VR, Saxton AM. Modifier QTL for fatty acid composition in soybean oil. Euphytica. 2006;152(1):67–73.
Monteros MJ, Burton JW, Boerma HR. Molecular mapping and confirmation of QTLs associated with oleic acid content in N00-3350 soybean. Crop Sci. 2008;48(6):2223–34.
Bachlava E, Dewey RE, Burton JW, Cardinal AJ. Mapping and comparison of quantitative trait loci for oleic acid seed content in two segregating soybean populations. Crop Sci. 2009;49(2):433–42.
Wang X, Jiang G-L, Green M, Scott RA, Hyten DL, Cregan PB. Quantitative trait locus analysis of saturated fatty acids in a population of recombinant inbred lines of soybean. Mol Breeding. 2012;30(2):1163–79.
Akond M, Liu S, Boney M, Kantartzi SK, Meksem K, Bellaloui N, et al. Identification of quantitative trait loci (QTL) underlying protein, oil, and five major fatty acids’ contents in soybean. Am J Plant Sci. 2014;5:158–67.
Ha B-K, Monteros MJ, Boerma HR. Development of SNP assays associated with oleic acid QTLs in N00-3350 soybean. Euphytica. 2010;176(3):403–15.
Bilyeu K, Palavalli L, Sleper DA, Beuselinck P. Molecular genetic resources for development of 1 % linolenic acid soybeans. Crop Sci. 2006;46(5):1913–8.
Pham A-T, Lee J-D, Shannon JG, Bilyeu KD. Mutant alleles of FAD2-1A and FAD2-1B combine to produce soybeans with the high oleic acid seed oil trait. BMC Plant Biol. 2010;10(1):195.
Bao Y, Vuong T, Meinhardt C, Tiffin P, Denny R, Chen S, et al. Potential of association mapping and genomic selection to explore PI 88788 derived soybean cyst nematode resistance. Plant Genome-US. 2014;7(3):1–13.
Zhang J, Song Q, Cregan PB, Nelson RL, Wang X, Wu J, et al. Genome-wide association study for flowering time, maturity dates and plant height in early maturing soybean (Glycine max) germplasm. BMC Genomics. 2015;16(1):217.
Wen Z, Tan R, Yuan J, Bales C, Du W, Zhang S, et al. Genome-wide association mapping of quantitative resistance to sudden death syndrome in soybean. BMC Genomics. 2014;15(1):809.
Vaughn JN, Nelson RL, Song Q, Cregan PB, Li Z. The genetic architecture of seed composition in soybean is refined by genome-wide association scans across multiple populations. G3-Genes, Genomes, Genet. 2014;4(11):2283–94.
Dhanapal AP, Ray JD, Singh SK, Hoyos-Villegas V, Smith JR, Purcell LC, et al. Genome-wide association study (GWAS) of carbon isotope ratio (δ13C) in diverse soybean [Glycine max (L.) Merr.] genotypes. Theor Appl Genet. 2015;128(1):73–91.
Hwang E-Y, Song Q, Jia G, Specht JE, Hyten DL, Costa J, et al. A genome-wide association study of seed protein and oil content in soybean. BMC Genomics. 2014;15:1.
Sonah H, O’Donoughue L, Cober E, Rajcan I, Belzile FO. Identification of loci governing eight agronomic traits using a GBS-GWAS approach and validation by QTL mapping in soya bean. Plant Biotechnol J. 2015;13:211–21.
Zhao X, Han Y, Li Y, Liu D, Sun M, Zhao Y, et al. Loci and candidate gene identification for resistance to Sclerotinia sclerotiorum in soybean (Glycine max L. Merr.) via association and linkage maps. Plant J. 2015: doi:10.1111/tpj.12810.
Iquira E, Humira S, François B. Association mapping of QTLs for sclerotinia stem rot resistance in a collection of soybean plant introductions using a genotyping by sequencing (GBS) approach. BMC Plant Biol. 2015;15(1):5.
Bastien M, Sonah H, Belzile F. Genome wide association mapping of resistance in soybean with a genotyping-by-sequencing approach. Plant Genome-US. 2014, 7(1):doi: 10.3835/plantgenome2013.3810.0030.
Zhou Z, Jiang Y, Wang Z, Gou Z, Lyu J, Li W, et al. Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nat Biotechnol. 2015: doi:10.1038/nbt.3096.
Qiu L-J, Xing L-L, Guo Y, Wang J, Jackson SA, Chang R-Z. A platform for soybean molecular breeding: the utilization of core collections for food security. Plant Mol Biol. 2013;83(1–2):41–50.
Li YH, Li W, Zhang C, Yang L, Chang RZ, Gaut BS, et al. Genetic diversity in domesticated soybean (Glycine max) and its wild progenitor (Glycine soja) for simple sequence repeat and single-nucleotide polymorphism loci. New Phytol. 2010;188:242–53.
Norusis MJ. PASW statistics 18 guide to data analysis. New York: Prentice Hall Press; 2010.
Li-Beisson Y, Shorrosh B, Beisson F, Andersson MX, Arondel V, Bates PD, et al. Acyl-lipid metabolism. In The Arabidopsis Book, vol. 11. The American Society of Plant Biologists; 2013.
Li YH, Zhou GY, Ma JX, Jiang WK, Jin LG, Zhang ZH, et al. De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits. Nature Biotechnol. 2014;32(10):1045–52.
Li YH, Zhao SC, Ma JX, Li D, Yan L, Li J, et al. Molecular footprints of domestication and improvement in soybean revealed by whole genome re-sequencing. BMC Genomics. 2013;14:579.
Li YH, Liu YL, Liu ZX, Liu B, Reif JC, Chang RZ, et al. Bi-parental re-sequencing coupled with SNP genotyping of the segregating population offers new insights into the landscape of recombination and identical by state regions in soybean. G3-Genes Genom Genet. 2014;4:553–60.
Choi I, Hyten D, Matukumalli L, Song Q, Chaky J, Quigley C, et al. A soybean transcript map: gene distribution, haplotype and single-nucleotide polymorphism analysis. Genetics. 2007;176(1):685–96.
Li H, Zhao T, Wang Y, Yu D, Chen S, Zhou R, et al. Genetic structure composed of additive QTL, epistatic QTL pairs and collective unmapped minor QTL conferring oil content and fatty acid components of soybeans. Euphytica. 2011;182(1):117–32.
Xie D, Han Y, Zeng Y, Chang W, Teng W, Li W. SSR-and SNP-related QTL underlying linolenic acid and other fatty acid contents in soybean seeds across multiple environments. Mol Breeding. 2012;30(1):169–79.
Li YH, Guan RX, Liu ZX, Ma YS, Wang LX, Li LH, et al. Genetic structure and diversity of cultivated soybean (Glycine max (L.) Merr.) landraces in China. Theor Appl Genet. 2008;117(6):857–71.
Liu K, Muse S. PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics. 2005;21(9):2128–9.
Falush D, Stephens M, Pritchard J. Inference of population structure using multilocus genotype data linked loci and correlated allele frequencies. Genetics. 2003;164(4):1567–87.
Li YH, Smulders MJM, Chang RZ, Qiu LJ. Analysis of SSRs uncovers hierarchical structure and genetic diversity in Chinese soybean landraces. J Integr Agr. 2010;9(12):1739–48.
Yu J, Pressoir G, Briggs WH, Bi IV, Yamasaki M, Doebley JF, et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet. 2005;38(2):203–8.
Zheng YZ, Gai JY, Zhao TJ, Zhou RB, Tian SZ. A study on variability of fat-related traits in cultivated and wild soybean germplasm in China. Sci Agri Sin. 2008;41(5):1283–90.
Pantalone V, Rebetzke G, Burton J, Wilson R. Genetic regulation of linolenic acid concentration in wild soybean Glycine soja accessions. J Am Oil Chem Soc. 1997;74:159–63.
Shibata M, Takayama K, Ujiie A, Yamada T, Abe J, Kitamura K. Genetic relationship between lipid content and linolenic acid concentration in soybean seeds. Breed Sci. 2008;58:361–6.
Xu X. The changes of the formation of the fatty acids in soybean fat in Hei Long Jiang. China Oils and Fats. 1996;21(5):50–2.
Li Y. Correlation and path-coefficient analysis of oil and its compositions in soybean. Soybean Sci. 1987;6(3):203–8.
Chi X, Yang Q, Lu Y, Wang J, Zhang Q, Pan L, et al. Genome-wide analysis of fatty acid desaturases in soybean (Glycine max). Plant Mol Biol Rep. 2011;29(4):769–83.
Ruddle Ii P, Whetten R, Cardinal A, Upchurch RG, Miranda L. Effect of a novel mutation in a Δ9-stearoyl-ACP-desaturase on soybean seed oil composition. Theor Appl Genet. 2013;126(1):241–9.
Jouhet J, Maréchal E, Block MA. Glycerolipid transfer for the building of membranes in plant cells. Prog Lipid Res. 2007;46(1):37–55.
Gillman J, Tetlow A, Hagely K, Boersma J, Cardinal A, Rajcan I, et al. Identification of the molecular genetic basis of the low palmitic acid seed oil trait in soybean mutant line RG3 and association analysis of molecular markers with elevated seed stearic acid and reduced seed palmitic acid. Mol Breeding. 2014;34(2):447–55.
Mensink R, Katan M. Effect of dietary trans fatty acids on high-density and low-density lipoprotein cholesterol levels in healthy subjects. N Engl J Med. 1990;323:439–45.
Spencer M, Pantalone V, Meyer E, Landau-Ellis D, Hyten D. Mapping the Fas locus controlling stearic acid content in soybean. Theor Appl Genet. 2003;106(4):615–9.
Goettel W, Xia E, Upchurch R, Wang M-L, Chen P, Yong-qiang CA. Identification and characterization of transcript polymorphisms in soybean lines varying in oil composition and content. BMC Genomics. 2014;15(1):299.
Kris-Etherton P, Yu S. Individual fatty acid effects on plasma lipids and lipoproteins: human studies. Am J Clin Nutr. 1997;65:1628S–44S.
Severin AJ, Woody JL, Bolon Y-T, Joseph B, Diers BW, Farmer AD, et al. RNA-Seq Atlas of Glycine max: a guide to the soybean transcriptome. BMC Plant Biol. 2010;10(1):160.
Li Q, Fan C, Zhang X, Wang X, Wu F, Hu R, et al. Identification of a soybean MOTHER OF FT AND TFL1 homolog involved in regulation of seed germination. PLoS ONE. 2014;9(6):e99642.
Upchurch R. Soybean fatty acid desaturation pathway: responses to temperature changes and pathogen infection. InTech: Croatia; 2011.
Gillman J, Stacey M, Cui Y, Berg H, Stacey G. Deletions of the SACPD-C locus elevate seed stearic acid levels but also result in fatty acid and morphological alterations in nitrogen fixing nodules. BMC Plant Biol. 2014;14:143.
Hyten D, Choi I, Song Q, Shoemaker R, Nelson R, Costa J, et al. Highly variable patterns of linkage disequilibrium in multiple soybean populations. Genetics. 2007;175:1937–44.
Chung W, Jeong N, Kim J, Lee W, Lee Y, Lee S, et al. Population structure and domestication revealed by high-depth resequencing of Korean cultivated and wild soybean genomes. DNA Res. 2014;21(2):153–67.
Ruddle Ii P, Whetten R, Cardinal A, Upchurch RG, Miranda L. Effect of Δ9-stearoyl-ACP-desaturase-C mutants in a high oleic background on soybean seed oil composition. Theor Appl Genet. 2014;127(2):349–58.
Ha BK, Kim HJ, Velusamy V, Vuong TD, Nguyen HT, Shannon JG, et al. Identification of quantitative trait loci controlling linolenic acid concentration in PI483463 (Glycine soja). Theor Appl Genet. 2014;127:1501–12.
Cardinal AJ, Whetten R, Wang S, Auclair J, Hyten D, Cregan P, et al. Mapping the low palmitate fap1 mutation and validation of its effects in soybean oil and agronomic traits in three soybean populations. Theor Appl Genet. 2014;127:97–111.
Li Z, Wilson R, Rayford W, Boerma H. Molecular mapping genes conditioning reduced palmitic acid content in N87-2122-4 soybean. Crop Sci. 2002;42:373–8.
Reinprecht Y, Poysa VW, Yu K, Rajcan I, Ablett GR, Pauls KP. Seed and agronomic QTL in low linolenic acid, lipoxygenase-free soybean (Glycine max (L.) Merr.) germplasm. Genome. 2006;49(12):1510–27.
Acknowledgments
This research was financed by the Agricultural Science and Technology Innovation Program (ASTIP) of the Chinese Academy of Agricultural Sciences, National Science and Technology Platform and the Platform of National Crop Germplasm Resources of China (nos. 2014–004 and 2015–004). The authors thank the China National GeneBank (www.nationalgenebank.org/) for seed of the Chinese materials, and the USDA-ARS Soybean Germplasm Collection (http://www.ars-grin.gov/npgs/) for seed of the international materials.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
L-J Qiu, R-Z Chang and Y-H Li designed the research; Y-H Li, Y-S Ma, H-L Hong and Z-X Liu performed the research and analyzed the data. Y-H Li, J C Reif and L-J Qiu wrote the manuscript. All authors read and approved the final manuscript.
Additional files
Additional file 1:
Information for the 421 worldwide soybean accessions analyzed. (XLS 106 kb)
Additional file 2:
Description of population genetic statistics of 1205 SNP markers in 421 soybean accessions. (XLSX 130 kb)
Additional file 3:
Palmitic acid bi-alleles of Map-6064 in three subpopulations. (TIFF 1235 kb)
Additional file 4:
The normalized transcription counts of 32 annotate genes with significant association signals. Fourteen tissues including tissues without seeds (root, nodule, young_leaf, flower, pod shell-10DAF (Days After Flowering) and pod shell-14DAF) and tissues with seeds (seeds-14DAF,-21DAF, −25DAF, −28DAF, −35DAF, −42DAF and one cm pod) were displayed in order. RNA Seq data was from Soybase database (http://www.soybase.org/soyseq/) [52]. (TIFF 927 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Li, Yh., Reif, J.C., Ma, Ys. et al. Targeted association mapping demonstrating the complex molecular genetics of fatty acid formation in soybean. BMC Genomics 16, 841 (2015). https://doi.org/10.1186/s12864-015-2049-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-015-2049-4