
Abstract
Background
This study aimed at (1) assessing the genomic stratification of experimental lines of Nelore cattle that have experienced different selection regimes for growth traits, and (2) identifying genomic regions that have undergone recent selection. We used a sample of 763 animals genotyped with the Illumina BovineHD BeadChip, among which 674 animals originated from two lines that are maintained under directional selection for increased yearling body weight and 89 animals from a control line that is maintained under stabilizing selection.
Results
Multidimensional analysis of the genomic dissimilarity matrix and admixture analysis revealed a substantial level of population stratification between the directional selection lines and the stabilizing selection control line. Two of the three tests used to detect selection signatures (FST, XP-EHH and iHS) revealed six candidate regions with indications of selection, which strongly indicates truly positive signals. The set of identified candidate genes included several genes with roles that are functionally related to growth metabolism, such as COL14A1, CPT1C, CRH, TBC1D1, and XKR4.
Conclusions
The current study identified genetic stratification that resulted from almost four decades of divergent selection in an experimental Nelore population, and highlighted autosomal genomic regions that present patterns of recent selection. Our findings provide a basis for a better understanding of the metabolic mechanism that underlies the growth traits, which are modified by selection for yearling body weight.
Similar content being viewed by others


Background
Human/animal interactions in livestock production have led to major phenotypic changes in animal traits, which are mainly due to the effect of selection [1]. Anthropogenic selection, along with natural selection and adaptation, have increased the frequency of favorable alleles and resulted in changes in the frequencies of their surrounding loci [2], thereby establishing genomic signatures of positive selection. In turn, the methods used to identify this type of signature of selection represent powerful tools to detect evidence of selection during the process of animal domestication or breed formation, and ultimately they aid in elucidating the mechanisms that underlie the morphological, productive and functional traits of a species [3].
Cattle are domestic animals used worldwide, with two divergent groups: taurine (Bos primigenious taurus) and indicine (Bos primigenious indicus) cattle, each of which includes several breeds, which have been established for different purposes by humans. One common strategy for investigating past selection in cattle involves the systematic comparison of the genomes of breeds that have evolved under different selection regimes, e.g., dairy versus beef breeds [4, 5]. The motivation behind such comparisons is to identify strongly differentiated genomic regions between different groups of animals. However, analysis of within-breed stratification may allow the identification of signatures of selection within a single breed for which genetically distinct lines have been established. One example of such a study in cattle involved Holstein lines that were divergently selected for milk yield, in which the identified signatures of selection co-localized with known quantitative trait loci (QTL) that affect milk traits [6, 7].
Nelore cattle (originally named Ongole in India) are an indicine breed, which is predominantly raised in Brazil for meat production. Apart from the national Nelore breeding programs, there is an experimental program with three selection lines that has been running since 1978 at the APTA Beef Cattle Center—Institute of Animal Science (IZ), Sertãozinho—Brazil. This program was initiated to demonstrate to producers the benefits of selection for growth traits and its economic impact on the bovine industry [8]. Briefly, three selection lines were established in 1980 by dividing an experimental herd representative of the main Nelore lineages that currently exist in Brazil. Since then, a line referred to as Nelore Control (NeC) has been maintained under stabilizing selection, in which animals with a yearling body weight (YBW) close to the average of the contemporary group are annually selected for reproduction. The two other lines, Nelore Selection (NeS) and Nelore Traditional (NeT), have independently undergone selection for increased YBW. While the NeS and NeC lines are maintained as closed lines, the NeT line has received sires from the two other lines (7 from NeC and 47 from NeT) and commercial herds.
After almost 40 years of selection, distinct phenotypic differences are observed between the lines undergoing stabilizing and directional selection, including differences in average body weight at various ages, body measurements, scrotal circumference and carcass quality [8,9,10]. Inbreeding level in these lines is controlled by adopting breeding strategies such as exclusion of siblings as sires, short mating time for each selected sire (only two seasons), and avoidance of mating between closely-related animals. In 2003, the observed coefficients of inbreeding in the three lines ranged from 4 to 4.3% [11], while in animals born in 2012 they ranged from 4.2 to 6.4%.
The existence of these experimental bovine lines has instigated studies on candidate genes, with the goal of detecting polymorphisms that show extreme differences in allele frequencies between lines, which may be linked to phenotypic differences [12,13,14]. However, genome-wide screening of high-density single nucleotide polymorphism (SNP) panels to detect selection is probably more suitable for revealing genomic regions that have undergone selection in this population and should help to understand the genetic mechanisms that underlie response to selection. Thus, the aims of this study were (1) to assess the effect of selection within this experimental Nelore population and (2) to scan for signatures of selection left by recent directional selection.
Methods
Description of the experimental selection program
Since the experimental program was established in 1980, the NeC, NeS and NeT lines are composed of 60, 120 and 170 cows with four, six and up to eight bulls, respectively, with 50% of the bulls at 2 years of age (first mating) and 50% at 3 years (second and last mating). The females that were initially assigned to each line were randomly chosen from an experimental herd of 350 cows, and since then, the annual culling rate of cows is approximately 20%, with replacement females for each line being selected based on their YBW at 550 days of age and raised on pasture. The bulls that were initially assigned to each line were chosen based on their YBW at 378 days of age, after 168-day feedlot performance tests. The bulls that were assigned to the NeC line had a YBW close to the average value for bulls with the same birth year (1977 or 1978), while those assigned to the NeS and NeT lines had a higher YBW. This bull selection scheme was maintained to the present day, by replacing annually the 50% older bulls within each line. Average annual selection intensities of 0.05 ± 0.24, 0.42 ± 0.29 and 0.56 ± 0.28 were applied to NeC, NeS and NeT females, respectively, while the corresponding values for males are equal to − 0.03 ± 0.20, 1.46 ± 0.30 and 1.60 ± 0.54.
Figure 1 shows the phenotypic differentiation in YBW between the lines obtained via selection. The individuals’ generation coefficients (GC) were determined by the method described by Brinks et al. [15], where the GC of each calf was the average of its parents’ GC values plus 1. The founder animals used for establishing the experimental lines were assigned a GC of 0.

Direct responses to selection for yearling body weight across generations. a and b Average yearling body weights (YBW) in Nelore Control (NeC), Nelore Selection (NeS) and Nelore Traditional (NeT) lines, within generation coefficient (GC) classes with intervals of 0.25, c and d 4 year old sires from the NeS (left) and NeC (right) lines
Sampling of animals and quality control procedures
We used a subsample of 782 animals born between 2004 and 2012 (92 NeC, 192 NeS and 498 NeT) that were previously genotyped because of phenotypes related to feed efficiency [16, 17]. The average GC for this subsample was equal to 6.1 (ranging from 4.4 to 7.7) and generation 6 had the broadest representation of animals from each line [see Additional file 1: Table S1]. The average estimated inbreeding coefficients in the genotyped subsample were equal to 0.05 ± 0.02 for NeC (ranging from 0.02 to 0.18), 0.03 ± 0.01 for NeS (ranging from 0.01 to 0.06) and 0.03 ± 0.01 for NeT (ranging from 0.01 to 0.08). Table 1 presents the average additive relationships between the genotyped animals of the three lines, which were estimated based on the full pedigree (see Silva et al. [17] for a complete pedigree description).
Each animal was genotyped using the Illumina® BovineHD BeadChip (San Diego, CA). Quality control (QC) procedures were performed using Plink (version 1.07) [18]: samples with call rates lower than 0.90 were removed, and SNPs with call rates lower than 0.95, non-autosomal, unmapped, and duplicated SNPs or those that were monomorphic in all lines were also discarded. In addition, SNPs that might be misplaced were removed based on the decay of r2 as a function of physical distance. Following an algorithm adapted from Bohmanova et al. [19], this QC procedure consisted of: (1) recording the distance between each SNP and the top 10 syntenic SNPs with the highest r2 values; (2) flagging a particular SNP once for each distance greater than 10 Mb within the list of its top 10 r2 and compiling a list of candidate misplaced SNPs for each chromosome; (3) excluding from further analysis the SNPs with the highest flag counts on each chromosome; and (4) repeating steps (1), (2) and (3) with the list of candidate misplaced SNPs, until the list is empty (an example of the removal of a misplaced SNP is in Figure S1 [see Additional file 2: Figure S1]).
Finally, 529,172 SNPs remained for further analyses [see Additional file 1: Table S2]. Next, imputation of missing genotypes and haplotype phasing were performed by combining all the samples together, regardless of their respective line, using Beagle software (version 3.3.2) [20].
Population structure analyses
Two different methods were used to assess population stratification. First, a multidimensional scaling (MDS) analysis was applied using the genomic distance matrix and the “cmdscale” function from the “stats” package in R software [21]. The genomic distance between two individuals was estimated as 1 minus the proportion of identical by state (IBS) alleles that they share. Second, an admixture analysis was performed to measure the proportion of individual ancestry from different numbers of hypothetical ancestral populations, using the ADMIXTURE software (version 1.04) [22]. Thus, an unsupervised model-based approach that relies on maximum likelihood was applied to the genotype matrix, using hypothetical ancestral population (K) numbers of 2 and 3, which reflect the different selection regimes and the number of lines in the subsample used for this study, respectively.
Wright’s fixation index (F ST)
Wright’s fixation index (FST) was computed for each pairwise combination of lines based on the estimator corrected for unequal sample sizes [23] implemented in the “fsthet” package [24] in R software [21], using all SNPs that had passed the QC procedures. Then, FST and heterozygosity averages were estimated for genomic windows of 100 kb across the autosomes, with an overlap of 75 kb. Putative signals of divergence were detected with two empirical strategies that are typically used in studies involving FST tests, one that identifies the top-ranking windows [25, 26] and another method that identifies outliers from the background level of FST-heterozygosity [24, 27]. Thus, windows were considered to show putative signatures of selection if they contained more than five SNPs, presented an average FST within the top 1%, and outside the 95% confidence interval of their heterozygosity bin in both cross-line comparisons of NeC with one of the lines under directional selection (“NeC vs. NeS” and “NeC vs. NeT”) [see Additional file 2: Figure S2].
Haplotype-based methods for assigning signatures of selection
The extended haplotype homozygosity (EHH) metric is defined as the probability of randomly choosing two identical extended haplotypes that surround a specific locus [28], for which higher values are expected for selected alleles with a frequency that increased rapidly due to selection. In our study, we applied two somewhat complementary EHH-derived methods to detect signatures of selection, the cross population EHH (XP-EHH) [29] and the integrated haplotype homozygosity score (iHS) [30], both of which were estimated using the SELSCAN software (version 1.1.0) [31]. Whereas XP-EHH is a cross population method that is useful to detect signals of divergent selection between lines, iHS is a within-population method that allows the identification of signals of ongoing positive selection.
The XP-EHH test was applied to the comparisons between NeC and each of the lines under directional selection (“NeC vs. NeS” and “NeC vs. NeT”). XP-EHH values were averaged for 100-kb windows with an overlap of 75 kb, and a cut-off threshold of 2.58 (p < 0.005) was applied to determine candidate signatures of selection. Only windows for which the average XP-EHH exceeded the upper cut-off in both cross-line comparisons and contained more than five SNPs were considered to harbor a signature of selection [see Additional file 2: Figure S3], which was characterized by the presence of some high-frequency haplotypes in the NeS and NeT lines but not in the NeC line.
The iHS test was performed by considering the lines under directional selection (NeS and NeT) together after removing SNPs with a minor allele frequency (MAF) lower than 0.01, since iHS is a within-population methodology that is not appropriate for analyzing SNPs close to fixation. One allele of each SNP was arbitrarily assigned as the “reference” allele, and absolute values of iHS (|iHS|) were then used to identify signatures of selection, as in Cohen-Zinder et al. [32]. The “reference” allele was randomly determined based on the high similarity observed between iHS values obtained from a subset of SNPs, with or without ancestral allele information retrieved from two cattle reference allele lists [33, 34] [see Additional file 2: Figure S4]. The |iHS| values of 494,372 SNPs were divided into 20 bins based on the frequency of the assigned ancestral allele, and the values were then standardized within each bin. The |iHS| values were averaged across 100-kb windows, with an overlap of 75 kb. Windows with |iHS| scores higher than 2.58 (p < 0.005) were considered as putative signatures of selection.
QTL and gene annotation
Known QTL that overlapped with each detected signature of selection were retrieved from the CattleQTL database [35]. Genes were identified within each putative selected region using the Ensembl cow gene set 84 [36]. Enrichment of genes in specific biological processes and molecular pathways was then analyzed using the DAVID database (version 6.7) [37], however since no significant pathway was detected, the results are not shown.
Results and discussion
Polymorphisms and population structure
After QC, the genotyped population was composed of 89, 189 and 485 samples from NeC, NeS, and NeT lines, respectively [see Additional file 1: Table S1]. The NeC line presented the largest number of monomorphic SNPs and the lowest heterozygosity (Table 2), which may be due to its smaller effective population size (Ne) relative to the population sizes of the other two lines. Current Ne values of 51, 88 and 177 were estimated for the NeC, NeS and NeT lines, respectively, based on the increase in individual inbreeding since the establishment of these lines, according to Gutiérrez et al. [38]. However, directional selection in successive generations, as experienced by the NeS and NeT lines, can result in the loss of rare alleles and an increase in apparent inbreeding, which may explain why these three lines that differ significantly in effective population size, present similar proportions of SNPs with a low MAF [see Additional file 2: Figure S5].
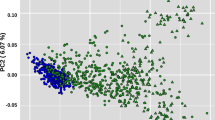
The first two principal coordinates obtained from multidimensional scaling (MDS) accounted for 7.88% of the total genomic variance in the subsample used in this study. PCO1 completely separates NeC from NeS and NeT, while PCO2 partially separates the NeS and NeT lines (Fig. 2a). The observed variation within each cluster was roughly proportional to its size, although the relatively higher diversity in NeT may be due to the gene flow that occurred in this line. The pattern of dispersion with the two selection lines being separated from NeC suggests that selection might be a significant force leading to genomic differentiation between these lines. However, since it is known that larger numbers of sires were transferred from NeS to NeT than from NeC to NeT, which results in NeS and NeT being more genetically connected, this might also have contributed to the observed pattern.

Population structure, as revealed by MDS and admixture analyses. a Depicts the distribution of samples based on the first two principal coordinates (PCo) provided by their genomic distance matrix. The variance explained by each PCo is indicated within brackets. b and c The results of admixture analyses with inferred numbers of clusters i.e. K = 2 (b) and K = 3 (c). White dashed lines separate the experimental lines. Each sample is represented by a vertical bar partitioned into stained segments according to its proportion of ancestry in each of the clusters
We performed an admixture analysis to estimate the proportions of ancestry in NeC, NeS and NeT with various numbers of clusters (K), which represent hypothetical ancestral populations. When K = 2, the clustering algorithm clearly separated NeC from NeS and NeT (Fig. 2b), and the gene flow into NeT became evident with K = 3 (Fig. 2c). Both MDS and admixture analyses identified NeT animals that showed a high degree of shared ancestry with the NeS cluster (Fig. 2a, c), which is likely due to the possible use of NeS sires for reproduction in the NeT line.
FST is a widely used metric for identifying population differentiation with values ranging from 0 to 1, where values close to 0 indicate more homogeneous populations and values close to 1 indicate more genetically diverged populations. The estimated average FST values in cross-line comparisons were equal to 0.05 ± 0.04 for “NeC vs. NeS”, 0.06 ± 0.04 for “NeC vs. NeT” and 0.02 ± 0.02 for “NeS vs. NeT”. These results reflect only subtle differentiation on a genome-wide scale, even after 40 years of intensive directional selection, which is consistent with the results of Flori et al. [4], who found minor genomic divergence between evolving lines of a single bovine breed. In spite of the low FST averages, their ranking was consistent with the population structure, with higher values being observed between NeC and one of the lines undergoing directional selection than between the two selection lines.
Signatures of selection
The FST test revealed windows with significant levels of differentiation on most of the bovine chromosomes, while XP-EHH and iHS only recognized signals on chromosomes 14 and 16 and on chromosomes 1, 3, 9, 14, 16 and 23, respectively (Fig. 3). Unusually negative XP-EHH scores, as observed on chromosome 12 (Fig. 3), may represent signatures of selection in the NeC line. However, we did not treat such scores as a signature of selection because stabilizing selection is expected to maintain haplotype diversity, rather than reduce it [39], thus producing subtle signatures of selection that cannot be detected by the XP-EHH test. Therefore, the low diversity in these regions is presumably due to genetic drift caused by the small Ne of the NeC line.

Genome-wide FST, XP-EHH and iHS scores averaged for 100-kb windows (75 kb overlapped). Blue lines indicate the significance threshold adopted for each test. The FST and XP-EHH values represent the means for the same windows in the “NeC vs. NeS” and “NeC vs. NeT” comparisons. Red dots represent windows that surpass the upper-cutoff windows in both comparisons, hence putative signatures of selection
After combining adjacent windows that displayed signatures of selection, 48, 7 and 17 genomic regions were identified by the FST, XP-EHH and iHS tests, respectively (see the full lists in Tables S3 and S4 [see Additional file 1: Tables S3 and S4]). In agreement with previous studies, most of the signals were detected by only one of the three approaches [40, 41]. Only six genomic regions were identified as containing signatures of selection based on two of the three metrics (Table 3).
For the signatures of selection that were identified based on two metrics, an identical extended haplotype had the highest frequency in the lines maintained under directional selection (Fig. 4), which suggests that selection for increased YBW improved the frequencies of those haplotypes in Nelore populations, thus shaping a special pattern in these regions. The signature of selection on BTA14:83,850,000–84,000,000, which was identified by both cross-line comparison metrics (FST and XP-EHH), overlaps with a QTL that affects efficiency of metabolizable energy utilization for maintenance and growth, as previously observed in Nelore cattle [49]. The COL14A1 (collagen type XIV alpha 1 chain) gene present in this region encodes a collagen isoform that plays a role in muscle development [50]. The signature of selection on BTA14:32,150,000–32,275,000, identified by both iHS and XP-EHH, overlaps with a QTL that affects several growth traits in beef breeds [47, 48, 51]. The most interesting candidate gene residing in this region, CRH (corticotropin releasing hormone), regulates appetite and controls the release of growth inhibitors in various organisms [51, 52]. Two additional candidate signatures of selection, detected via iHS and XP-EHH, co-localized on chromosome 16, and overlapped with a QTL reported to affect calving ease [42].

Haplotype frequencies for the four signatures of selection detected by two methods. Genomic regions were detected by both FST and iHS (a, b), XP-EHH and iHS (c, e, f) or both FST and XP-EHH (d). Different colors, except the grey color, refer to haplotypes with a frequency higher than 0.1 in at least one of the three lines
In addition to the two coincident signals detected by both iHS and FST and the signal identified by both iHS and XP-EHH, within the 21–32 Mb region on chromosome 14, several other narrow regions were identified exclusively based on one of the tests (Fig. 5). In beef breeds, several studies have reported signatures of selection within this genomic interval, which are mainly in close vicinity to the PLAG1 gene (BTA14:25,007,291–25,009,296) [5, 53, 54], which is one of the major genes that affect bovine stature and other growth traits [55]. This genomic segment also includes QTL that were previously detected in Nelore cattle and affect sexual precocity, meat quality, body and carcass weight [43, 56,57,58]. Fortes et al. [44] reported a significant association between several SNPs residing in this region and IGF1 serum levels in Brahman cattle. The segregation of this QTL in the Nelore breed could explain the signatures of selection that were detected in the lines undergoing directional selection for increased YBW, since IGF1 is a growth stimulator.

Graphical representation of signatures of selection detected via XP-EHH and iHS within a hotspot region on chromosome 14. Grey and blue represent XP-EHH and iHS scores, respectively, for 100-kb windows; light green, orange and yellow boxes indicate signals detected via iHS, FST and XP-EHH, respectively. The dark green boxes indicate overlap between two tests
Some interesting genomic regions were detected exclusively based on one of the three metrics. For instance, the signature of selection on BTA6:58,700,000–58,925,000 that was revealed by the FST test overlaps with a previously reported signature of selection in Limousin cattle [5]. The candidate gene located in this region, TBC1D1 (TBC1 domain family member 1), encodes a well-known body weight regulator in several species, including humans, mice, chickens and pigs [59,60,61,62,63], that is directly related to insulin-stimulated glucose uptake in muscle cells [64]. Another candidate signature of selection identified via FST, BTA6:118,650,000–118,800,000, is linked to a QTL for average daily gain that was previously detected in the same population as that used here [16]. This signature of selection could reflect the influence of the selection for YBW on the daily rate of weight gain. This region includes the PSAPL1 (prosaposin-like 1) gene and coincides with a copy number variant (CNV) region reported in Nelore cattle [65]. Previous studies have indicated a high level of coincidence between signatures of selection and CNV regions in cattle, which suggests that CNV may have a role in recent selection [66, 67].
Three of the signatures of selection detected via XP-EHH in our study (BTA14:77,975,000–78,875,000, BTA14:80,525,000–84,675,000 and BTA16:69,100,000–70,900,000) correspond to signals of recent positive selection that were previously reported in Nelore cattle [68]. In addition, the XKR4 (XK related 4) gene resides in the signature of selection on BTA14:24,150,000–24,300,000, which is a well-characterized gene associated with growth traits in cattle, including Nelore birth weight and meat tenderness [43, 57].
In addition to the signatures of selection that could be associated with directional selection for growth, the iHS test revealed signatures of selection that represent historical selection for attributes that have become fixed in the Nelore breed. For instance, the signature of selection on BTA23:25,450,000–25,625,000 overlaps with the Bola DR-alpha gene, which is involved in the bovine immune system and may be related to adaptation and high resistance to parasites. In addition, the signature of selection on BTA14:30,850,000–31,650,000 coincides with a QTL that was reported in the Nelore breed [69] and affects alignment and appropriate angles of feet and legs, which are phenotypes that have been strongly selected for in animals registered with the Brazilian breed association ever since the breed was first established in the country.
Conclusions
We describe a genomic scan in an indicine breed that has been selected for growth traits and is crucial for beef production in tropical environments. Our findings demonstrate that genetic stratification has occurred in this experimental Nelore population due to almost four decades of divergent selection. Three complementary approaches for the detection of signatures of selection were used to identify candidate regions on the autosomes, most of which overlapped with or were near known QTL and candidate genes that affect essential economic traits of beef cattle. The panel of identified loci includes well-known genes related to growth traits, such as TBC1D1, CRH, and XKR4, as well as novel candidates, such as COL14A1, PSAPL1 and CPT1C1. The candidate signatures of selection and the list of candidate genes presented here provide a basis to better understand the metabolic mechanisms that underlie the growth traits, which have been modified by selection for YBW. Moreover, these results provide candidate genes for further fine-mapping studies.
References
Lush JL. Animal breeding plans. 3rd ed. Ames: Iowa State College Press; 1945.
Smith JM, Haigh J. The hitch-hiking effect of a favourable gene. Genet Res. 1974;23:23–35.
Qanbari S, Simianer H. Mapping signatures of positive selection in the genome of livestock. Livest Sci. 2014;166:133–43.
Flori L, Gonzatti MI, Thevenon S, Chantal I, Pinto J, Berthier D, et al. A quasi-exclusive European ancestry in the Senepol tropical cattle breed highlights the importance of the slick locus in tropical adaptation. PLoS One. 2012;7:e36133.
Zhao F, McParland S, Kearney F, Du L, Berry DP. Detection of selection signatures in dairy and beef cattle using high-density genomic information. Genet Sel Evol. 2015;47:49.
Sonstegard TS, Ma L, Van Tassell CP, Kim ES, Cole JB, Wiggans GR, et al. Forty years of artificial selection in US Holstein cattle had genome-wide signatures. In Proceedings of the 9th World Congress on Genetics Applied to Livestock Production: 1–6 August 2010; Leipzig. 2010.
Kim ES, Cole JB, Huson H, Wiggans GR, Van Tassell CP, Crooker BA, et al. Effect of artificial selection on runs of homozygosity in US Holstein cattle. PLoS One. 2013;8:e80813.
Mercadante MEZ, Packer IU, Razook AG, Cyrillo JNSG, Figueiredo LA. Direct and correlated responses to selection for yearling weight on reproductive performance of Nelore cows. J Anim Sci. 2003;81:376–84.
Cyrillo JNSG, Razook AG, Figueiredo LA, Bonilha Neto LM, Ruggieri AC, Tonhati H. Efeitos da Seleção para Peso Pós-desmame sobre Medidas Corporais e Perímetro Escrotal de Machos Nelore de Sertãozinho (SP). Braz J Anim Sci. 2000;29:403–12.
Bonilha SFM, Tedeschi LO, Packer IU, Razook AG, Alleoni GF, Nardon RF, et al. Evaluation of carcass characteristics of Bos indicus and tropically adapted Bos taurus breeds selected for postweaning weight. J Anim Sci. 2008;86:1770–80.
Mercadante MEZ, Razook AG. Programa de melhoramento genético do Instituto de Zootecnia. In: Pires AV. Bovinocultura de Corte. 1st ed. Piracicaba: FEALQ. 2010. pp. 1508.
Ayres DR, Souza FRP, Mercadante MEZ, Fonseca LFS, Tonhati H, Cyrillo JNSG, et al. Evaluation of TFAM and FABP4 gene polymorphisms in three lines of Nellore cattle selected for growth. Genet Mol Res. 2010;9:2050–9.
Cardoso DF, de Souza FRP, de Camargo GMF, da Fonseca PD, Fonseca LFS, Braz CU, et al. Polymorphism analysis in genes of the somatotropic axis in Nellore cattle selected for growth. Gene. 2014;545:215–9.
de Souza FRP, Chiquitelli MG, da Fonseca LFS, Cardoso DF, da Silva Fonseca PD, de Camargo GMF, et al. Associations of FASN gene polymorphisms with economical traits in Nellore cattle (Bos primigenius indicus). Mol Biol Rep. 2012;39:10097–104.
Brinks JS, Rice FJ, Clark RT. Estimation of genetic trends in beef cattle. J Anim Sci. 1961;20:903.
Olivieri BF, Mercadante MEZ, Cyrillo JN, Branco RH, Bonilha SFM, de Albuquerque LG, et al. Genomic regions associated with feed efficiency indicator traits in an experimental Nellore cattle. PLoS One. 2016;11:e0164390.
Silva RMO, Fragomeni BO, Lourenco DAL, Magalhães AFB, Irano N, Carvalheiro R, et al. Accuracies of genomic prediction of feed efficiency traits using different prediction and validation methods in an experimental Nelore cattle population. J Anim Sci. 2016;94:3613–23.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Bohmanova J, Sargolzaei M, Schenkel FS. Characteristics of linkage disequilibrium in North American Holsteins. BMC Genomics. 2010;11:421.
Browning SR, Browning BL. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet. 2007;81:1084–97.
R Core Team R. A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing. http://www.r-project.org/ (2013). Accessed 1 Feb 2018.
Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–64.
Cockerham CC, Weir BS. Estimation of gene flow from F-statistics. Evolution. 1993;47:855–63.
Flanagan SP, Jones AG. Constraints on the FST–heterozygosity outlier approach. J Hered. 2017;108:561–73.
Gholami M, Erbe M, Gärke C, Preisinger R, Weigend A, Weigend S, et al. Population genomic analyses based on 1 million SNPs in commercial egg layers. PLoS One. 2014;9:e94509.
Yang B, Cui L, Perez-Enciso M, Traspov A, Crooijmans RPMA, Zinovieva N, et al. Genome-wide SNP data unveils the globalization of domesticated pigs. Genet Sel Evol. 2017;49:71.
Beaumont MA, Nichols RA. Evaluating loci for use in the genetic analysis of population structure. Proc R Soc Lond B Biol Sci. 1996;263:1619–26.
Sabeti PC, Reich DE, Higgins JM, Levine HZP, Richter DJ, Schaffner SF, et al. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419:832–7.
Sabeti PC, Varilly P, Fry B, Lohmueller J, Hostetter E, Cotsapas C, et al. Genome-wide detection and characterization of positive selection in human populations. Nature. 2007;449:913–8.
Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72.
Szpiech ZA, Hernandez RD. selscan: an efficient multithreaded program to perform EHH-based scans for positive selection. Mol Biol Evol. 2014;31:2824–7.
Cohen-Zinder M, Donthu R, Larkin DM, Kumar CG, Rodriguez-Zas SL, Andropolis KE, et al. Multisite haplotype on cattle chromosome 3 is associated with quantitative trait locus effects on lactation traits. Physiol Genomics. 2011;43:1185–97.
Rocha D, Billerey C, Samson F, Boichard D, Boussaha M. Identification of the putative ancestral allele of bovine single-nucleotide polymorphisms. J Anim Breed Genet. 2014;131:483–6.
Utsunomiya YT, Pérez O’Brien AM, Sonstegard TS, Van Tassell CP, do Carmo AS, Mészáros G, et al. Detecting loci under recent positive selection in dairy and beef cattle by combining different genome-wide scan methods. PLoS One. 2013;8:e0064280.
Hu ZL, Park CA, Reecy JM. Developmental progress and current status of the Animal QTLdb. Nucleic Acids Res. 2015;44:D827–33.
Yates A, Akanni W, Amode MR, Barrell D, Billis K, Carvalho-Silva D, et al. Ensembl 2016. Nucleic Acids Res. 2016;44:D710–6.
Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57.
Gutiérrez JP, Cervantes I, Goyache F. Improving the estimation of realized effective population sizes in farm animals. J Anim Breed Genet. 2009;126:327–32.
Vitti JJ, Grossman SR, Sabeti PC. Detecting natural selection in genomic data. Annu Rev Genet. 2013;47:97–120.
Evans LM, Slavov GT, Rodgers-Melnick E, Martin J, Ranjan P, Muchero W, et al. Population genomics of Populus trichocarpa identifies signatures of selection and adaptive trait associations. Nat Genet. 2014;46:1089–96.
He Q, Kim KW, Park YJ. Population genomics identifies the origin and signatures of selection of Korean weedy rice. Plant Biotechnol J. 2017;15:357–66.
Saatchi M, Schnabel RD, Taylor JF, Garrick DJ. Large-effect pleiotropic or closely linked QTL segregate within and across ten US cattle breeds. BMC Genomics. 2014;15:442.
Magalhães AFB, de Camargo GMF, Fernandes GA, Gordo DGM, Tonussi RL, Costa RB, et al. Genome-wide association study of meat quality traits in Nellore cattle. PLoS One. 2016;11:e0157845.
Fortes MRS, Reverter A, Hawken RJ, Bolormaa S, Lehnert SA. Candidate genes associated with testicular development, sperm quality, and hormone levels of inhibin, luteinizing hormone, and insulin-like growth factor 1 in Brahman bulls. Biol Reprod. 2012;87:58.
Ramayo-Caldas Y, Fortes MRS, Hudson NJ, Porto-Neto LR, Bolormaa S, Barendse W, et al. A marker-derived gene network reveals the regulatory role of PPARGC1A, HNF4G, and FOXP3 in intramuscular fat deposition of beef cattle. J Anim Sci. 2014;92:2832–45.
Veneroni-Gouveia G, Meirelles SL, Grossi DA, Santiago AC, Sonstegard TS, Yamagishi MEB, et al. Whole-genome analysis for backfat thickness in a tropically adapted, composite cattle breed from Brazil. Anim Genet. 2012;43:518–24.
Lee SH, Choi BH, Lim D, Gondro C, Cho YM, Dang CG, et al. Genome-wide association study identifies major loci for carcass weight on BTA14 in Hanwoo (Korean cattle). PLoS One. 2013;8:e74677.
McClure MC, Ramey HR, Rolf MM, McKay SD, Decker JE, Chapple RH, et al. Genome-wide association analysis for quantitative trait loci influencing Warner-Bratzler shear force in five taurine cattle breeds. Anim Genet. 2012;43:662–73.
de Oliveira PSN, Cesar ASM, do Nascimento ML, Chaves AS, Tizioto PC, Tullio RR, et al. Identification of genomic regions associated with feed efficiency in Nelore cattle. BMC Genet. 2014;15:100.
Tono-Oka S, Tanase S, Miike T, Tanaka H. Transient expression of collagen type XIV during muscle development and its reappearance after denervation and degeneration. J Histochem Cytochem. 1996;44:907–18.
Buchanan FC, Thue TD, Yu P, Winkelman-Sim DC. Single nucleotide polymorphisms in the corticotrophin-releasing hormone and pro-opiomelancortin genes are associated with growth and carcass yield in beef cattle. Anim Genet. 2005;36:127–31.
Sevane N, Armstrong E, Wiener P, Pong-Wong R, Dunner S. GemQual Consortium. Polymorphisms in twelve candidate genes are associated with growth, muscle lipid profile and meat quality traits in eleven European cattle breeds. Mol Biol Rep. 2014;41:4721–31.
Kemper KE, Saxton SJ, Bolormaa S, Hayes BJ, Goddard ME. Selection for complex traits leaves little or no classic signatures of selection. BMC Genomics. 2014;15:246.
Urbinati I, Stafuzza NB, Oliveira MT, Chud TCS, Higa RH, Regitano LC, et al. Selection signatures in Canchim beef cattle. J Anim Sci Biotechnol. 2016;7:29.
Karim L, Takeda H, Lin L, Druet T, Arias JAC, Baurain D, et al. Variants modulating the expression of a chromosome domain encompassing PLAG1 influence bovine stature. Nat Genet. 2011;43:405–13.
Fernandes Júnior GA, Costa RB, de Camargo GMF, Carvalheiro R, Rosa GJM, Baldi F, et al. Genome scan for postmortem carcass traits in Nellore cattle. J Anim Sci. 2016;94:4087–95.
Utsunomiya YT, do Carmo AS, Carvalheiro R, Neves HH, Matos MC, Zavarez LB, et al. Genome-wide association study for birth weight in Nellore cattle points to previously described orthologous genes affecting human and bovine height. BMC Genet. 2013;14:52.
Costa RB, Camargo GMF, Diaz IDPS, Irano N, Dias MM, Carvalheiro R, et al. Genome-wide association study of reproductive traits in Nellore heifers using Bayesian inference. Genet Sel Evol. 2015;47:67.
Stone S, Abkevich V, Russell DL, Riley R, Timms K, Tran T, et al. TBC1D1 is a candidate for a severe obesity gene and evidence for a gene/gene interaction in obesity predisposition. Hum Mol Genet. 2006;15:2709–20.
Meyre D, Farge M, Lecoeur C, Proenca C, Durand E, Allegaert F, et al. R125 W coding variant in TBC1D1 confers risk for familial obesity and contributes to linkage on chromosome 4p14 in the French population. Hum Mol Genet. 2008;17:1798–802.
Chadt A, Leicht K, Deshmukh A, Jiang LQ, Scherneck S, Bernhardt U, et al. Tbc1d1 mutation in lean mouse strain confers leanness and protects from diet-induced obesity. Nat Genet. 2008;40:1354–9.
Wang Y, Xu HY, Gilbert ER, Peng X, Zhao XL, Liu YP, et al. Detection of SNPs in the TBC1D1 gene and their association with carcass traits in chicken. Gene. 2014;547:288–94.
Fontanesi L, Galimberti G, Calò DG, Fronza R, Martelli PL, Scotti E, et al. Identification and association analysis of several hundred single nucleotide polymorphisms within candidate genes for back fat thickness in Italian Large White pigs using a selective genotyping approach. J Anim Sci. 2012;90:2450–64.
Dokas J, Chadt A, Nolden T, Himmelbauer H, Zierath JR, Joost HG, et al. Conventional knockout of Tbc1d1 in mice impairs insulin- and AICAR-stimulated glucose uptake in skeletal muscle. Endocrinology. 2013;154:3502–14.
da Silva JM, Giachetto PF, da Silva LO, Cintra LC, Paiva SR, Yamagishi MEB, et al. Genome-wide copy number variation (CNV) detection in Nelore cattle reveals highly frequent variants in genome regions harboring QTLs affecting production traits. BMC Genomics. 2016;17:454.
Pérez O’Brien AM, Utsunomiya YT, Mészáros G, Bickhart DM, Liu GE, Van Tassell CP, et al. Assessing signatures of selection through variation in linkage disequilibrium between taurine and indicine cattle. Genet Sel Evol. 2014;46:19.
Teo YY, Fry AE, Bhattacharya K, Small KS, Kwiatkowski DP, Clark TG. Genome-wide comparisons of variation in linkage disequilibrium. Genome Res. 2009;19:1849–60.
Somavilla AL, Sonstegard TS, Higa RH, Rosa AN, Siqueira F, Silva LOC, et al. A genome-wide scan for selection signatures in Nellore cattle. Anim Genet. 2014;45:771–81.
Vargas G, Neves HHR, Camargo GMF, Cardoso V, Munari DP, Carvalheiro R. Genome-wide association study and functional analysis of feet and leg conformation traits in Nellore cattle. J Anim Sci. 2018 (accept for publication).
Authors’ contributions
Conceived and designed the study: HT, FB, LGA, JNSGC and MEZM. Funding acquisition: LGA. Formal analysis: DFC. Support for data analysis: GCV, CR, DCBS, SQ, ME, GMFC, AVN. Guided the research: HT and HS. Original draft: DFC. All authors contributed to the interpretation of the results. All authors read and approved the final manuscript.
Acknowledgements
The authors are grateful to the APTA Beef Cattle Center - Institute of Animal Science (IZ) for providing the data and biological material used in this study.
Competing interests
The authors declare that they have no competing interests.
Ethics approval
This study was approved by the Ethics Committee on Animal Use (CEUA) of the Faculty of Agrarian and Veterinary Sciences, Sao Paulo State University (UNESP), under Protocol Number 18340/16.
Funding
This work was supported by the Sao Paulo Research Foundation (FAPESP), with Grant 2009/16118-5 for financial support; and Grants 2012/24600-4 and 2014/13445-3 for the first author.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Table S1.
Distribution of genotyped animals by line and generation. Table S2. Distribution of SNPs after quality control. Table S3. Genomic regions presenting extreme FST values with candidate genes. Table S4. Genomic regions presenting extreme XP-EHH and |iHS| values with candidate genes.
Additional file 2: Figure S1.
Decay of r2 as a function of physical distance on chromosomes 11 and 12. Figure S2. Genome-wide FST scores in overlapping windows for the “NeC vs. NeS” (upper) and “NeC vs. NeT” (bottom) comparisons. Figure S3. Genome-wide XP-EHH scores in overlapping windows for the “NeC vs. NeS” (upper) and “NeC vs. NeT” (bottom) comparisons. Figure S4. Correlation between SNP iHS values when considering ancestral allele definitions from available lists (|iHS|_1) and randomly defining one allele as ancestral (|iHS|_2). Figure S5. Minor allele frequency distributions in three selection lines.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Cardoso, D.F., de Albuquerque, L.G., Reimer, C. et al. Genome-wide scan reveals population stratification and footprints of recent selection in Nelore cattle. Genet Sel Evol 50, 22 (2018). https://doi.org/10.1186/s12711-018-0381-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12711-018-0381-2