
Abstract
Background
Peranakan Ongole (PO) is a major Indonesian Bos indicus breed that derives from animals imported from India in the late 19th century. Early imports were followed by hybridization with the Bos javanicus subspecies of cattle. Here, we used genomic data to partition the ancestry components of PO cattle and map loci implicated in birth weight.
Results
We found that B. javanicus contributes about 6-7 % to the average breed composition of PO cattle. Only two nearly fixed B. javanicus haplotypes were identified, suggesting that most of the B. javanicus variants are segregating under drift or by the action of balancing selection. The zebu component of the PO genome was estimated to derive from at least two distinct ancestral pools. Additionally, well-known loci underlying body size in other beef cattle breeds, such as the PLAG1 region on chromosome 14, were found to also affect birth weight in PO cattle.
Conclusions
This study is the first attempt to characterize PO at the genome level, and contributes evidence of successful, stabilized B. indicus x B. javanicus hybridization. Additionally, previously described loci implicated in body size in worldwide beef cattle breeds also affect birth weight in PO cattle.
Similar content being viewed by others

Background
The humped Ongole or Nellore cattle are indigenous to the Nellore-Ongole region in Prakasam District (Andhra Pradesh State), Southeastern coast of India. Molecular evidence supports a pure Bos indicus origin to the modern Indian Ongole population [1]. The history of Ongole cattle is only partially documented, but it is believed to date back to the late Bronze Ages (some 4000 years ago) when pastoral nomad Aryan tribes migrated to India, bringing different types of cattle that rapidly spread throughout the country [2]. These imports probably contributed to the formation of white/gray cattle breeds over the centuries, such as the Ongole. Heat tolerance, disease resilience, and draft power have made Ongole cattle attractive for beef production in low-input systems, which stimulated imports of Ongole bulls to several tropical countries in the late 19th century, including South America [3] and Indonesia.
Ongole cattle importation to Indonesia from the Nellore province of India was carried out by Dutch colonials, with massive imports from 1905 to 1920. In 1912, the Dutch government designated the Sumba Island as the official location for maintaining imported Ongole animals, which led to the formation of the Sumba Ongole (SO) population. From 1915 to 1929, the “Ongolization program” was responsible for distributing SO animals to several regions of Indonesia, including the Java Island, where SO cattle were crossed with local Bos javanicus cattle (also known as Java or Banteng cattle), and formed the Ongole-grade or Peranakan Ongole (PO) breed [4, 5]. This historical hybridization is supported by microsatellite and mitochondrial DNA data [6]. To date, PO cattle are one of the most popular breeds in Indonesia, spreading almost evenly throughout the country, ranging from Sumatra, Java and Sulawesi Islands. Yet, the breed remains genetically uncharacterized and poorly selected for production traits.
The uncontrolled use of bulls together with non-systematic breeding has undermined the genetic progress of PO cattle in Indonesia over the years. Consequently breeders have recently started to produce crossbreds between PO and exotic taurine breeds, such as Limousin and Simmental, in an attempt to rapidly improve beef production, thus threatening the conservation of the breed. The identification of quantitative trait loci (QTLs) underlying traits of interest, such as body size and weight, may be of help to encourage breeders to further explore the genetic potential of the breed.
Here, we used a panel of 54,609 SNPs (50k) to genetically characterize PO cattle by comparison with B. taurus, B. indicus, B. javanicus and composite B. taurus x B. indicus breeds. We aimed at: i) assessing the genetic relationships between PO and other cattle breeds; ii) estimating the contribution of B. javanicus to PO breed composition; iii) partitioning the ancestral origins of the B. indicus component of the PO genome; iv) map putative B. javanicus haplotypes segregating in PO; and 5) investigating whether major QTLs for body size previously found in other breeds also segregate in PO.
Methods
Ethical statement
All animal procedures related to PO and BALI samples were approved by the Indonesian Agency for Agricultural Research and Development, Ministry of Agriculture, KKP3N activity 70/PL.220/I.1/3/2014. Genotypes for the remaining breeds were obtained from previously published data [7].
Genotypes, phenotypes and quality control
A total of 48 Indonesian PO male steers were genotyped for 54,609 SNPs using the Illumina® BovineSNP50 v2 Genotyping BeadChip assay (50K), according to the manufacturer's protocol. These animals were selected based on their extreme phenotypic values for birth weight (used here as a proxy for body size), representing the lower (n = 24, mean = 21.31 ± 1.61 kg) and upper (n = 24, mean = 28.75 ± 2.82 kg) tails of the phenotypic distribution. Additionally, 18 B. javanicus animals from the Bali Island (BALI) were genotyped with the 50k panel.
The following Illumina® BovineHD genotypes (HD - 786,799 SNPs) were available from previously published data [7]: European B. taurus Holstein (HOL, n = 59), African B. taurus N'Dama (NDA, n = 24), Brazilian B. indicus of Indian origin Nellore (NEL, n = 35) and Gyr or Gir (GIR, n = 30), admixed B. indicus Brahman (BRM, n = 25) and modern B. taurus x B. indicus composites Santa Gertrudis (SGT, n = 24) and Beef Master (BMA, n = 24). The later two were included because both have B. taurus and B. indicus contributions to their genomes: Ongole was one of the breeds directly used in the formation of SGT, whereas the creation of the BMA composite was intermediated by the use of Brahman bulls. On the other hand, Brahman is a B. indicus breed developed using Gir, Nellore and Guzerat, which has been crossed with B. taurus breeds.
PO cattle genotypes were merged with the remaining samples into a single dataset by overlapping the common set of markers between the HD and the 50K panels. Both data sets had genomic coordinates annotated in the UMD v3.1 reference assembly. The final data comprised 287 samples and 49,915 SNPs. Markers and individuals were removed from the dataset using PLINK v1.07 [8] if they did not present call rate of at least 90 %. We decided to not trim the data by minor allele frequency (MAF) in the breed level, as markers with private alleles may be highly informative in comparative analyses. Instead, we excluded SNPs that were monomorphic across all breeds. Summary statistics such as pairwise FST and heterozygosity were also computed and made available as supplementary data (Additional file 1).
Genotype clustering and admixture analysis
Genetic relationships between PO and other worldwide cattle breeds were determined using both distance-based and model-based genotype clustering analyses. In the distance-based method, we performed a Classical Multidimensional Scaling (CMDS) analysis using PLINK v1.07 and customized scripts in R v3.1.2 (available at: http://www.r-project.org/). First, a similarity matrix was constructed from the proportion of alleles shared identically by state between each possible pair of samples. Then, CMDS was applied to the similarity matrix, and the first two principal coordinates were used to obtain a two-dimensional graphical representation of relationships among individuals.
ADMIXTURE v1.23 was used for a model-based unsupervised clustering of individuals via maximum likelihood [9]. We assumed different models where the individuals’ genome could be partitioned in K clusters, assuming they may derive from K different ancestral populations. We started with a model assuming that genome fragments derive from one of four ancestral populations (K = 4): European B. taurus, African B. taurus, B. indicus or B. javanicus. Then, we fitted models with increasing number of clusters up to the number of breeds (K = 5 to K = 9).
Although the model implemented in ADMIXTURE v1.23 allows for estimating ancestry proportions, it does not provide means to formally test for the presence of admixture. Therefore, we used the threepop program of the TreeMix v1.12 package [10] to compute f 3 statistics [11]. Standard deviation scores (Z-scores) for f 3 were computed for all possible population triplets. Significant admixture was declared when Z < −3. Additionally, TreeMix v1.12 was also used to generate a maximum likelihood phylogeny with migration edges. The tree was rooted on B. javanicus and computed using blocks of 1000 markers. The assumed number of migration events was chosen to match the number of admixed breeds in the model-based clustering analysis.
Detection of PO haplotypes inherited from Bos javanicus
Probability of B. javanicus haplotypic ancestry was estimated following Bolormaa et al. (2011) [12]. We estimated haplotype phase and imputed missing genotypes using SHAPEIT2 [13]. Haplotype frequencies were computed for overlapping windows of five consecutive markers using customized scripts in R. Then, for each haplotype found in PO, the probability of B. javanicus ancestry was estimated as:
where \( {\mathit{\mathsf{p}}}_{\mathit{\mathsf{B}}\mathit{\mathsf{j}}} \) and \( {\mathit{\mathsf{p}}}_{\mathit{\mathsf{Bi}}} \) are the haplotype frequencies in B. javanicus and B. indicus, respectively. Haplotype frequency in B. javanicus was estimated using BALI samples. For B. indicus, we used NEL and GIR samples to compute haplotype frequencies. Haplotypes with a frequency of at least 10 % in PO cattle presenting \( {\mathit{\mathsf{p}}}_{\mathit{\mathsf{B}}\mathit{\mathsf{j}}} > \mathsf{0.8} \) in all comparisons were considered as candidate identical-by-descent (IBD) B. javanicus haplotypes.
Haplotypes were further filtered and divided into three groups: group A, comprising haplotypes that were absent in B. indicus breeds but present in B. javanicus; group B, including haplotypes with high frequency in PO and B. javanicus (>10 %) but low frequency in B. indicus (<5 %); group C, haplotypes with moderate-to-high frequencies in B. indicus (>5 %) but that were more frequent in B. javanicus; and group D, including all remaining haplotypes. Group A represented a proxy for B. javanicus private haplotypes. Group B was likely to contain haplotypes derived from B. javanicus that are identical-by-state (IBS) with B. indicus rare haplotypes. Group C was similar to Group B, except that the IBS haplotypes in B. indicus had higher frequencies. Additionally, nearly fixed (>80 %) B. javanicus haplotypes were considered candidates under selection, regardless of the groups mentioned above.
Genome-wide mapping of loci affecting PO birth weight
Putative QTLs for birth weight were detected using the following regression model:
where \( \mathit{\mathsf{y}} \) is the vector of birth weights, 1 n is a vector of 1's, \( \mathit{\mathsf{\mu}} \) is the overall mean, \( \mathit{\mathsf{x}} \) is an incidence vector for birth season, \( \mathit{\mathsf{b}} \) is the fixed effect of birth season, \( \mathit{\mathsf{g}}=\mathit{\mathsf{Z}}\mathit{\mathsf{a}} \) is the vector of random direct genomic values (i.e., sum of the effects of genome-wide markers), \( \mathit{\mathsf{Z}} \) is the matrix of centered genotypes, \( \mathit{\mathsf{a}} \) is the vector of random marker effects, and e is the vector of residual effects.
de Los Campos et al. (2009) [14] demonstrated that this model can be re-parameterized by \( \mathit{\mathsf{g}}=\mathit{\mathsf{K}}\mathit{\mathsf{c}} \), where \( \mathit{\mathsf{K}} \) is the kernel matrix of additive genetic relationships between pairs of individuals, \( \mathit{\mathsf{c}} \) is a random vector distributed as \( \mathit{\mathsf{N}}\left(0,{\mathit{\mathsf{K}}}^{-1}{\mathit{\mathsf{\sigma}}}_{\mathit{\mathsf{c}}}^2\right) \), and \( {\mathit{\mathsf{\sigma}}}_{\mathit{\mathsf{c}}}^2 \) is the variance attributed to \( \mathit{\mathsf{c}} \). The additive kernel matrix can be computed as \( \mathit{\mathsf{K}}=\mathit{\mathsf{Z}}\mathit{\mathsf{D}}\mathit{\mathsf{Z}}\hbox{'}\mathit{\mathsf{q}} \) [15], where the scaling parameter \( \mathit{\mathsf{q}} \) is \( 1/{\displaystyle \sum {2\mathrm{p}}_{\mathit{\mathsf{i}}}}\left(1-{\mathit{\mathsf{p}}}_{\mathit{\mathsf{i}}}\right),\ {\mathit{\mathsf{p}}}_{\mathit{\mathsf{i}}} \) is the reference allele frequency at marker \( \mathit{\mathsf{i}} \), and \( \mathit{\mathsf{D}} \) is a diagonal matrix of marker weights. This parametrization avoids fitting a model with as many predictors as markers by fitting as many predictors as samples. Marker effects can be back-transformed from estimates \( \widehat{\mathit{\mathsf{c}}} \) as in the Genomic Best Linear Unbiased Predictor (GBLUP) method [16]:
Under the assumption of equal variance across markers, \( \mathit{\mathsf{D}}=\mathit{\mathsf{I}} \) and
From the definitions above, \( \widehat{\mathit{\mathsf{g}}}=\mathit{\mathsf{K}}\widehat{\mathit{\mathsf{c}}} \), then:
Since \( {\mathit{\mathsf{K}}}^{-1}\mathit{\mathsf{K}}=\mathit{\mathsf{I}} \), we have:
Model parameters were estimated using the Gibbs sampling algorithm implemented in the BGLR v1.0.3 package in R v.3.1.2 [17]. Normal priors were assigned to random effects and flat priors were assigned to the overall mean and birth season. Variance components were assumed a priori scaled inverse chi-squared distributed. A single Markov chain with a length of 1,000,000 iterations was used. The burn-in period was set at 10,000 iterations and the thinning interval at 100 iterations. Only polymorphic autosomal SNPs presenting Fisher's exact test p-value for Hardy-Weinberg equilibrium (HWE) greater than 1 × 10−20 were included in this analysis. Posterior samples for the variance explained by genome-wide SNPs were obtained as \( \frac{{\widehat{\sigma_c}}^2}{{\widehat{\sigma_c}}^2+{\widehat{\sigma_e}}^2} \) and the point estimate was derived from the average of these samples. The 95 % credible interval was defined as the 2.5 % and 97.5 % percentiles of the posterior distribution.
The variance attributed to overlapping chromosome segments encompassed by 20 consecutive SNPs was computed as \( \mathit{\mathsf{v}}\mathit{\mathsf{a}}\mathit{\mathsf{r}}\left({\displaystyle \sum_{\mathit{\mathsf{i}}=1}^{20}{\mathit{\mathsf{z}}}_{\mathit{\mathsf{i}}}}{\widehat{\mathit{\mathsf{a}}}}_{\mathit{\mathsf{i}}}\right) \) [18]. As this analysis was underpowered due to small sample size (n = 48), we considered candidate QTLs only the top scoring SNP windows with variance above \( 10\mathit{\mathsf{I}}\mathit{\mathsf{Q}}\mathit{\mathsf{R}}+\mathit{\mathsf{Q}}\mathsf{3} \) [19], where \( \mathit{\mathsf{I}}\mathit{\mathsf{Q}}\mathit{\mathsf{R}} \) and \( \mathit{\mathsf{Q}}\mathsf{3} \) are the interquartile range and the third quartile of the distribution of SNP window variances, respectively.
Results and discussion
Genetic relationships between Peranakan Ongole and other cattle breeds
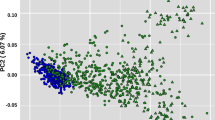
When all breeds were analyzed simultaneously using CMDS (Fig. 1a), the first coordinate (C1, x-axis) explained the genetic differences between B. taurus, B. indicus and B. javanicus. The second coordinate (C2, y-axis) separated breeds by geographical origin, explaining genetic differences between African and non-African cattle. These findings are consistent with the previously reported clustering behavior of 50k genotypes in worldwide cattle breeds [20, 21]. As B. indicus breeds were poorly separated in comparison to the B. taurus breeds as a result of ascertainment bias [22] (see Additional file 1), the relationships among B. indicus breeds were assessed by re-running the analysis without B. taurus and B. javanicus genotypes (Fig. 1b). This analysis revealed that GIR, BRM, PO and NEL cluster as distinct populations, with PO and NEL exhibiting greater similarity. This is not unexpected, provided PO and NEL were believed to derive from the same ancestral population.

Multidimensional scaling analysis. When all breeds are simultaneously analyzed (a), the differences between European B. taurus (HOL), African B. taurus (NDA) and B. indicus breeds (PO, BRM, GIR and NEL) are well demonstrated. However, B. indicus breeds are poorly distinguishable due to ascertainment bias. The analysis of B. indicus breeds alone (b) resolves the relationships among BRM, GIR, NEL and PO cattle, highlighting a closer proximity between the later two. See Material and Methods for breed abbreviations
Ancestry components in the Peranakan Ongole genome
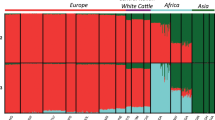
Results from the model-based clustering analysis are found in Fig. 2. When K = 4 was assumed, European B. taurus, African B. taurus, B. javanicus and B. indicus breeds were assigned to different clusters. SGT and BMA presented an average B. indicus contribution of 34 %, whereas BRM exhibited an average of 6 % of B. taurus ancestry. Interestingly, backcrossing to Ongole bulls over the course of 100 years was not capable of eliminating all B. javanicus introgression in PO cattle, which presented a mean B. javanicus ancestry of approximately 6 %. Historical hybridization with B. javanicus cattle seems to also extend to other Indonesian B. indicus populations, such as the Brebes [21] and Madura [21, 23] breeds. This is in contrast with the historical B. taurus introgression in NEL and GIR [3], which seems to have been consistently eliminated by intensive backcrossing [20, 21, 24]. This suggests that either B. javanicus haplotypes were kept by selective forces, or backcrossing in Indonesia was not as strong as in Brazil, and B. javanicus haplotypes are just drifting in PO cattle.

Model-based clustering of cattle breeds assuming different numbers of ancestral populations (K). Each individual is represented by a vertical bar that can be partitioned into colored fragments with length proportional to cluster contribution. K = 4 approximates the ancestral European B. taurus, African B. taurus, B. indicus and B. javanicus populations. K = 7 distinguishes between the two zebu ancestors that generated GIR and NEL, and PO exhibits contributions from both ancestral populations. See Material and Methods for breed abbreviations
Bos indicus and B. taurus have karyotypes consisting of 29 pairs of acrocentric autosomes, whereas B. javanicus has 25 pairs of acrocentric and two pairs of submetacentric autosomes. Based on cross-species fluorescence in-situ hybridization analysis, Ropiquet and colleagues [25] suggested that the bi-armed autosomes in B. javanicus are equivalent to Robertsonian translocations of autosomes 1–29 and 2–28 in B. taurus. It is still unclear how the first generations of Indonesian B. indicus × B. javanicus hybrids coped with chromosome number imbalance, but the preservation of sequence homology between these species of cattle seems to have guaranteed successful hybridization.
The f 3 statistics [11] provided further support for the evidence of B. javanicus hybridization in the PO genome. As expected, only SGT, BMA, BRM and PO presented Z-scores lower than −3, suggesting no significant mixture in HOL, NDA, NEL, GIR and BALI. The lowest scores for SGT (Z = −10.12) and BMA (Z = −9.51) were obtained with the comparisons HOL-NEL and HOL-GIR, respectively, reproducing the known crossbreeding in these populations. In the case of BRM, the lowest score (−9.18) was obtained by contrasting with HOL-GIR, also highlighting the B. taurus introgression in this breed. The mixture between B. javanicus and B. indicus in PO was well supported with a score of −17.88 for the sister group BALI-NEL.
We carried out further admixture analyses assuming different numbers of ancestral populations in order to dissect the B. indicus component of the PO cattle genome. In spite of over 100 years of isolation, NEL and PO share a very similar history of importation [3] and are deemed to derive from the same Indian Ongole population. Therefore, it was expected that all B. indicus ancestry in PO pertained to the same origin of NEL. Surprisingly, at K = 7 (Fig. 2), GIR separated from NEL, revealing that PO has contributions from both ancestral pools, with an average of 20.3 % of the PO genome descending from the same ancestral population that originated GIR. Two hypotheses can be formulated from this finding: 1) the first imports of Ongole to Brazil comprised purebred animals, whereas Indonesian imports comprised admixed animals; 2) both Brazilian and Indonesian imports included admixed animals, and systematic breeding in Brazil promoted selection against the GIR component.
Together, these results predicted that the phylogenetic analysis should cluster PO to the B. indicus clade, and a migration edge should be drawn from the B. javanicus branch towards PO. Therefore, we constructed a maximum likelihood tree using TreeMix [10], assuming four migration events representing mixtures in SGT, BMA, BRM and PO. Indeed, the estimated phylogeny behaved as predicted (Fig. 3). As expected, the remaining three estimated migrations were European B. taurus introgressions into SGT, BMA and BRM.

Estimated phylogenetic tree of cattle breeds. The scale bar represents 10 times the average standard error of the estimated entries in the sample covariance matrix. Migration edges were heat-colored according to the weight of contribution from the parental migrant population. Migration edges show B. javanicus hybridization into PO cattle, and European B. taurus introgression into SGT, BMA and BRM. Vertex a approximates the divergence between Bos primigenius and B. javanicus. Vertex b bifurcates the ancestral B. primigenius into the ancestors of B. taurus and B. indicus. Vertex c is an artificial bifurcation of modern composite breeds SGT and BMA. Vertex d approximates the divergence between European and African B. taurus cattle
Bos javanicus haplotypes in Peranakan Ongole
We examined a total of 113,665 PO haplotypes with a frequency of at least 10 %. From these, 8010 haplotypes presented \( \mathit{\mathsf{Pr}}\left(\mathit{\mathsf{B}}\mathit{\mathsf{j}}\right) > \mathsf{0.8} \) (Additional file 2). These stringent frequency and probability thresholds were adopted in order to minimize the false positive rate in our sample, considering that low frequency haplotypes could have been generated due to genotyping or phasing errors, and because we were particularly interested in finding high frequency private B. javanicus haplotypes in PO cattle. We considered only NEL and GIR as reference B. indicus because these breeds are representative of the ancestral populations that originated PO, and because BRM presented B. taurus introgression in the clustering analysis. Remarkably, the proportion of putative B. javanicus haplotypes in respect to all detected haplotypes was approximately 7 %, which is consistent with the average ancestry estimated from the admixture analysis.
A total of 779, 1480, 3286 and 2465 haplotypes were categorized in groups A (private B. javanicus haplotypes), B (B. javanicus haplotypes IBS with B. indicus rare haplotypes), C (B. javanicus haplotypes IBS with B. indicus common haplotypes), and D (remaining candidates), respectively (Additional file 1). Although groups A and B were of primary interest, these categories can also emerge from the ascertainment bias of the SNP panel. Group C is less prone to false positives induced by SNP bias, but is also more susceptible to ancestry mis-assignment because frequent IBS haplotypes are found in B. javanicus and B. indicus.
Only two genomic regions presented nearly fixed B. javanicus haplotypes (frequency > 80 %): 5:106.1-106.5Mb and 16:33.0-33.4Mb. The highest five markers haplotype frequency within the chromosome 5 region (97.9 %) was also fixed in B. javanicus, but absent in B. indicus. This segment contained four protein-coding genes: fibroblast growth factor 6 (FGF6), fibroblast growth factor 23 (FGF23), fructose-2,6-bisphosphatase (FR2BP, also known as TP53-induced glycolysis and apoptosis - TIGAR), and cyclin D2 (CCND2). The chromosome 16 region contained nearly fixed haplotypes in PO and B. javanicus (>91 %), but with modest frequencies in B. indicus (<12 %). Five protein-coding genes mapped to this region: EF-hand calcium binding domain 2 (EFCAB2), heterogeneous nuclear ribonucleoprotein U (HNRNPU), COX20 (FAM36A), peptidase domain containing 1 (PPPDE1) and human chromosome 1 open reading frame 101 (C1ORF101). Additionally, one small nucleolar RNA and one U6 spliceosomal RNA also mapped to this chromosome 16 domain.
Besides the possibility of false positives due to ascertainment bias, these haplotypes may have achieved nearly fixation either because an advantageous B. javanicus allele in this chromosome segment has been subjected to positive selection in PO, or because this B. javanicus haplotype hitchhiked with a selected B. indicus variant. The small number of nearly fixed B. javanicus haplotypes suggests that most of these are just drifting in PO, but we also do not discard balancing selection.
Candidate QTLs for birth weight in Peranakan Ongole
The genotype filters reduced the SNP panel to 34,482 markers, which were estimated to explain 33.5 % of the variance in birth weights, with a 95 % credible interval of 12.0 % - 67.6 %. These estimates must be taken with caution, as reliable variance components inference often requires the analysis of thousands of records. A clear limitation of the QTL mapping analysis presented here is the use of a small sample size (n = 48), rendering our investigation prone to high false discovery rates and limited to the detection of major QTLs. Nevertheless, we extensively compared the results obtained here with the literature in order to determine whether some of the detected loci have been previously associated with body size traits in other cattle breeds.
A total of 34,221 windows of 20 consecutive SNPs were inspected, and in spite of the small number of PO samples, positional candidates for birth weight were found on chromosomes (CHR) 1, 3, 11, 14, 15, 18, 24 and 29 (Fig. 4). Remarkably, the majority of these candidate loci have already been described as associated with body size in beef cattle.

Manhattan plot of birth weight variance attributed to overlapping windows of 20 consecutive SNPs. The dashed horizontal line represents the \( 10\mathit{\mathsf{I}}\mathit{\mathsf{Q}}\mathit{\mathsf{R}}+\mathit{\mathsf{Q}}\mathsf{3} \) threshold, where \( \mathit{\mathsf{I}}\mathit{\mathsf{Q}}\mathit{\mathsf{R}} \) and \( \mathit{\mathsf{Q}}\mathsf{3} \) are the interquartile range and the third quartile of the distribution of SNP window variances, respectively
The positional candidate locus on CHR14 (peaking at 25 Mb) maps to the PLAG1 (pleiotropic adenoma gene 1) chromosomal domain, and was previously found to be associated with birth weight in NEL cattle [26], as well as with a number of body size [27–30] and reproductive traits [19, 31, 32] in several B. taurus and B. indicus breeds. The mechanism by which PLAG1 may affect fetal growth and reproduction is most likely associated with the trans-acting regulation of the expression of insulin-like growth factors, particularly IGF2 [33].
In Brahmans, the C allele of rs109231213 in the vicinity of PLAG1 is associated with positive effects on body size but negative effects on fertility, and also presents evidence of selection and B. taurus origin [29]. We hypothesized that a similar phenomenon could underly birth weight variance in PO, namely selective pressures on the PLAG1 region potentially involving B. javanicus haplotypes.Interestingly, this CHR14 segment indeed presented large LD blocks (data not shown). High LD in this chromosome domain has also been observed in NEL [19], and a possible event of balancing selection has been hypothesized in that breed [26]. However, this long range LD can be partially explained by our sampling strategy, which privileged extremes from the phenotypic distribution. Furthermore, we found no evidence of differential effects between B. javanicus and B. indicus haplotypes for this or any other candidate QTLs (data not shown), suggesting that variants of B. indicus origin are responsible for differences in birth weight in PO.
The putative QTL on CHR3 (116.4 to 116.8 Mb) is in the vicinity of associations for ribeye area in Shorthorn [28] and calf size in Holstein cattle [34], and contains genes that are involved in early embryo development and growth, such as gastrulation brain homeobox 2 (GBX2) and constitutive photomorphogenic homolog subunit 8 (COPS8). The CHR15:44.6-45.0 Mb region is nearby the ribosomal protein L27a gene (RPL27A) and coincides with QTLs for mature weight and ribeye area in Hereford [28] and mature height, ribeye area and carcass weight in Angus [35]. The 30.9 to 32.2 Mb region on CHR18 is in a gene desert but flanks QTLs for longissimus dorsi area and body weight in Angus [35], and carcass weight in Angus x Brahman crosses [36]. In contrast, the locus on CHR29 (29.2 to 30.7 Mb) contains 24 protein-coding genes, and also overlaps QTLs for carcass, weaning and yearling weights in Maine-Anjou cattle [28]. Interestingly, the adoption of the highly stringent Bonferroni correction prevented the detection of this QTL in a genome-wide association study for birth weight in NEL [26].
The candidate QTLs on CHR1 (28.9 to 29.2 Mb), CHR11 (97.8 to 98.5 Mb), and CHR24 (61.4 to 62.0 Mb) did not overlap any previously identified QTLs for body size or related traits, and are either novel QTLs or false discoveries. However, these loci harbor interesting candidate genes. For instance, the CHR1 region shelters the 1,4-alpha-glucan branching enzyme gene (GBE1), responsible for increasing the solubility of glycogen molecules to allow its accumulation in the liver and muscle cells, and could be involved in somatic growth and muscle development. The CHR11 region contains the somatic cytochrome c gene (CYCS), previously shown to affect embryonic growth [37]. Finally, the CHR24 locus shelters the PH domain and leucine rich repeat protein phosphatase 1 gene (PHLPP1). Disruption of this gene in mice has been recently found to lead to decreased bone mass [38].
Conclusions
We found molecular evidence of B. javanicus × B. indicus hybridization in the genomes of Peranakan Ongole steers, consistent with historical records. Haplotype analyses detected only two candidate B. javanicus haplotypes that may have been subjected to positive selection, suggesting that the majority of the B. javanicus contribution is either drifting or under balancing selection. We also estimated that the B. indicus ancestry component of Peranakan Ongole animals derived from two distinguishable genetic pools that are closely related to Brazilian Nellore and Gir cattle. Quantitative trait loci underlying body size in other beef cattle populations were shown to also segregate in Peranakan Ongole cattle, representing an opportunity for selection and improvement of this breed.
Availability of supporting data
The original data sets supporting the results of this article are available in Additional file 3. They are accessible by password request from the authors.
Abbreviations
- PO:
-
Peranakan Ongole
- SO:
-
Sumba Ongole
- NEL:
-
Nellore
- GIR:
-
Gyr or Gir
- HOL:
-
Holstein
- NDA:
-
N'Dama
- SGT:
-
Santa Gertrudis
- BMA:
-
Beefmaster
- CMDS:
-
Classical multidimensional scaling
- SNP:
-
Single-nucleotide polymorphism
- CHR:
-
Chromosome
- MAF:
-
Minor allele frequency
- 50K:
-
Illumina® BovineSNP50 v2 Genotyping BeadChip assay
- HD:
-
Illumina® BovineHD BeadChip assay
- QTL:
-
Quantitative trait locus
- HWE:
-
Hardy-Weinberg equilibrium
References
Kumar P, Freeman AR, Loftus RT, Gaillard C, Fuller DQ, Bradley DG. Admixture analysis of South Asian cattle. Heredity (Edinb). 2003;91:43–50.
Karthickeyan SMK, Kumarasamy P, Sivaselvam SN, Saravanan PT R. Analysis of microsatellite markers in Ongole breed of cattle. Indian J Biotechnol. 2008;7(January):113–6.
Ajmone-Marsan P, Garcia JG, Lenstra JA. On the origin of cattle: how aurochs became cattle and colonized the world. Evol Anthropol Issues, News, Rev. 2010;19:148–57.
Hardjosubroto W. Aplikasi Pemuliabiakan Ternak Di Lapangan. Jakarta, Indonesia: PT. Gramedia Widiasarana; 1994. p. 284.
Astuti M. Potensi dan keragaman sumberdaya genetik sapi Peranakan Ongole (PO). Wartazoa. 2004;14:30–9.
Mohamad K, Olsson M, van Tol HTA, Mikko S, Vlamings BH, Andersson G, et al. On the origin of Indonesian cattle. PLoS One 2009;4:e5490.
Porto-Neto LR, Sonstegard TS, Liu GE, Bickhart DM, Da Silva MVB, Machado MA, et al. Genomic divergence of zebu and taurine cattle identified through high-density SNP genotyping. BMC Genomics. 2013;14:876.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–64.
Pickrell JK, Pritchard JK: Inference of Population Splits and Mixtures from Genome-Wide Allele Frequency Data. PLoS Genet 2012, 8:e1002967.
Reich D, Thangaraj K, Patterson N, Price AL, Singh L. Reconstructing Indian population history. Nature. 2009;461:489–94.
Bolormaa S, Hayes BJ, Hawken RJ, Zhang Y, Reverter A, Goddard ME. Detection of chromosome segments of zebu and taurine origin and their effect on beef production and growth. J Anim Sci. 2011;89:2050–60.
O’Connell J, Gurdasani D, Delaneau O, Pirastu N, Ulivi S, Cocca M, et al. A general approach for haplotype phasing across the full spectrum of relatedness. PLoS Genet. 2014;10:e1004234.
De Los Campos G, Gianola D, Rosa GJM. Reproducing kernel Hilbert spaces regression: a general framework for genetic evaluation. J Anim Sci. 2009;87:1883–7.
VanRaden PM. Efficient methods to compute genomic predictions. J Dairy Sci. 2008;91:4414–23.
Strandén I, Garrick DJ. Technical note: derivation of equivalent computing algorithms for genomic predictions and reliabilities of animal merit. J Dairy Sci. 2009;92:2971–5.
Pérez P, de Los Campos G: Genome-Wide Regression & Prediction with the BGLR Statistical Package. Genetics. 2014;198:483–95.
Wang H, Misztal I, Aguilar I, Legarra A, Fernando RL, Vitezica Z, et al. Genome-wide association mapping including phenotypes from relatives without genotypes in a single-step (ssGWAS) for 6-week body weight in broiler chickens. Front Genet 2014, 5:134.
Utsunomiya YT, Carmo AS, Neves HHR, Carvalheiro R, Matos MC, Zavarez LB, et al. Genome-wide mapping of loci explaining variance in scrotal circumference in nellore cattle. PLoS One. 2014;9.
The Bovine HapMap Consortium. Genome-wide survey of SNP variation uncovers the genetic structure of cattle breeds. Science (80-). 2009;324:528–32.
Decker JE, McKay SD, Rolf MM, Kim JW, Molina Alcalá A, Sonstegard TS, et al. Worldwide patterns of ancestry, divergence, and admixture in domesticated cattle. PLoS Genet. 2014;10.
Decker JE, Pires JC, Conant GC, McKay SD, Heaton MP, Chen K, et al. Resolving the evolution of extant and extinct ruminants with high-throughput phylogenomics. Proc Natl Acad Sci U S A. 2009;106:18644–9.
Nijman IJ, Otsen M, Verkaar ELC, de Ruijter C, Hanekamp E, Ochieng JW, et al. Hybridization of banteng (Bos javanicus) and zebu (Bos indicus) revealed by mitochondrial DNA, satellite DNA, AFLP and microsatellites. Heredity (Edinb). 2003;90:10–6.
Utsunomiya YT, Bomba L, Lucente G, Colli L, Negrini R, Lenstra JA, et al. Revisiting AFLP fingerprinting for an unbiased assessment of genetic structure and differentiation of taurine and zebu cattle. BMC Genet. 2014;15:47.
Ropiquet A, Gerbault-Seureau M, Deuve JL, Gilbert C, Pagacova E, Chai N, et al. Chromosome evolution in the subtribe Bovina (Mammalia, Bovidae): the karyotype of the Cambodian banteng (Bos javanicus birmanicus) suggests that Robertsonian translocations are related to interspecific hybridization. Chromosom Res. 2008;16:1107–18.
do Carmo AS, Utsunomiya YT, Carvalheiro R, Neves HHR, Matos MC, Zavarez LB, et al. Genome-wide association study for birth weight in Nellore cattle points to previously described orthologous genes affecting human and bovine height. BMC Genet. 2013;14:52.
Karim L, Takeda H, Lin L, Druet T, Arias JAC, Baurain D, et al. Variants modulating the expression of a chromosome domain encompassing PLAG1 influence bovine stature. Nat Genet. 2011;43:405–13.
Saatchi M, Schnabel RD, Taylor JF, Garrick DJ. Large-effect pleiotropic or closely linked QTL segregate within and across ten US cattle breeds. BMC Genomics. 2014;15:442.
Fortes MRS, Kemper K, Sasazaki S, Reverter A, Pryce JE, Barendse W, et al. Evidence for pleiotropism and recent selection in the PLAG1 region in Australian Beef cattle. Anim Genet. 2013;44:636–47.
Bolormaa S, Pryce JE, Reverter A, Zhang Y, Barendse W, Kemper K, et al. A Multi-Trait, Meta-analysis for Detecting Pleiotropic Polymorphisms for Stature, Fatness and Reproduction in Beef Cattle. PLoS Genet. 2014;10:e1004198.
Fortes MRS, Reverter A, Kelly M, Mcculloch R, Lehnert SA. Genome-wide association study for inhibin, luteinizing hormone, insulin-like growth factor 1, testicular size and semen traits in bovine species. Andrology. 2013;1:644–50.
Fortes MRS, Lehnert SA, Bolormaa S, Reich C, Fordyce G, Corbet NJ, et al. Finding genes for economically important traits: Brahman cattle puberty. Anim Prod Sci. 2012;52:143–50.
Voz ML, Agten NS, Van De Ven WJM, Kas K. PLAG1, the main translocation target in pleomorphic adenoma of the salivary glands, is a positive regulator of IGF-II. Cancer Res. 2000;60:106–13.
Höglund JK, Guldbrandtsen B, Lund MS, Sahana G: Analyzes of genome-wide association follow-up study for calving traits in dairy cattle. BMC Genetics 2012;13:71.
McClure MC, Morsci NS, Schnabel RD, Kim JW, Yao P, Rolf MM, et al. A genome scan for quantitative trait loci influencing carcass, post-natal growth and reproductive traits in commercial Angus cattle. Anim Genet. 2010;41:597–607.
Kim J, Farnir F, Savell J, Taylor JF. Detection of quantitative trait loci for growth and beef carcass fatness traits in a cross between Bos taurus (Angus) and Bos indicus (Brahman) cattle 1. J Anim Sci. 2003;81:1933–42.
Li K, Li Y, Shelton JM, Richardson JA, Spencer E, Chen ZJ, et al. Cytochrome c deficiency causes embryonic lethality and attenuates stress-induced apoptosis. Cell. 2000;101:389–99.
Bradley EW, Carpio LR, Newton AC, Westendorf JJ: Deletion of the PH-domain and leucine rich repeat protein phosphatase 1 (Phlpp1) increases fibroblast growth factor (Fgf) 18 expression and promotes chondrocyte proliferation. J Biol Chem 2015 [epub ahead of print].
Acknowledgments
The authors are grateful to the Indonesian Agency for Agricultural Research and Development, Ministry of Agriculture through KKP3N activity in 2013 and 2014 that has facilitated and funded the research activities. YTU is supported by the São Paulo Research Foundation (FAPESP) - process 2014/01095-8. Mention of trade name proprietary product in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the authors or their respective institutions.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JFG and M conceived and coordinated the study. H and M conducted the genotyping and collected phenotypes of Peranakan Ongole samples. H and J conducted the genotyping of Bali samples. TSS contributed to the study design and provided genotypes for the remaining breeds. H and YTU performed the data analysis and drafted the manuscript. All authors read and approved the final manuscript.
Hartati Hartati and Yuri Tani Utsunomiya contributed equally to this work.
Additional files
Additional file 1:
Heterozygosity and F ST analysis.
Additional file 2:
List of putative Bos javanicus haplotypes in Indonesian Peranakan Ongole.
Additional file 3:
Supporting data (temporary browsable access): https://drive.google.com/file/d/0B4MpcTN6ypdyOXhGaE9CXzFvTHc/view?usp=sharing .
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Hartati, H., Utsunomiya, Y.T., Sonstegard, T.S. et al. Evidence of Bos javanicus x Bos indicus hybridization and major QTLs for birth weight in Indonesian Peranakan Ongole cattle. BMC Genet 16, 75 (2015). https://doi.org/10.1186/s12863-015-0229-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-015-0229-5