
Abstract
Background
Patients with long QT syndrome due to rare loss-of-function mutations in the human ether-á-go-go-related gene (hERG) have prolonged QT interval, risk of arrhythmias, increased secretion of insulin and incretins and impaired glucagon response to hypoglycemia. This is caused by a dysfunctional Kv11.1 voltage-gated potassium channel. Based on these findings in patients with rare variants in hERG, we hypothesized that common variants in hERG may also lead to alterations in glucose homeostasis. Subsequently, we aimed to evaluate the effect of two common gain-of-function variants in hERG (rs36210421 and rs1805123) on QT interval and plasma levels of glucagon-like peptide-1 (GLP-1), glucose-dependent insulinotropic polypeptide (GIP), insulin and glucagon during an oral glucose tolerance test (OGTT).
We used two population-based cohorts for evaluation of the effect of common variants in hERG on QT-interval and circulation levels of incretins, insulin and glucagon. The Danish population-based Inter99 cohort (n = 5895) was used to assess the effect of common variants on QT-interval. The Danish ADDITION-PRO cohort was used (n = 1329) to study genetic associations with levels of GLP-1, GIP, insulin and glucagon during an OGTT.
Results
Carriers of either the minor A-allele of rs36210421 or the minor G-allele of rs1805123 had ~ 2 ms shorter QT interval per risk allele (p = 0.025 and p = 1.9 × 10− 7). Additionally, both variants were associated with alterations in pancreatic and gut hormone release among carriers. The minor A- allele of rs36210421 was associated with increased GLP-1 and decreased GIP response to oral glucose stimulation, whereas the minor G-allele of rs1805123 is associated with decreased fasting plasma insulin and glucagon release. A genetic risk score combining the two gene variants revealed reductions in glucose-stimulated GIP, as well as suppressed glucagon response to increased glucose levels during an OGTT.
Conclusions
Two common missense polymorphisms of the Kv11.1 voltage-gated hERG potassium channel are associated with alterations in circulating levels of GIP and glucagon, suggesting that hERG potassium channels play a role in fasting and glucose-stimulated release of GIP and glucagon.
Trial registration
ClinicalTrials.gov (NCT00289237). Trial retrospectively registered at February 9, 2006. Studies were approved by the Ethical Committee of the Central Denmark Region (journal no. 20080229) and by the Copenhagen County Ethical Committee (KA 98155).
Similar content being viewed by others


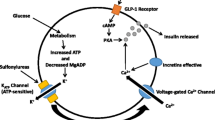
Background
Loss-of-function mutations in the human ether-á-go-go related (hERG) gene result in a dysfunctional Kv11.1 voltage-gated potassium channel causing delayed cardiac repolarization, and long QT syndrome (LQTS) with increased risk of cardiac arrhythmias and sudden death [1,2,3,4]. LQTS is characterized by a prolongation of the QT-interval. The QT-interval is the time between the start of the Q-wave and the end of the T-wave in the ECG. The QT-interval reflects the depolarization and repolarization of the ventricles in the heart. Aside from cardiac muscle cells, voltage-gated hERG-encoded Kv11.1 potassium channels are expressed in a number of tissues throughout the body including endocrine intestinal K- and L-cells and pancreatic α- and β-cells [5,6,7,8]. This has led to speculations that hERG channels may play a role in secretion of hormones from endocrine cells [9].
Glucose homeostasis is tightly regulated by interaction between glucose-regulating hormones from endocrine intestinal cells and pancreatic islet cells. A well-regulated interplay between the incretins, glucagon-like peptide-1 (GLP-1) and gastric inhibitory polypeptide (GIP) from intestinal L- and K-cells, and the secretion of insulin and glucagon from the pancreatic islets ensures glucose homeostasis by limiting glucose excursions, by facilitating uptake and storage of glucose during fed states, and by regulating glucose production for glucose-dependent tissues during fasting conditions. Secretion from the pancreatic islets cells and L-cells is determined by their electrical activity which, among others, is regulated by different types of voltage-gated potassium channels, and the hERG channel is involved in repolarization of the membrane potential [6,7,8,9,10,11,12].
Recently, Hyltén-Cavallius et al. [9] reported that patients with LQTS due to impaired function of the hERG-encoded Kv11.1 potassium channel not only have alterations in their cardiac conduction, but also exhibit increased insulin, GLP-1 and GIP secretion with risk of hypoglycemia, as well as decreased fasting and hyperglycemia induced levels of glucagon. Based on the findings in carriers of rare variants, we hypothesized that common variants may also cause alterations in QT-interval and in circulating levels of incretins, insulin and glucagon levels. We selected non-synonymous coding variants with a minor allele frequency > 3% in hERG (rs36210421 and rs1805123). We used data from two large cohorts; the Inter99 cohort (n = 5487) to evaluate the effect of common variants in hERG on QT-interval and the ADDITION-PRO cohort (n = 1329) to evaluate the effect on common variants on circulating plasma levels of incretins, insulin and glucagon.
Methods
Populations
ADDITION-PRO is a cohort study of individuals at low to high risk of type 2 diabetes, nested within the population-based ADDITION-Denmark [13]. Overall, 1329 individuals were included - 708 normal glucose tolerance (NGT); 254 had isolated impaired fasting glucose (IFG); 103 had isolated impaired glucose tolerance (IGT); 116 had both IFG and IGT, and 148 had screen detected type 2 diabetes. Participants (48% females) had a mean age of 66.3 ± 6.9 and a mean BMI of 27.1 ± 4.6. All individuals who fasted < 8 h before the blood samples were taken, or who used diabetes medication, or individuals with already known diabetes were excluded (n = 22). Characteristics of the ADDITION-PRO participants included in the present study are given in Additional file 1: Table S1.
Inter99 is a population-based randomized intervention study of 6784 participants aiming at preventing ischemic heart disease by non-pharmacologic intervention (ClinicalTrials.gov, NCT00289237). The Inter99 study has been described in details previously [14]. We included 5487 individuals (51%) with a mean age of 46.2 ± 7.9 and a BMI of 26.2 ± 4.5. More characteristics on the study participants can be found in the Additional file 1: Table S1.
The ADDITION-PRO study was approved by the Ethical Committee of the Central Denmark Region (journal no. 20080229) and the Inter99 study was approved by the Copenhagen County Ethical Committee (KA 98155) and the National Board of Health. Both studies were conducted in accordance with the principles of the Helsinki Declaration. All study participants provided written informed consent.
Biochemical measures
ADDITION-PRO and Inter99
Baseline venous blood samples were drawn after an overnight fast (≥8 h). Participants underwent a standard 75 g oral glucose tolerance test (OGTT) with blood samples drawn at 0, 30 and 120 min. Measurements of circulating levels of glucose, insulin, GLP-1, GIP, and glucagon were collected during the OGTT. Serum insulin was measured by immunoassay (AutoDELFIA, Perkin Elmer, Massachusetts, United States). Plasma glucose was measured using the Hitachi 912 system (Roche Diagnostics, Mannheim, Germany) or the Vitros 5600 system (Ortho Clinical Diagnostics, Illkirch Cedex, France). Blood samples for the measurement of GLP-1, GIP and glucagon were obtained in tubes containing EDTA and put on ice immediately, before centrifugation at 4 °C. Plasma was stored at − 80 °C. Total plasma GLP-1 (intact GLP-1 plus the metabolite GLP-1 (9–36) amide), total plasma GIP (the sum of intact GIP plus the metabolite GIP 3–42) and plasma glucagon were determined using radio-immunological assays, as previously described [15,16,17].
Electrocardiogram measures (ECG)
All ECGs were obtained on inclusion into the Inter99 cohort and were digitally recorded and stored in the MUSE Cardiology Information System (GE Healthcare, Wauwatosa, WI, USA) and processed using Marquette 12SL algorithm, version 21. ECG measurements were assessed and analyzed digitally for heart rate and mean QTc interval. QT- interval was used for assessment of the impact of genetic variants on cardiac conduction. QT interval was corrected for heart rate using Fridericias formula (QTcF = QT / 3√RR) [18]. The RR interval refers to the time between R waves.
Genetics
Non-synonymous coding variants in hERG were included in this study. The variants were missense variants in hERG with a minor allele frequency > 3% causing amino acid substitutions with a significant effect on the hERG channel function [19,20,21]. Two variants were chosen, rs36210421 and rs1805123. Previous functional in vitro and in vivo studies [19,20,21] and population-based studies [20,21,22,23,24,25] have demonstrated that the minor A-allele of rs36210421 (R1047L) and minor G-allele of rs1805123 (K897 T) have gain-of-function effects on the hERG channel.
Addition-pro
We genotyped 1657 participants of the ADDITION-PRO cohort applying the Illumina Infinium HumanCoreExome Beadchip (Illumina, San Diego, CA). We excluded individuals which were first degree relatives, duplicates, ethnic outliers, or individuals who had extreme inbreeding coefficients, mislabeled gender, or call rate < 95%, leaving 1342 individuals who passed quality control criteria [26]. A total of 1329 individuals had information on GLP1, GIP, glucagon, fasting glucose and fasting serum insulin levels. Additional genotypes were imputed (rs36210421) with high quality (proper_info > 0.95) into the 1000 genomes phase 1 panel using IMPUTE2. All variants were in Hardy Weinberg equilibrium (P > 0.05) [27].
INTER-99
In the Inter99 cohort, 6161 individuals were genotyped with the Illumina Human Exome BeadChip v1.0 genotyping array (“the exome chip”), as previously described [28].
Statistical analyses
Statistical analyses were performed using R, version 3.1.3 (https://www.r-project.org/). The trapezoidal method was used for calculation of total area under the curve (AUC), incremental area under the curve (iAUC) for GLP-1, GIP, insulin and glucagon. Calculations can be found in Additional file 1. We used multiple linear regression analysis to evaluate the association of QTcF, fasting and stimulated levels of glucose, insulin, GLP-1, GIP, and glucagon (as dependent variables) with the minor A-allele of rs36210421 and the minor G-allele of rs1805123 SNPs/ GRS (as independent variable). Effect sizes (β coefficients) per copy of the risk alleles of the SNPs investigated were estimated by linear regression analysis adjusted for age, sex and BMI. In order to adjust for population stratification, relatedness and to limit the type 1 error we calculated genome-wide principal components (PC) and adjusted our model for the first three PCs along with other covariates. P values < 0.05 were considered significant.
Genetic risk score (GRS)
To evaluate the combined effect of the two gene variants, rs36210421 and rs1805123, on QT-interval and metabolic phenotypes, an additive model was used to construct an unweighted genetic risk score. The GRS was calculated by summation of the number of risk alleles across the two gene variants.
Results
The variants rs36210421 and rs1805123 in hERG were present in individuals from the ADDITION-PRO cohort with a minor allele frequency (MAF) of 3.4% (A-allele) and 24% (G-allele) respectively, and in Inter99 with a MAF of 3.1% and 22%, respectively. We found a low degree of linkage disequilibrium between rs36210421 and rs1805123 (R2 = 0.005 and D prime = 0.241). Demographic description of the Inter99 and the ADDITION-PRO cohort can be seen in Additional file 1: Table S1.
Association of gene variants with duration of QT interval
The minor A-allele of rs36210421 (R1047L) and the minor G-allele of rs1805123 (K897 T) were significantly associated with shorter QTcF interval (β = − 2.4 ms, p = 0.016 and β = − 2.1 ms, p = 1.3 × 10− 7) in the Inter99 study (Table 1).
Associations of gene variants with fasting and stimulated levels of glucose, insulin, GLP-1, GIP, and glucagon
In the ADDITION-PRO cohort the minor G-allele of rs1805123 (K897 T) was significantly associated with 3.18 pmol/L (CI -5.87;-0.49)) lower fasting insulin (p = 0.025). The minor G-allele was associated with 0.95 pmol/L (− 1.52; − 0.38)) lower fasting glucagon (p = 0.003) (Additional file 1: Table S3). Additionally, we found decreased glucagon AUC at 0–30 and 0–120 min (β = − 2.73 (− 4.73, − 0.70), p = 0.009 and β = − 2.27 (− 4.25, − 0.24), p = 0.029, respectively) (Additional file 1: Table S3). The minor A-allele of rs36210421 was associated with decreased fasting GIP (p = 0.04) and decreased GIP120min (p = 0.04), as well as increased GLP-1 iAUC at 30 and 120 min (p = 0.02 and p = 0.04). In Inter99, plasma glucose and serum insulin levels were not associated with the two gene variants (Table 1).
We calculated an unweighted genetic risk score (GRS), assessing the additive effect of the minor A-allele of rs36210421 and the minor G-allele of rs1805123 on metabolic measures (Table 2, Fig. 1 and Additional file 1: Table S2, and Fig. S2). The GRS was significantly associated with lower fasting insulin (β = − 3.77 (− 6.36, − 1.18), p = 0.004), lower fasting GIP (β = − 0.60 (− 1.16, − 0.04), p = 0.041) and lower fasting glucagon (β = − 1.08 (− 1.78, − 0.38), p = 0.005). Additionally, the GRS was significantly associated with GIP AUC at 0–30 min was significantly decreased (β = − 1.96 (− 4.10, − 0.19), p = 0.044).The GRS was further associated with a decrease in glucagon AUC at 0–30 min (β = − 2.27 (− 4.21, − 0.29), p = 0.025), although not significant at 0–120 min (β = − 1.85 (− 3.78, 0.12, p = 0.066). Additionally, there was higher decremental glucagon AUC at 0–30 min, p = 0.011 and at 0–120 min, p = 0.003). Changes in plasma levels of glucagon during an OGTT according to number of risk alleles are shown in Fig. 1 and Additional file 1: Figure S2.

Incremental glucagon levels according to number of risk alleles. Mean and SEM of decremental glucagon levels according to genetic risk score (GRS) from (rs36210421 and rs1805123). GRS = 0 indicates no risk alleles, GRS = 1 indicates carriers of one risk allele of either rs1805123 or rs36210421, GRS = 2 indicates two or more risk alleles of either rs1805123 or rs36210421. P values indicate statistical significance, obtained from multiple linear regression adjusted for age, sex and bmi. *P < 0.05; **P < 0.01
Discussion
In this study, we demonstrate that common variants in hERG, the minor A-allele of rs36210421 (R1047L) and the minor G-allele of rs1805123 (K897 T), have pleotropic effects, which not only cause shortening of QT-interval, but also alterations in pancreatic and gut hormone release among carriers. We report that the minor A- allele of rs36210421 is associated with increased GLP-1 and decreased GIP response to oral glucose stimulation, whereas the minor G-allele of rs1805123 is associated with decreased fasting plasma insulin and glucagon release. Furthermore, we demonstrate that the additive effect of both variants cause increased GIP secretion and suppressed glucagon secretion, suggesting that hERG potassium channels play an important role in regulation of incretin and pancreatic hormone release.
We report that the minor A-allele of rs36210421 and the minor G-allele of rs1805123 were associated with significantly shorter QT interval among healthy individuals, indicating a mild gain-of-function of both variants in cardiac muscle cells, supporting the findings of previous studies [19, 20, 23, 24]. The hERG ion channel is formed by the KCNH2 protein that consists of six transmembrane alpha helices, a pore helix and cytoplasmically located N- and C-termini. The pore helix is thought to act as a voltage-sensitive sensor [5]. The helices are formed by bindings between amino acids and it is possible that amino acids changes (caused by rs1805123 or rs36210421) could lead to altered folding of the pore helix causing alterations in sensing or in opening/closing of the channel.
Hylten-Cavallius et al. recently reported that rare loss-of-function mutations in hERG cause prolonged QT-interval and increased GIP and GLP-1 secretion during an OGTT [9]. We report that the two examined gain-of-function hERG variants are associated with decreased fasting GIP and GIP secretion during an OGTT, supporting a role of hERG in GIP release from K-cells.
Furthermore, we assessed the effect of the variants in hERG on glucose-stimulated insulin secretion. We report that non-diabetic carriers of common variants in hERG have lower fasting levels of insulin, but no alterations in glucose-stimulated insulin release during an OGTT in the ADDITION-PRO study. We did not replicate lower fasting insulin levels among carriers of hERG variants in the Inter99 study. Thus further studies are needed to clarify whether common hERG variants have a role in beta cell function.
The directionality of effect of the common gain-of-function variants in hERG seems to be clear in K-cells causing gain-of-function effect leading to decreased secretion of GIP. It is more challenging to assess the effect of the common variants in hERG on the alpha-cells in pancreas. Alpha-cells secrete glucagon secondary to action potential firing and elevation of cytoplasmic Ca2+ concentration [29]. These are regulated by fluctuations in glucose and nutrient levels, and perhaps by autocrine and paracrine control by insulin, GABA and Zn2+ secreted from adjacent beta-cells and in particular by somatostatin from the delta cells [30,31,32]. It is challenging to deduce how the common variants in hERG influence all these factors. Moreover, the variants in hERG may very well have diverging effects in alpha-cells compared to K-cells.
In our study, we found lower fasting glucagon levels and smaller decrements in glucose-stimulated glucagon secretion in hERG gain-of-function carriers. The carriers did, however, have normal glucose levels at fasting and during oral glucose stimulation. Blocking of hERG channels in α-cells has been shown to decrease glucagon secretion [6]. We therefore expected that the common gain-of-function variants would cause alpha cells to increase glucagon secretion. Carriers of the gain-of-function variants had lower fasting levels of glucagon, but this might be a consequence of the interplay with insulin and plasma glucose. The mean glucagon levels during OGTT (at 30 and 120 min) were not different from non-carriers, indicating that the suppression of glucagon during glucose-stimulation was probably intact. Supporting this hypothesis, Hardy et al. previously demonstrated that at low glucose levels, blockage of the hERG channel yields a initially stimulatory effect, but then depolarizes the alpha-cells sufficiently to limit action potential firing and as a consequence less glucagon is secreted [6]. hERG channels may therefore be important regulators of glucagon secretion, but detailed in vitro functional studies and/or hyperglycemic clamp studies with arginine stimulation in carriers of common gain-of-function hERG variants are needed to pinpoint the exact role of hERG channels in glucagon secretion.
Conclusions
We demonstrate that common amino acid polymorphisms in the hERG (Kv11.1) voltage-gated potassium channel lead to considerable alterations in plasma GIP and glucagon release, suggesting that voltage-gated hERG potassium channels may play an important role in the regulation of the release of incretin and pancreatic hormones.
Abbreviations
- AUC:
-
Area under the curve
- GIP:
-
Gastric inhibitory polypeptide
- GLP-1:
-
Glucagon-like peptide-1
- GRS:
-
Genetic risk score
- hERG:
-
Human ether-a-go-go channel
- hERG :
-
Human ether-a-go-go gene
- iAUC:
-
Incremental area under the curve
- LQTS:
-
Long QT syndrome
- OGTT:
-
Oral glucose tolerance test
- SNP:
-
Single nucleotide polymorphism
References
Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, et al. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102:1178–85.
Dekker JM, Schouten EG, Klootwijk P, Pool J, Kromhout D. Association between QT interval and coronary heart disease in middle-aged and elderly men. The Zutphen study. Circulation. 1994;90:779–85.
Schouten EG, Dekker JM, Meppelink P, Kok FJ, Vandenbroucke JP, Pool J. QT interval prolongation predicts cardiovascular mortality in an apparently healthy population. Circulation. 1991;84:1516–23.
Algra A, Tijssen JG, Roelandt JR, Pool J, Lubsen J. QTc prolongation measured by standard 12-lead electrocardiography is an independent risk factor for sudden death due to cardiac arrest. Circulation. 1991;83:1888–94.
Vandenberg JI, Perry MD, Perrin MJ, Mann SA, Ke Y, Hill AP. hERG K(+) channels: structure, function, and clinical significance. Physiol Rev. 2012;92:1393–478.
Hardy AB, Fox JEM, Giglou PR, Wijesekara N, Bhattacharjee A, Sultan S, et al. Characterization of erg K+ channels in alpha- and beta-cells of mouse and human islets. J Biol Chem. 2009;284:30441–52.
Rogers GJ, Tolhurst G, Ramzan A, Habib AM, Parker HE, Gribble FM, et al. Electrical activity-triggered glucagon-like peptide-1 secretion from primary murine L-cells. J Physiol. 2011;589:1081–93.
Rosati B, Marchetti P, Crociani O, Lecchi M, Lupi R, Arcangeli A, et al. Glucose- and arginine-induced insulin secretion by human pancreatic beta-cells: the role of HERG K(+) channels in firing and release. FASEB J Off Publ Fed Am Soc Exp Biol. 2000;14:2601–10.
Hyltén-Cavallius L, Iepsen EW, Wewer Albrechtsen NJ, Svendstrup M, Lubberding AF, Hartmann B, et al. Patients with long QT syndrome due to impaired hERG-encoded Kv11.1 Potassium Channel have exaggerated endocrine pancreatic and incretin function associated with reactive hypoglycemia. Circulation. 2017;
Mühlbauer E, Bazwinsky I, Wolgast S, Klemenz A, Peschke E. Circadian changes of ether-a-go-go-related-gene (erg) potassium channel transcripts in the rat pancreas and beta-cell. Cell Mol Life Sci CMLS. 2007;64:768–80.
Torekov SS, Iepsen E, Christiansen M, Linneberg A, Pedersen O, Holst JJ, et al. KCNQ1 long QT syndrome patients have hyperinsulinemia and symptomatic hypoglycemia. Diabetes. 2014;63:1315–25.
Hyltén-Cavallius L, Iepsen EW, Christiansen M, Graff C, Linneberg A, Pedersen O, et al. Glucose ingestion causes cardiac repolarization disturbances in type 1 long QT syndrome patients and healthy subjects. Heart Rhythm. 2017;
Johansen NB, Hansen A-LS, Jensen TM, Philipsen A, Rasmussen SS, Jørgensen ME, et al. Protocol for ADDITION-PRO: a longitudinal cohort study of the cardiovascular experience of individuals at high risk for diabetes recruited from Danish primary care. BMC Public Health. 2012;12:1078.
Glümer C, Jørgensen T, Borch-Johnsen K. Inter99 study. Prevalences of diabetes and impaired glucose regulation in a Danish population: the Inter99 study. Diabetes Care. 2003;26:2335–40.
Orskov C, Jeppesen J, Madsbad S, Holst JJ. Proglucagon products in plasma of noninsulin-dependent diabetics and nondiabetic controls in the fasting state and after oral glucose and intravenous arginine. J Clin Invest. 1991;87:415–23.
Orskov C, Rabenhøj L, Wettergren A, Kofod H, Holst JJ. Tissue and plasma concentrations of amidated and glycine-extended glucagon-like peptide I in humans. Diabetes. 1994;43:535–9.
Deacon CF, Mannucci E, Ahrén B. Glycaemic efficacy of glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors as add-on therapy to metformin in subjects with type 2 diabetes-a review and meta analysis. Diabetes Obes Metab. 2012;14:762–7.
Fridericia LS. The duration of systole in an electrocardiogram in normal humans and in patients with heart disease. 1920. Ann. Noninvasive Electrocardiol. Off. J. Int. Soc. Holter noninvasive Electrocardiol. Inc. 2003;8:343–51.
Bezzina CR, Verkerk AO, Busjahn A, Jeron A, Erdmann J, Koopmann TT, et al. A common polymorphism in KCNH2 (HERG) hastens cardiac repolarization. Cardiovasc Res. 2003;59:27–36.
Anson BD, Ackerman MJ, Tester DJ, Will ML, Delisle BP, Anderson CL, et al. Molecular and functional characterization of common polymorphisms in HERG (KCNH2) potassium channels. Am J Physiol Heart Circ Physiol. 2004;286:H2434–41.
Paavonen KJ, Chapman H, Laitinen PJ, Fodstad H, Piippo K, Swan H, et al. Functional characterization of the common amino acid 897 polymorphism of the cardiac potassium channel KCNH2 (HERG). Cardiovasc Res. 2003;59:603–11.
Newton-Cheh C, Guo C-Y, Larson MG, Musone SL, Surti A, Camargo AL, et al. Common genetic variation in KCNH2 is associated with QT interval duration: the Framingham heart study. Circulation. 2007;116:1128–36.
Pfeufer A, Jalilzadeh S, Perz S, Mueller JC, Hinterseer M, Illig T, et al. Common variants in myocardial ion channel genes modify the QT interval in the general population: results from the KORA study. Circ Res. 2005;96:693–701.
Marjamaa A, Newton-Cheh C, Porthan K, Reunanen A, Lahermo P, Väänänen H, et al. Common candidate gene variants are associated with QT interval duration in the general population. J Intern Med. 2009;265:448–58.
Gouas L, Nicaud V, Berthet M, Forhan A, Tiret L, Balkau B, et al. Association of KCNQ1, KCNE1, KCNH2 and SCN5A polymorphisms with QTc interval length in a healthy population. Eur J Hum Genet EJHG. 2005;13:1213–22.
Justesen JM, Allin KH, Sandholt CH, Borglykke A, Krarup NT, Grarup N, et al. Interactions of lipid genetic risk scores with estimates of metabolic health in a Danish population. Circ Cardiovasc Genet. 2015;8:465–72.
Mahendran Y, Jonsson A, Have CT, Allin KH, Witte DR, Jørgensen ME, et al. Genetic evidence of a causal effect of insulin resistance on branched-chain amino acid levels. Diabetologia. 2017;60:873–8.
Andersson EA, Pilgaard K, Pisinger C, Harder MN, Grarup N, Faerch K, et al. Type 2 diabetes risk alleles near ADCY5, CDKAL1 and HHEX-IDE are associated with reduced birthweight. Diabetologia. 2010;53:1908–16.
Walker JN, Ramracheya R, Zhang Q, Johnson PRV, Braun M, Rorsman P. Regulation of glucagon secretion by glucose: paracrine, intrinsic or both? Diabetes Obes Metab. 2011;13(Suppl 1):95–105.
Unger RH, Orci L. Paracrinology of islets and the paracrinopathy of diabetes. Proc Natl Acad Sci U S A. 2010;107:16009–12.
Rorsman P, Berggren PO, Bokvist K, Ericson H, Möhler H, Ostenson CG, et al. Glucose-inhibition of glucagon secretion involves activation of GABAA-receptor chloride channels. Nature. 1989;341:233–6.
Ishihara H, Maechler P, Gjinovci A, Herrera P-L, Wollheim CB. Islet beta-cell secretion determines glucagon release from neighbouring alpha-cells. Nat Cell Biol. 2003;5:330–5.
Acknowledgements
The authors thank the ADDITION-PRO cohort and the Inter99 cohort, the staff and the participants for their contributions to this study. The authors thank the Danish Diabetes Academy and the Novo Nordisk Foundation Center for Basic Metabolic Research for making this study possible.
Funding
This work was supported by research grants from The Danish Diabetes Academy, The Novo Nordisk Foundation Center for Basic Metabolic Research, which is an independent research center at the University of Copenhagen partially funded by an unrestricted donation from the Novo Nordisk Foundation (www.cbmr.ku.dk). Also, this project has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement No 667191.
Availability of data and materials
The authors confirm that, for approved reasons, some access restrictions apply to the data underlying the findings. Data are available from the Novo Nordisk Foundation Center for Basic Metabolic Research, section of Metabolic Genetics whose authors may be contacted at torben.hansen@sund.ku.dk.
Author information
Authors and Affiliations
Contributions
TH, JKK, SST and YM planned the study. YM and AGP performed the statistical analyses. LE, YM, HV, AJ, KF, PW, MEJ, DRW, TJ, NG, OP, JJH, SST, JKK and TH interpreted the data. JJH provided data. LE wrote the first draft. All authors participated in editing and revising the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The Inter99 cohort (ClinicalTrials.gov, NCT00289237) was approved by the Copenhagen County Ethical Committee (KA 98155) and the ADDITION-PRO study were approved by the Ethical Committee of the Central Denmark Region (journal no. 20080229). Both studies were approved by the National Board of Health and were conducted in accordance with the principles of the Helsinki Declaration. All study participants provided both oral and written informed consent. No participants were under the age of 18 years.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Calculations of AUC and iAUC. Information on how AUC and iAUC was calculated in the cohorts. Table S1: Participant characteristics in the ADDITION-PRO cohort and Inter99. Anthropometric measures of individuals in the ADDITION-PRO and Inter99 cohort. Table S2. Association of variants (rs36210421 and rs1805123) with QT interval and with metabolic and incretin levels in the Inter99 cohort (n = 5487). Table with measures of QTcF interval and glucose levels in the Inter99 cohort sorted by genetic variant and combined in a genetic risk score. Table S3. Association of KCNH2 variant rs36210421 with metabolic and incretin levels in ADDITION-PRO cohort (N = 1324) (Non diabetes and newly diagnosed T2D). Table with measures of glucose, insulin GIP, GLP-1 and glucagon levels according to carrier status for rs1805123 and rs36210421. Figure S1. Levels of incretins and glucagon according to number of risk alleles. Levels of GLP-1, GIP and Glucagon during an OGTT according to number of risk alleles in the GRS. (DOCX 122 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Engelbrechtsen, L., Mahendran, Y., Jonsson, A. et al. Common variants in the hERG (KCNH2) voltage-gated potassium channel are associated with altered fasting and glucose-stimulated plasma incretin and glucagon responses. BMC Genet 19, 15 (2018). https://doi.org/10.1186/s12863-018-0602-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-018-0602-2