
Abstract
Background
Recent technological advances in genomics have allowed the genotyping of cattle through single nucleotide polymorphism (SNP) panels. High-density SNP panels possess greater genome coverage and are useful for the identification of conserved regions of the genome due to selection, known as selection signatures (SS). The SS are detectable by different methods, such as the extended haplotype homozygosity (EHH); and the integrated haplotype score (iHS), which is derived from the EHH. The aim of this study was to identify SS regions in Canchim cattle (composite breed), genotyped with high-density SNP panel.
Results
A total of 687,655 SNP markers and 396 samples remained for SS analysis after the genotype quality control. The iHS statistic for each marker was transformed into piHS for better interpretation of the results. Chromosomes BTA5 and BTA14 showed piHS > 5, with 39 and nine statistically significant SNPs (P < 0.00001), respectively. For the candidate selection regions, iHS values were computed across the genome and averaged within non-overlapping windows of 500 Kb. We have identified genes that play an important role in metabolism, melanin biosynthesis (pigmentation), and embryonic and bone development.
Conclusions
The observation of SS indicates that the selection processes performed in Canchim, as well as in the founder breeds (i.e. Charolais), are maintaining specific genomic regions, particularly on BTA5 and BTA14. These selection signatures regions could be associated with Canchim characterization.
Similar content being viewed by others

Background
Canchim cattle has excellent meat yield and quality and has perfomed well when raised on natural pastures of Brazil, but it still has a small inventory with 28–30 thousand registered animals and represents about 3 % of the crossbred animals reared in the country [1]. The Canchim breed was developed in the early 1960’s aiming to combine fitness traits from zebu to the higher reproduction efficiency and meat quality from the Charolais breed [2–4]. The genetic makeup of this composite breed was a 62.5 % Charolais and 37.5 % zebu proportion. The genetic group called MA (resulting of mating between Charolais bulls and ½ Canchim + ½ zebu dams) is widely used by Canchim breeders, mainly to expand the genetic basis of the breed and present an expected proportion of 65.6 % Charolais and 34.4 % zebu [5]. Many studies were carried out in this breed, mostly on quantitative genetics [6–10].
Single nucleotide polymorphisms (SNPs) panels are applied in several studies, including selection signatures, in which regions of the genome maintained from generation to generation due to selection are identified. When positive selection occurs, it is expected that some loci adjacent to favorable mutations on a chromosome region segregates in a dependent manner, resulting in high and non-random allelic frequencies in specific regions [11]. The recent positive selection is characterized by increased linkage disequilibrium and decreased genetic variability in the population [12], which is caused by the rapid fixation of mutations favorable to the selected traits [13]. When positively selected alleles achieve high frequency, other alleles in linkage disequilibrium also increase their frequencies (hitchhike), retaining not only the favorable allele, but a region of the genome next to it (selective sweep effect) [14].
The identification of selection signatures could be used to assess the genome regions that control the quantitative traits under selection, and genes associated with phenotypic variations of the traits of interest, allowing to understand the biological mechanisms involved in the phenotypic manifestation. Therefore, the aim of our study was to identify and characterize selection signatures in Canchim using high density SNP data by means of the integrated haplotype score method.
Methods
Ethics statement
This study was approved by the Embrapa Southeast Livestock Ethical Committee for Animal Use (CEUA-CPPSE) under protocol 02/2009.
Animals and genotyping data
Genotypic data on 285 Canchim animals and 114 MA animals were used in this study. The animals were born between 1999 and 2005, being 205 females and 194 males. All animals were genotyped with the Illumina BeadChip BovineHD panel, consisting of 786,799 SNP markers.
According to Mokry et al. [15], the persistence of linkage disequilibrium phase between Canchim and MA genetic group was consistent and high, suggesting that both groups could be considered together in genetic evaluation programs. Thus, we have decided to treat both Canchim and MA as one dataset for further analyses and refer to all of these animals as Canchim.
Quality control of genotypes and imputation
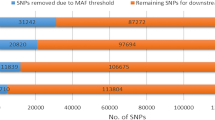
The genotype quality control (QC) was performed using the snpStats package [16] in R statistical software [17]. Only SNPs on autosomal chromosomes with defined position according to the UMD_3.1 bovine genome assembly [18], were used in the analyses. SNPs with genotype calling score below 0.20 were unreliable and coded as missing genotypes. Minor allele frequencies (MAF) below 1 %, call rate for SNP below 95 %, and call rate for samples below 90 % were excluded.
The software BEAGLE v.3.3.2 [19] was used for inference of linkage phase, haplotype construction, and imputation of missing genotypes. These steps are required for the study of selection signatures.
Identification of selection signatures
The integrated haplotype score (iHS), a statistical methodology proposed by Voight et al. [20] and derived from the extended haplotype homozygosity (EHH) methodology [21], was used to identify the selection signatures. The EHH methodology, also known as long-range haplotype, detects recent positive selection by analyzing the haplotypes structure of individuals in a population. The method accounts for the allelic frequencies and the extent of linkage disequilibrium. Linkage disequilibrium refers to the non-random association between alleles at two loci [22], whereas the EHH methodology measures the association between a single core allele at one locus with multiple loci at different distances.
The integrated haplotype score (iHS) was calculated as:
in which iHH A and iHH D represent the integrated EHH score for ancestral (A) and derived (D) core alleles; and E and SD are, respectively, expectation and standard deviation of \( \left(\frac{iH{H}_A}{iH{H}_D}\right) \).
Values of iHS were standardized so that they followed a standard normal distribution (iHS ~ N (0,1)). Thus, the iHS values of different SNPs are directly comparable, regardless of allelic frequencies of these markers. The rehh package [23] was used to calculate the iHS for each allele (ancestral and derivative). The online database available in the supplementary material of Utsunomiya et al. [24] was used as source to determine the ancestor alleles. These authors defined the ancestral alleles using genotyped data of species considered common founders of the Bovinae subfamily (Bos gaurus, Bubalus bubalis and Bos grunniens), in which the alleles were fixed at MAF equal to zero. Furthermore, SNPs with lower MAF represents alleles near fixation (i.e. background ancestry) [24]. Therefore, these are not useful for the iHS method, as this enables identifying only recent selection. Thus, SNPs with MAF greater than or equal to 0.05 were used to represent the selection signature regions in Canchim cattle (Fig. 1).

Distribution of integrated haplotype score (iHS) and piHS of each SNP per chromosome. a integrated haplotype score (iHS); b transformed iHS scores in piHS = − log10[1 − 2|Φ iHS − 0.5|] wherein Φ iHS is the Gaussian cumulative distribution function of iHS. Markers with statistically significant piHS values (P < 0.00001) are above the dashed line. Only SNPs with MAF ≥ 0.05 were considered
The iHS statistical values were transformed into piHS = − log10[1 − 2|Φ iHS − 0.5|] in which Φ iHS is the cumulative distribution Gaussian function of iHS. This transformation allows better visualization and comparison of regions with selection signatures [25]. Assuming that iHS statistics are normally distributed, piHS can be interpreted as \( { \log}_{10}\frac{1}{P} \), in which P is the P-value associated with the null hypothesis (no selection). Values of piHS greater than or equal to five were considered statistically significant (P <0.00001), thus rejecting the null hypothesis.
The linkage disequilibrium between genetic markers was obtained for the SNPs within selection signature regions by means of the r2 measure proposed by Hill and Robertson [22]. The software plink [26] was used to conduct the linkage disequilibrium analyses.
Survey for associated genes
The SNPs with statistically significant iHS (P < 0.00001) were inspected using the National Center for Biotechnology Information [27] and Ensembl Genome Browser [28] databases to identify genes or surrounding genes. Panther [29] and DAVID 6.7 [30, 31] databases were used to survey biological processes and metabolic pathways in which the identified genes are involved.
For the candidate selection regions, statistically significant (P < 0.00001) iHS values were computed across the genome and averaged within non-overlapping windows of 500 kb. The QTL Animal database [32] was used to identify quantitative trait loci (QTL) described in the literature for each window.
Results
After the QC, a total of 396 animals and 687,655 SNPs remained for the selection signature study. Fixed alleles in the population (3,270 SNPs) were excluded from the analysis. The iHS distribution was approximately normal (iHS ~ N (0,1)), thus the markers and the chromosomes can be compared. In Fig. 1a are presented the dispersion of the iHS by chromosome, while in Fig. 1b the same markers are presented transformed into piHS.
The statistically significant regions (P < 0.00001) were identified on BTA5 and BTA14 (Fig. 1b). In Fig. 2 are presented the distributions of piHS for SNPs (Fig. 2a, b) and windows (Fig. 2c, d) on BTA5 and BTA14. In Table 1 are presented the descriptive statistics for BTA5 and BTA14 according to SNPs and windows. Despite being distributed in nine windows, the SNPs on BTA5 are concentrated in a region of about 18 Mb (between the 54,155,228 bp and 72,410,862 bp positions), which represents 15 % of the chromosome total size. On BTA14, all significant SNPs (P < 0.00001) are concentrated in an interval of only 1 Mb (window 25).

Values of piHS for SNPs (a and b) and windows (c and d) on BTA5 and BTA14. Statistically significant regions (P < 0.00001), respective positions in base pairs (bp), with statistically significant SNPs and windows (P < 0.00001) highlighted in black
There were 39 and nine significant SNPs found on BTA5 and BTA14, respectively (Table 2). In order to identify selection signature regions which could be associated to any of the phenotypic traits used in the traditional selection, we considered the significance level of P < 0.00001 for piHS. None of the identified SNPs were located in protein coding regions, 17 SNPs were observed in intronic regions, one in 3′ untranslated region (UTR), one in 5′ UTR, and 20 in intergenic regions of the BTA5. All SNPs of the BTA14 were mapped into intergenic regions.
The analysis using Panther database identified biological processes related to apoptotic process (GO:0006915), biological adhesion (GO:0022610), biological regulation (GO:0065007), cellular component organization or biogenesis (GO:0071840), cellular process (GO:0009987), developmental process (GO:0032502), immune system process (GO:0002376), localization (GO:0051179), metabolic process (GO:0008152), multicellular organismal process (GO:0032501), and response to stimulus (GO:0050896).
Candidate regions located in windows presented 14 genes and one pseudogene (Table 3). The largest candidate region was observed in window 55 of BTA5, with 11 significant SNPs, two genes (LRIG3 and LOC101907520), and one pseudogene (LOC785078) were identified. The smallest candidate region, also on BTA5, totaled 7,180 bp and had only one annotated gene (TESPA1). The region with the most of the significant SNPs (13 markers) was observed in window 58 of BTA5 and presented eight genes (IKZF4, DGKA, RAB5B, PYM1, SUOX, PMEL, CDK2, and RPS26).
The windows 55 (11 SNPs) and 58 (13 SNPs) identified on BTA5, were located in QTL (quantitative trait loci) regions reported for resistance to ticks, fat thickness, marbling, body weight at birth and yearling, preweaning and average daily gain, and calving ease [33–40]. In the candidate region on BTA14 (window 25), between 25,505,663 and 25,877,586 bp, QTL were reported as associated with resistance to ticks, carcass weight, fat thickness and ribeye area, birth weight and average body weight, average pre-weaning daily gain, calving ease, and gestational age [39–44].
The SNPs with piHS with significance of P < 0.0001 are shown in Additional file 1. In this significance level, 296 statistically significant SNPs (P < 0. 0001) were observed covering 24, three, two, five and one windows on BTA5, BTA6, BTA8, BTA14, and BTA16, respectively. In the candidate regions, between the first and last significant SNP of these 35 windows, 167 genes or pseudogenes were identified. There are 157 genes or pseudogenes in the BTA5 candidate regions, five in BTA8 regions, four in BTA14 regions and one in the BTA16 region.
Pathway analyses for the genes presented in the Additional file 1 revealed androgen/estrogene/progesterone biosynthesis, gonadotropin releasing hormone receptor pathway, fibroblast growth factor (FGF) signaling pathway, DNA replication, integrin signaling pathway, B cell activation, T cell activation, interleukin signaling pathway, inflammation mediated by chemokine and cytokine signaling pathway, endocytosis, cell cycle, oocyte meiosis, progesterone-mediated oocyte maturation, among others.
The average linkage disequilibrium on BTA5 and BTA14 were equal to 0.65 and 0.52, respectively. The extent of the linkage disequilibrium in each of the candidate selection regions are presented in the Additional file 2. According to Pérez O’Brien et al. [45] high linkage disequilibrium over an extended region is likely the result of recent selection.
Discussion
The selection signatures herein identified are located in important QTL regions of beef cattle, which includes productive (birth, weaning and yearling weights), reproductive, and conformation traits. These traits are also included in the Canchim selection index. Machado et al. [3] found in Canchim cattle, QTL associated with birth weight and body weight at 365 days old. Genome-wide association for scrotal circumference measured at long yearling age was identified on BTA5, between 45.24 and 45.35 Mb, in Canchim cattle and between 41.98 and 78.59 Mb on BTA14 [46].
In a study by Bolormaa et al. [47], genome-wide association on BTA5 and BTA14 were observed for stature, fatness, and reproduction in beef cattle using multi-trait model. These authors used taurine and zebu breeds and composite animals in the study. Pérez O’Brien et al. [45] used methodology based on regions with lower genetic variability and specific regional linkage disequilibrium patterns to the study selection signatures in Nelore, Gyr, Angus and Brown Swiss breeds. These authors observed genes with potential adaptive and productive importance on BTA5 and BTA14. The BTA14 has been widely explored for genes related to important economic traits in beef and dairy cattle [48].
The iHS statistic gives an idea of how unusual haplotypes around a given SNP are, relative to the genome as a whole, or how different the selected region is in relation to a region subject to the consequences of random mating in the population. Thus, iHS values equal or near zero indicate EHH decay rates similar to ancestral and derivatives core alleles, negative values indicate long haplotypes around a derivative core allele, and positive values indicate long haplotypes around an ancestral core allele, as seen in Fig. 1a. Both iHS extremes are indicative of conserved regions, due to direct selection on derivative alleles, and hitchhike effect, caused by the linkage disequilibrium between ancestral alleles with selected regions [20]. Highly conserved regions over the recent generations in this population are concentrated on the observed peaks (signals) (Fig. 1b). Thus, it is presumed that highly conserved regions are maintained through selection favoring this population.
Kemper et al. [49] used methodologies based on the haplotype homozygosity and found evidence of selection in regions on BTA5, BTA14 and BTA16 in Charolais animals, which corroborate with the results of our study. On BTA5 and BTA14, these authors found selection signatures between 52.8 and 64.75 Mb and 19.75 and 29.55 Mb, respectively. Somavilla et al. [50] investigated selection signatures in Nelore animals using the EHH methodology and verified significant regions in the FABP4 gene (fatty acid binding protein 4, adipocyte) located on BTA14, this gene plays important role in lipid metabolism and adipocytes homeostasis [51], while the SNPs in this gene have been described as associated with marbling and subcutaneous fat deposit in Wagyu × Limousin F2 crosses [52] and intramuscular fat levels in crossbred Bos taurus cattle [53]. In our study, the FABP4 gene was not found in the candidate regions, perhaps because of the significance level (P < 0.00001) considered and sliding windows approach.
The selection conducted in Canchim has transmitted and preserved some regions of the founder breeds across generations. Utsunomiya et al. [24] found regions of recent selection signatures on BTA1, BTA2, and BTA18. We can assume that, as the genetic proportion of zebu is low in the formation of Canchim cattle, greater resemblance of Canchim with Charolais are expected. According to Porto-Neto et al. [54], the linkage disequilibrium is higher in taurine than in zebu cattle. This fact corroborates the results observed in our study, because adjacent loci in greater linkage disequilibrium (probably from the Charolais breed contribution) provide more extended and, apparently, more conserved haplotypes among Canchim and Charolais animals. The majority of detected signals may have arisen during breed formation [24] and large conserved regions could also refer to the selection that occurred in the founder breeds of Canchim.
The region on BTA5 harbored many genes including PMEL, SUOX, SLC16A7, TESPA1, SPIC, RAB5B, DGKA, PYM1, TXNRD1, and LARGE. These genes are involved in biological functions, such as coat color, amino acids metabolism, transport of glucose, development of immune cells, glicerolipids and phospholipids metabolism, fatty acids metabolism, and biosynthesis of glycoproteins.
The PMEL (premelanosome protein) gene, identified in the candidate region of window 58 on BTA5, is involved in the melanin biosynthesis process, in which the primary function is the pigmentation of hair, skin, eyes, and mucous membranes. According to Gutiérrez-Gil et al. [55], mutations described in this gene are partly responsible for the dilution of pigmentation in Charolais cattle, resulting in the white colored coat of the breed. This white coat is an important trait of Charolais cattle and this breed standard is retained in Canchim, through the selection of animals with light colored coat. The light colored coat used to characterize the Canchim, may have conserved the mutation for pigment dilution, resulting in selection of the alleles on PMEL gene and extended conservation to gene neighboring regions, due to linkage disequilibrium. This fact can be seen in this study, in which the most significant SNPs in window 58 on BTA5 had negative iHS values (10 out of 13 SNPs). Thus, core alleles are derivative alleles, i.e., originating from migration or gene mutation.
The TESPA1 (thymocyte expressed, positive selection associated 1) gene in the window 60 and SPIC (Spi-C transcription factor) gene in the window 66, are involved in the development of immune cells, related to innate immunity (red pulp macrophages development - [56] and acquired immunity (T-cell development - [57]; and B cells differentiation - [58]). Genes of the immune system are often related to positive selection due to the continued resistance to pathogens [59]. The zebu breeds, crossbreds animals, and composite breeds are characterized by resistance to endo- and ectoparasites [39] and, therefore, it is speculated that the identified selection signatures associated with the immune system genes reflects these selected traits.
On BTA14, we identified the IMPAD1 (inositol monophosphatase domain containing 1) gene that plays a role in the bone-cartilage system. This gene participates in sulfur and inositol phosphate metabolism, and mutations identified in this gene are associated with chondrodysplasia and abnormal joint development in humans [60]. In a genome-wide association study, Fortes et al. [61] observed association of SNPs in this gene with age at detection of the first corpus luteum and scrotal circumference in Brahman cattle. In Canchim cattle, selection has been practiced for scrotal circumference due to its association with reproductive and productive performance and because of its pleiotropic effect with female reproductive traits [7].
Recent studies have identified selection signature in a candidate region on BTA14, which comprises the PLAG1 gene [62, 63]. This gene has association with stature and body weight [64], reproductive [65], and carcass traits [66] in cattle. In our study, we identified two regions (24,425,758–24,473,841 bp and 25,505,663–25,987,996 bp) on BTA14 surrounding the PLAG1 gene region (25,555,459–25,052,403 bp). We believe that PLAG1 was not observed in the windows herein determined due to methodological differences and different sliding windows approach in the consulted literature. Considering the associations of PLAG1 gene described in the literature and the proximity to the candidate regions observed in our study, this gene could be an interesting target of selection in Canchim beef cattle.
Conclusion
The selection signature regions were detected in Canchim cattle, suggesting that the selection processes conducted in this breed (human driven selection during breed formation) and its founders led to the genomic conservation in some regions on BTA5 and BTA14. These selection signatures regions could be associated with Canchim characterization, which sought to unite desirable characteristics of zebu and taurine breeds.
Abbreviations
- Bp:
-
base pairs
- BTA:
-
Bos taurus chromosome
- EHH:
-
extended haplotype homozygosity
- iHS:
-
integrated haplotype score
- Kb:
-
kilobase
- Mb:
-
megabase pairs
- QC:
-
quality control
- QTL:
-
quantitative trait loci
- SNP:
-
single nucleotide polymorphism
- SS:
-
selection signature
References
Raça Canchim - O precoce brasileiro. 2008. http://canchim.com.br/canchimnamidia/mercadodacarne_set2008/mercado_da_carne_set2008.pdf dodacarne_set2008/mercado_da_carne_set2008.pdf. Accessed 25 Nov 2015.
Buzanskas ME, Grossi DA, Ventura RV, Schenkel FS, Sargolzaei M, Meirelles SLC, et al. Genome-wide association for growth traits in canchim beef cattle. PLoS One. 2014;9:e94802.
Machado MBB, Alencar MM, Pereira AP, Oliveira HN, Casas E, Coutinho LL, et al. QTL affecting body weight in a candidate region of cattle chromosome 5. Genet Mol Biol. 2003;26:259–65.
Alencar MM. Bovino - Raça Canchim: Origem e Desenvolvimento. Brasília: Embrapa-DMU; 1988.
Andrade PC, Grossi DA, Paz CCP, Alencar MM, Regitano LCA, Munari DP. Association of an insulin-like growth factor 1 gene microsatellite with phenotypic variation and estimated breeding values of growth traits in Canchim cattle. Anim Genet. 2008;39:480–5.
Silva AM, Alencar MM, Freitas AR, Barbosa RT, Barbosa PF, Oliveira MCS, et al. Herdabilidades e Correlações Genéticas para Peso e Perímetro Escrotal de Machos e Características Reprodutivas e de Crescimento de Fêmeas, na Raça Canchim. Rev Bras Zootec. 2000;29:2223–30.
Buzanskas ME, Grossi DA, Baldi F, Barrozo D, Silva LOC, Torres Júnior RAA, et al. Genetic associations between stayability and reproductive and growth traits in Canchim beef cattle. Livest Sci. 2010;132:107–12.
Baldi F, Albuquerque LG, Alencar MM. Random regression models on Legendre polynomials to estimate genetic parameters for weights from birth to adult age in Canchim cattle. J Anim Breed Genet. 2010;127:289–99.
Mattar M, Silva LOC, Alencar MM, Cardoso FF. Genotype x environment interaction for long-yearling weight in Canchim cattle quantified by reaction norm analysis. J Anim Sci. 2011;89:2349–55.
Barichello F, Alencar MM, Torres Júnior RAA, Silva LOC. Efeitos ambientais e genéticos sobre peso, perímetro escrotal e escores de avaliação visual à desmama em bovinos da raça Canchim. Rev Bras Zootec. 2011;40:286–93.
Goddard ME, Wray NR, Verbyla K, Visscher PM. Estimating Effects and Making Predictions from Genome-Wide Marker Data. Stat Sci. 2009;24:517–29.
Wiener P, Burton D, Ajmone-Marsan P, Dunner S, Mommens G, Nijman IJ, et al. Signatures of selection? Patterns of microsatellite diversity on a chromosome containing a selected locus. Heredity. 2003;90:350–8.
Kim Y, Nielsen R. Linkage disequilibrium as a signature of selective sweeps. Genetics. 2004;167:1513–24.
McVean G. The structure of linkage disequilibrium around a selective sweep. Genetics. 2007;175:1395–406.
Mokry FB, Buzanskas ME, Mudadu MA, Grossi DA, Higa RH, Ventura RV, et al. Linkage disequilibrium and haplotype block structure in a composite beef cattle breed. BMC Genomics. 2014;15:S6.
Clayton D. snpStats: SnpMatrix and XSnpMatrix classes and methods. 2014. p. 78.
R Development Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2015.
Zimin AV, Delcher AL, Florea L, Kelley DR, Schatz MC, Puiu D, et al. A whole-genome assembly of the domestic cow, Bos taurus. Genome Biol. 2009;10:r42.
Browning SR, Browning BL. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet. 2007;81:1084–97.
Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72.
Sabeti PC, Reich DE, Higgins JM, Levine HZP, Richter DJ, Schaffner SF, et al. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419:832–7.
Hill WG, Robertson A. Linkage disequilibrium in finite populations. Theor Appl Genet. 1968;38:226–31.
Gautier M, Vitalis R. rehh: an R package to detect footprints of selection in genome-wide SNP data from haplotype structure. Bioinformatics. 2012;28:1176–7.
Utsunomiya YT, Pérez O’Brien AM, Sonstegard TS, Van Tassell CP, Carmo AS, Mészáros G, et al. Detecting loci under recent positive selection in dairy and beef cattle by combining different genome-wide scan methods. PLoS One. 2013;8:e64280.
Gautier M, Naves M. Footprints of selection in the ancestral admixture of a New World Creole cattle breed. Mol Ecol. 2011;20:3128–43.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
National Center for Biotechnology Information. 2016. http://www.ncbi.nlm.nih.gov/snp/. Accessed 11 Feb 2016
Ensembl Genome Browser. 2016. http://www.ensembl.org/index.html. Accessed 12 Jan 2016.
Mi H, Thomas P. PANTHER Pathway: An Ontology-Based Pathway Database Coupled with Data Analysis Tools. Methods Mol Biol. 2009;563:123–40.
Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2008;4:44–57.
Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13.
AnimalQTLdb. 2016. http://www.animalgenome.org. Accessed 15 Jan 2016.
Casas E, Shackelford SD, Keele JW, Stone RT, Kappes SM, Koohmaraie M. Quantitative trait loci affecting growth and carcass composition of cattle segregating alternate forms of myostatin. J Anim Sci. 2000;78:560–9.
Casas E, Shackelford SD, Keele JW, Koohmaraie M, Smith TPL, Stone RT. Detection of quantitative trait loci for growth and carcass composition in cattle. J Anim Sci. 2003;81:2976–83.
Kim JJ, Farnir F, Savell J, Taylor JF. Detection of quantitative trait loci for growth and beef carcass fatness traits in a cross between Bos taurus (Angus) and Bos indicus (Brahman) cattle. J Anim Sci. 2003;81:1933–42.
Casas E, Lunstra DD, Stone RT. Quantitative trait loci for male reproductive traits in beef cattle. Anim Genet. 2004;35:451–3.
Li C, Basarab J, Snelling WM, Benkel B, Murdoch B, Hansen C, et al. Assessment of positional candidate genes MYF5 and IGF1 for growth on bovine chromosome 5 in commercial lines of Bos taurus. J Anim Sci. 2004;82:1–7.
Li C, Basarab J, Snelling WM, Benkel B, Kneeland J, Murdoch B, et al. Identification and fine mapping of quantitative trait loci for backfat on bovine chromosomes 2, 5, 6, 19, 21, and 23 in a commercial line of Bos taurus. J Anim Sci. 2004;82:967–72.
Gasparin G, Miyata M, Coutinho LL, Martinez ML, Teodoro RL, Furlong J, et al. Mapping of quantitative trait loci controlling tick [Riphicephalus (Boophilus) microplus] resistance on bovine chromosomes 5, 7 and 14. Anim Genet. 2007;38:453–9.
McClure MC, Morsci NS, Schnabel RD, Kim JW, Yao P, Rolf MM, et al. A genome scan for quantitative trait loci influencing carcass, post-natal growth and reproductive traits in commercial Angus cattle. Anim Genet. 2010;41:597–607.
Kneeland J, Li C, Basarab J, Snelling WM, Benkel B, Murdoch B, et al. Identification and fine mapping of quantitative trait loci for growth traits on bovine chromosomes 2, 6, 14, 19, 21, and 23 within one commercial line of Bos taurus. J Anim Sci. 2004;82:3405–14.
Mizoshita K, Watanabe T, Hayashi H, Kubota C, Yamakuchi H, Todoroki J, et al. Quantitative trait loci analysis for growth and carcass traits in a half-sib family of purebred Japanese Black (Wagyu) cattle. J Anim Sci. 2004;82:3415–20.
Mizoshita K, Takano A, Watanabe T, Takasuga A, Sugimoto Y. Identification of a 1.1-Mb region for a carcass weight QTL onbovine Chromosome 14. Mamm Genome. 2005;16:532–7.
Maltecca C, Weigel KA, Khatib H, Cowan M, Bagnato A. Whole-genome scan for quantitative trait loci associated with birth weight, gestation length and passive immune transfer in a Holstein x Jersey crossbred population. Anim Genet. 2009;40:27–34.
Pérez O’Brien AM, Utsunomiya YT, Mészáros G, Bickhart DM, Liu GE, Van Tassell CP, et al. Assessing signatures of selection through variation in linkage disequilibrium between taurine and indicine cattle. Genet Sel Evol. 2014;46:19.
Buzanskas ME, Grossi DA, Ventura RV, Chud TCS, Urbinati I, Meirelles SLC, et al. Genome-Wide Association Study on Long-Yearling Scrotal Circumference in Canchim Cattle. 10th World Congr. Genet. Appl. to Livest. Prod. Genome-Wide 17–22 August 2014. Vancouver. 2014.
Bolormaa S, Pryce JE, Reverter A, Zhang Y, Barendse W, Kemper K, et al. A multi-trait, meta-analysis for detecting pleiotropic polymorphisms for stature, fatness and reproduction in beef cattle. PLoS Genet. 2014;10:e1004198.
Wibowo TA, Gaskins CT, Newberry RC, Thorgaard GH, Michal JJ, Jiang Z. Genome assembly anchored QTL map of bovine chromosome 14. Int J Biol Sci. 2008;4:406–14.
Kemper KE, Saxton SJ, Bolormaa S, Hayes BJ, Goddard ME. Selection for complex traits leaves little or no classic signatures of selection. BMC Genomics. 2014;15:246.
Somavilla AL, Sonstegard TS, Higa RH, Rosa AN, Siqueira F, Silva LOC, et al. A genome-wide scan for selection signatures in Nellore cattle. Anim Genet. 2014;45:771–81.
Hoashi S, Hinenoya T, Tanaka A, Ohsaki H, Sasazaki S, Taniguchi M, et al. Association between fatty acid compositions and genotypes of FABP4 and LXR-alpha in Japanese black cattle. BMC Genet. 2008;9:84.
Michal JJ, Zhang ZW, Gaskins CT, Jiang Z. The bovine fatty acid binding protein 4 gene is significantly associated with marbling and subcutaneous fat depth in Wagyu x Limousin F2 crosses. Anim Genet. 2006;37:400–2.
Pannier L, Mullen M, Hamill RM, Stapleton PC, Sweeney T. Association analysis of single nucleotide polymorphisms in DGAT1, TG and FABP4 genes and intramuscular fat in crossbred Bos taurus cattle. Meat Sci. 2010;85:515–8.
Porto-Neto LR, Kijas JW, Reverter A. The extent of linkage disequilibrium in beef cattle breeds using high-density SNP genotypes. Genet Sel Evol. 2014;46:22.
Gutiérrez-Gil B, Wiener P, Williams JL. Genetic effects on coat colour in cattle: dilution of eumelanin and phaeomelanin pigments in an F2-Backcross Charolais x Holstein population. BMC Genet. 2007;8:56.
Kohyama M, Ise W, Edelson BT, Wilker PR, Hildner K, Mejia C, et al. Role for Spi-C in the development of red pulp macrophages and splenic iron homeostasis. Nature. 2009;457:318–21.
Wang D, Zheng M, Lei L, Ji J, Yao Y, Qiu Y, et al. Tespa1 is involved in late thymocyte development through the regulation of TCR-mediated signaling. Nat Immunol. 2012;13:560–8.
Schweitzer BL, Huang KJ, Kamath MB, Emelyanov AV, Birshtein BK, DeKoter RP. Spi-C Has Opposing Effects to PU.1 on Gene Expression in Progenitor B Cells. J Immunol. 2006;177:2195–207.
Li W-H. Molecular Evolution. Sinauer Associates, United States of America: Sunderland; 1997.
Vissers LELM, Lausch E, Unger S, Campos-Xavier AB, Gilissen C, Rossi A, et al. Chondrodysplasia and abnormal joint development associated with mutations in IMPAD1, encoding the Golgi-resident nucleotide phosphatase, gPAPP. Am J Hum Genet. 2011;88:608–15.
Fortes MRS, Lehnert SA, Bolormaa S, Reich C, Fordyce G, Corbet NJ, et al. Finding genes for economically important traits: Brahman cattle puberty. Anim Prod Sci. 2012;52:143–50.
Zhao F, McParland S, Kearney F, Du L, Berry DP. Detection of selection signatures in dairy and beef cattle using high-density genomic information. Genet Sel Evol. 2015;47:49.
Randhawa IA, Khatkar M, Thomson P, Raadsma H. Composite selection signals can localize the trait specific genomic regions in multi-breed populations of cattle and sheep. BMC Genet. 2014;15:34.
Karim L, Takeda H, Lin L, Druet T, Arias JAC, Baurain D, et al. Variants modulating the expression of a chromosome domain encompassing PLAG1 influence bovine stature. Nat Genet. 2011;43:405–13.
Fortes MRS, Reverter A, Kelly M, McCulloch R, Lehnert SA. Genome-wide association study for inhibin, luteinizing hormone, insulin-like growth factor 1, testicular size and semen traits in bovine species. Androl. 2013;1:644–50.
Hoshiba H, Setoguchi K, Watanabe T, Kinoshita A, Mizoshita K, Sugimoto Y, et al. Comparison of the effects explained by variations in the bovine PLAG1 and NCAPG genes on daily body weight gain, linear skeletal measurements and carcass traits in Japanese Black steers from a progeny testing program. Anim Sci J. 2013;84:529–34.
Acknowledgements
The authors are thankful to the Brazilian Agricultural Research Corporation (EMBRAPA) for providing the data used in this study. IU, MTO, TCSC, and MEB would like to thank the São Paulo Research Foundation (FAPESP) (grant 2013/09050-0, 2014/02253-6, 2015/08939-0, and 2013/19335-2, respectively). NBS, LCAR, MMA, and DPM were supported by a fellowship from the National Council of Technological and Scientific Development (CNPq).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
IU and MEB: contributed for the development of this research with data analysis, interpretation, figure compositions, manuscript writing and revision. RHH: data analysis, manuscript discussion and revision. MMA: experimental design. NBS, MTO, TCSC and DPM: data interpretation. LCAR: experimental design, interpretation, and manuscript revision. All authors read and approved the final manuscript.
Additional files
Additional file 1:
Candidate regions identified for piHS = 4 (P < 0.0001). (DOC 61 kb)
Additional file 2:
Extent of linkage disequilibrium in candidate selection regions present on BTA5 (A) and BTA14 (B). (TIFF 1970 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Urbinati, I., Stafuzza, N.B., Oliveira, M.T. et al. Selection signatures in Canchim beef cattle. J Animal Sci Biotechnol 7, 29 (2016). https://doi.org/10.1186/s40104-016-0089-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40104-016-0089-5