
Abstract
Cancer biomarkers have transformed current practices in the oncology clinic. Continued discovery and validation are crucial for improving early diagnosis, risk stratification, and monitoring patient response to treatment. Profiling of the tumour genome and transcriptome are now established tools for the discovery of novel biomarkers, but alterations in proteome expression are more likely to reflect changes in tumour pathophysiology. In the past, clinical diagnostics have strongly relied on antibody-based detection strategies, but these methods carry certain limitations. Mass spectrometry (MS) is a powerful method that enables increasingly comprehensive insights into changes of the proteome to advance personalized medicine. In this review, recent improvements in MS-based clinical proteomics are highlighted with a focus on oncology. We will provide a detailed overview of clinically relevant samples types, as well as, consideration for sample preparation methods, protein quantitation strategies, MS configurations, and data analysis pipelines currently available to researchers. Critical consideration of each step is necessary to address the pressing clinical questions that advance cancer patient diagnosis and prognosis. While the majority of studies focus on the discovery of clinically-relevant biomarkers, there is a growing demand for rigorous biomarker validation. These studies focus on high-throughput targeted MS assays and multi-centre studies with standardized protocols. Additionally, improvements in MS sensitivity are opening the door to new classes of tumour-specific proteoforms including post-translational modifications and variants originating from genomic aberrations. Overlaying proteomic data to complement genomic and transcriptomic datasets forges the growing field of proteogenomics, which shows great potential to improve our understanding of cancer biology. Overall, these advancements not only solidify MS-based clinical proteomics’ integral position in cancer research, but also accelerate the shift towards becoming a regular component of routine analysis and clinical practice.
Similar content being viewed by others


Background
Cancer is the second leading cause of death and poses a major problem to healthcare systems worldwide. The prevalence of cancer remains stable with an estimated 1.7 million new cases, resulting in 600,000 new deaths, in 2018 in the United States alone [1]. Currently, clinical practices are being improved by research on early detection methods, appropriate classification of risk groups and treatment efficacies. Much of this research has characterized tumours at the molecular level using a systems biology approach aimed at biomarker discovery. The National Cancer Institute (NCI) defines a biomarker as a biological molecule found in blood, other body fluids, or tissues that provides an indication of a normal or abnormal process, or of a condition or of a disease. They are used in the early detection, diagnosis, prognosis and treatment selection in the oncology clinic. The routine measurement of biomarkers and better treatment options in oncology clinics have led to a gradual reduction in cancer mortality rates with an estimated 1.5% annual decline, amounting to a 26% decrease over the past three decades [1].
Other fields of clinical research attempt to elucidate molecular differences between cancer cases and healthy controls or different stages of cancers as the disease progresses. These include genomics and transcriptomics that have identified numerous cancer-driving genes. While these omics datasets have demonstrated the ability to compare and contrast different clinical cancer groups, one limitation is that these changes do not necessarily directly translate to our understanding of disease biology. On the other hand, proteins are the biomolecules that directly carry out most biological processes suggesting they are ideal predictors of disease progression [2]. Additionally, proteins are the active targets of most cancer therapeutics including the growing field of immunotherapies. This makes clinical proteomics a growing field in molecular clinical research: the large-scale study of proteins, including their expression, functions and structure, and applying the findings to improve patient care.
Multiple studies have shown that globally mRNA expression is positively, but weakly, correlated with protein expression [3,4,5,6]. This may be one reason why results from transcriptomic studies have translated to the clinic with mixed results and support the implementation of additional (and complementary) research in clinical proteomics. This discordance arises from the highly dynamic and complex nature of proteome regulation. Protein expression is affected by alternative splicing, SNP’s (which translate to different proteoforms) and transcript degradation, as well as protein-level processes such as protein–protein interactions, degradation rates and post-translational modifications (PTMs) [7, 8]. Accurate protein detection techniques are required for routine clinical analysis.
There currently exists a strong bias towards antibody-based techniques for the detection of clinically-relevant proteins. ELISA is commonly used to quantify protein biomarkers in a variety of biofluids, with ongoing improvements, such as Prostate-specific antigen (PSA) in the blood of suspected prostate cancer (PCa) patients as low as one hundred picograms per millilitre [9]. Immunohistochemistry (IHC) stains tissues to provide spatial information regarding well-established cancer markers. For example, the protein markers HER2, ER and PgR are used to classify breast cancer subtypes which has significant implications in selecting an appropriate treatment. ER and PgR-positive tumors are treated by endocrine therapy while HER2-positive status is a prerequisite for targeted immunotherapy [10]. IHC is also useful in discerning tissue physiologies associated with poor prognosis or treatment response. These include tissue hypoxia using markers such as HIF1α [11], or staining for infiltrating immune cells such CD69+ activated lymphocytes in melanoma [12]. Fluorescence activated cell sorting (FACS) uses antibodies to detect a small panel of protein markers and determine heterogeneity amongst a population of cells. The main advantage of antibodies is the detection specificity they provide but their application comes with several disadvantages: cost of development, availability, quality, and ability to be multiplexed. The need for higher-throughput techniques that capture a wider swath of the cancer proteomic landscape have opened the door for mass spectrometry (MS)-based techniques in the oncology clinic. Rapid technological advancements on multiple fronts including sample preparation, peptide separation, MS-detection, and data analysis have all been essential for the robust quantitation of proteins from complex clinical samples.
In this review, we highlight relevant literature related to MS-based clinical proteomics with a specific focus on cancer research. We specifically focus on clinical sample types, sample preparation techniques, MS configurations and protein quantitation strategies. To limit this review to a more manageable scope we further describe notable studies that specifically investigate the proteomes of cancer tissues and bodily fluids. While we attempted to be as complete and inclusive as possible, we apologize to authors whose papers were not cited as part of this review.
Recent advances in clinical proteomic methodologies
Clinical sample preparation methods
A wide array of sample types has been analyzed by clinical proteomics. First and foremost, larger cohorts of primary patient materials in the form of tissue samples are becoming increasingly feasible, due to improvements in biobanking and proteomics technologies. As a result, the direct proteomic investigation of clinical tissues is becoming increasingly popular. Preservation of the tissue’s proteome dynamics is critical from time of surgical resection to the protein digestion stage, and there are a few methods of doing so: fresh frozen (FF), formalin-fixed paraffin embedded (FFPE), and optimal cutting temperature embedded (OCT). One important caveat for consideration in clinical tissue-based proteomics is that surgical procedures could possibly take hours from the time a patient is admitted to the operating room to the point of sample retrieval and preservation. How this affects a tissue proteome is currently poorly understood. More rapid procedures such as needle biopsies have the potential to overcome some of these complications but provide significantly lower amounts of tissue for proteome analysis. While FF is the preservation method of choice from a proteome coverage perspective, FFPE tissues have been banked for decades, providing extensive clinical follow-up and an invaluable resource for clinical proteomics. Previously, cross-linking-based modifications produced insufficient proteomic coverage for global proteomic studies of FFPE tissues. Improvements in sample preparation have led to more efficient de-crosslinking of fixed proteins, and as a result, greater protein availability for digestion. Tissue samples can be further prepared by laser-capture microdissection (LCM) to add an element of spatial resolution. This allows different regions of the same tissue sample to be compared, whether normal adjacent or tumorous.
While tissue samples have the potential to provide novel biological insights, many clinical proteomics studies have aimed to discover novel biomarkers, ideally in clinical samples that are obtainable in a non-invasive or minimally invasive manner (i.e. liquid biopsies). The most commonly analyzed biofluids are blood (plasma, serum) and urine, but other biofluids that have been analyzed by proteomics include post-digital rectal examination urine [13], expressed prostatic secretions [14], saliva [15], tears [16], cerebrospinal fluid (CSF) [17], and ascites [18, 19] to name a few. The promise of analyzing patients’ body fluids is that disease-relevant changes in molecular markers such as cfDNA, RNA, proteins, lipids and metabolites are reflected in the fluid sample. Therefore, a liquid biopsy sample can be collected in a renewable manner for longitudinal studies that monitor cancer progression and a patient’s response to treatment.
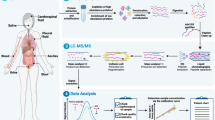
Clinical sample cohorts of the past were often underpowered due to biobanking limitations (i.e. availability of high-quality, richly annotated samples). As a result, various model systems have been developed to facilitate the discovery of new biomarkers or aid in characterizing proteins of interest, as in Fig. 1. These models include transgenic animal models, immortalized cancer cell lines, primary cell lines and xenograft models. Cell lines of various cancer subtypes can be grown in 2D on cell culture dishes, either directly on plastic or on various matrices (i.e. collagen), or under more sophisticated 3D conditions (i.e. embedded in Matrigel). More recently, primary cells have been established as organoid models that more accurately mimic three-dimensional tumour development [13]. Alternatively, cancer cell lines or patient-derived tumors can also be engrafted into immunocompromised mice to generate so-called cell line-derived xenografts (CDX) or patient-derived xenografts (PDX), respectively [20,21,22]. These xenograft models are thought to more accurately recapitulate human tumor specimens, due to the presence of matrix components and stromal cells (i.e. vascular cells, fibroblasts, etc.), but still lack important contributions from the immune system. Model systems play a critical role in allowing for an expansion of clinical proteomic studies to gain more functional insights. As such, data from representative model systems can act as a complement to data obtained from clinical samples.

Overview of clinical cancer proteomics strategies. a Various sample types are used for clinical proteomics. These include solid tumor tissues, patient body fluids, animal models and cell-based systems. Tumor tissues are obtained either as surgically resected samples or are biopsy based. There are a number of tissue processing approaches available, which include the analysis of “bulk” tissue or preferentially after pathological inspection, tissue macro-dissection or laser capture microdissection (LCM). Patient fluids are a popular source for the discovery of biomarkers. The most commonly used patient body fluids include blood (processed to plasma or serum) and urine. Animal models are a popular in vivo model system for clinical proteomics. The most common models include transgenic disease models and patient-derived xenografts (PDX). Cell-based systems continue to be popular model systems in cancer biology. They include immortalized cancer cell lines or more sophisticated organoid systems that are established using defined culture conditions and primary patient material. Samples obtain from these sources are homogenized and proteolytically digested prior to proteomic analyses (i.e. bottom-up proteomics). b Proteomic analyses can use several well-established workflows. These include label-free proteomics (LFQ), isobaric labelling strategies or the specific enrichment of post-translational modification such as phosphorylation, ubiquitination, glycosylation, etc. c Integration of proteomics data with publicly available resources such as the CPTAC proteomics data or transcriptional profiles from GTEx, CCLE and TCGA can be used for biomarker prioritization. d Bioinformatics analyses (clustering, enrichment, pathways, etc.) are used to extract biological content or further prioritize candidates for targeted proteomics validation, using multiple reaction monitoring (MRM) and Parallel reaction monitoring (PRM)
Sample preparation plays an important role in the proteomic characterization of clinical samples and rigorous standard operating procedures need to be established in order to get relevant information on the complex biological processes that lead to cancer progression. There is no universal protocol for proteomic sample preparation, but rather the selected strategy should be optimized/selected based on the proteomic complexity, the available quantity of sample and the goal of the study. The first step in sample preparation for MS includes lysis and extraction of proteins from the clinical samples. This includes extraction reagents such as different organic solvents and detergents followed by tissue disruption techniques such as freeze–thaw cycles, sonication or mechanical disruption to maximise the protein extraction and solubilisation. Organic solvents solubilise and denature proteins and can be easily removed by evaporation using lyophilization. TFE (2,2,2-Trifluoroethanol) based lysis and extraction on nano-scale (30 µg) and macro-scale (> 100 µg) input materials gave comparable protein detection rates relative to the traditional detergent-based methods [23]. While the use of other organic solvents such as acetonitrile have been reported, TFE-based sample preparation in clinical studies have been shown in the study of ovarian cancer [24] and PCa tissues [6].
Denaturants (urea and guanidine HCl), ionic detergents (SDS, SDC), and non-ionic detergents (Triton X-100, NP-40) act to efficiently lyse cells and solubilise protein complexes, especially membrane proteins. The disadvantage of detergent use is their difficult removal from samples for downstream MS applications that could lead to peptide ion suppression. Detergents also tend to deposit in the electrospray emitters and liquid chromatography lines, C18 chromatography columns, and in the MS instrument front-end causing added maintenance. Many MS-compatible, commercially available detergents have been reported and widely used including Rapigest (Waters), ProteaseMax (Promega), Invitrosol (Thermo). These detergents degrade with the addition of heat or acidic pH conditions; hence reducing problems described above. Recently different protein purification techniques have been developed with increased efficiency, reduce protein digestion time requirements and minimized sample losses as described below.
FASP, MStern and S-trap
The anionic surfactant sodium dodecyl sulfate (SDS) is an excellent agent to solubilise proteins but possess limited compatibility with MS applications. The removal of SDS from the peptide sample has proven to be a major barrier with conventional methods. Recently developed sample preparation methods focus on using SDS and other denaturants as the solubilization agent and their removal through various membrane-based protein capture techniques. One of these developed methods is filter aided sample preparation (FASP) first described by Manza et al. [25] and further characterized by Wisniewski et al. [26]. FASP uses molecular weight (MW) filtration to bind proteins to a nitrocellulose filter, while lower MW analytes pass through the filter. Consecutive urea washes facilitate SDS removal, followed by on-filter digestion and peptide elution. This technique reduces sample preparation time and sample loss while maintaining the advantages of using SDS for improved proteome coverage. One of FASP’s limitations is a reduced binding efficiency with small quantities of starting material with greater sample losses [27]. Additionally, the small membrane pore size in FASP requires higher spinning speeds which makes it time consuming in the 96-well format. Distler et al. [28] made minor modifications to the FASP protocol to minimize potential losses of input material and reduce processing time. Alkylation and reduction were performed on-filter and further washes with MS-compatible volatile salts such as ammonium bicarbonate leading to highly purified samples. FASP has been utilised in different clinical tissue proteomics studies including colorectal cancer (CRC) FFPE samples [29, 30], PCa FFPE tissue samples [31] and FFPE samples of PCa bone metastases [32], to name a few.
Clinical proteomics studies consist of increasingly large cohorts which require efficient and timely sample preparation; which has been facilitated by the development of 96-plate format techniques. The MStern method was developed to overcome the problem of slow liquid transfer through nitrocellulose membrane. MStern uses hydrophobic PVDF membranes with significantly larger pore sizes allowing for improved liquid transfer and more efficient protein adsorption relative to nitrocellulose [33]. A vacuum system is used for passing the samples through the membrane more effectively than centrifugation. Similarly to FASP, MStern involves reduction, alkylation and digestion on the same membrane. Apart from the speed and efficiency of MStern, peptides are not eluted with a high salt concentration. Rather, they are eluted by acetonitrile and formic acid which limits the need for extra desalting steps. The one limitation of MStern is that the binding capacity of each well is 25 µg compared to < 400 μg for FASP. A study by Berger et al. compared MStern and FASP techniques using urine, CSF and whole cell lysate samples with varying amounts of starting material. MStern showed a comparable number of protein detections while saving 9.5 h of processing time [33].
Another recently developed membrane-based method that uses a similar principle as FASP is called suspension trapping (S-trap) [34]. S-Trap packed filters consist of quartz fibers packed with a larger pore size compared to FASP. The other protocol modifications include the use of higher SDS concentrations (5%) in the lysis method. The addition of methanol and phosphoric acid causes the formation of protein particulates which are trapped by the filter. Similarly, to FASP and MStern, reduction, alkylation and digestion are done directly on the filters. The comparative study to evaluate the overall efficiency of S-Trap, MStern and FASP showed that S-Trap and FASP provided the greatest number of protein detections compared to the polyvinylidene difluoride (PVDF) method. The digestion efficiency was greatest for S-trap which reported the lowest number of missed cleavages [35]. Another study showed that S-Trap outperformed FASP in terms of protein detection due to a higher digestion efficiency [36].
SP3 and iST
Clinical tissue samples are sometimes challenging to process due to their small size, particularly LCM samples. These samples require efficient sample processing techniques to ensure limited sample losses and maximal extraction of the proteins to maximize proteome coverage. The solid-phase-enhanced sample preparation (SP3) method was developed with these limitations in mind. Originally described by Hughes et al. [37] the method uses paramagnetic beads which are coated with hydrophilic carboxylate groups. The beads are compatible with various detergents and organic solvents including SDS, urea, TFE and acetonitrile (ACN) [27, 38]. The proteins are immobilized to the charged carboxylate groups in the presence of an organic solvent with acidic or basic pH. After the immobilisation of the proteins, detergents are removed with high organic content washes, followed by on-bead digestion. Eluted peptides can be directly introduced into the MS without the need for desalting. SP3 protocol has been further modified to improve the efficiency and its reproducibility. These studies showed that binding efficiency of proteins are lower in acidic conditions as compared to neutral pH [39]. Furthermore, using ethanol in neutral pH during protein binding resulted in better protein recovery compared to ACN in acidic pH [40]. The strength of SP3 beads lie in providing a platform for efficient protein binding from minute amounts of starting material; with all sample preparation steps happening in one tube to limit potential losses. This method has been scaled to an 96-well automated robot liquid handling platform for robust reproducibility and through-put [41]. Several clinical studies using patient derived samples have used the SP3 method, including ovarian cancer [42], CRC [43].
The in-StageTip (iST) method developed by Kulak et al. [44] in 2014 is another sample preparation workflow, which is compatible with low input material. The method focuses on using a single stage tip enclosed by a barrier to perform multiple sample processing steps to minimize sample losses and to provide better proteome coverage. In the iST workflow, a pipette tip is inserted with a reversed-phase membrane barrier at the bottom. The sample in the tip is introduced from the top, where they can be lysed through heating or sonication. The sample is then denatured, alkylated and digested. The membrane at the bottom of the tip is then used for peptide clean-up. Alternatively, samples can also be fractionated on the same tip. The iST workflow shows high performance handling ultra-low amount of material, but due to the reverse-phase membrane barrier the iST is incompatible to use detergents (SDS) and organic solvents (TFE) for lysis [37].
The work by Sielaff et al. [39] showed an independent comparison between commercially available iST, FASP and SP3 sample preparation strategies using minute amounts of starting material. Initially, varying amounts of HELA lysates (1–20 µg) were used to process these samples. The results showed that all three methods performed similarly with respect to the numbers of protein detections and reproducibility. Reduction of the input < 10 µg resulted in SP3 and iST providing similar proteome coverage, whereas FASP showed a decrease in performance. Furthermore, clinically relevant FACS immune cells (25,000) were processed in triplicates using these three methods. The highest number of proteins were detected through SP3 method giving an average detection of 3152, followed by 2343 proteins by iST and FASP detecting 109 proteins. These results suggest that SP3 and iST methods are suitable for low input starting material while FASP may not be feasible for ultra-low amounts of starting material.
Front-end MS developments
The field of clinical proteomics has seen advancements at the sample preparation stages, as well as in MS technology. MS-based proteomics’ emergence can be attributed to the development of soft ionization techniques such as electrospray ionization (ESI) and matrix-assisted laser desorption ionization (MALDI). Further advancements in front end technologies have propelled clinical proteomics to further depths of the human proteome. ESI continues to rely on well-established reversed-phase nano-LC technologies, or combined with capillary electrophoresis [45], and is hence more practical for discovery-based experiments. Orthogonal peptide separation techniques have grown in popularity amongst clinical proteomics research applications. For increased proteome coverage over a single-shot experiment, a peptide pool can be fractionated by basic reverse-phase LC or strong cation exchange chromatography. For example, peptides from lung cancer cell lines resistant and sensitive to tyrosine-inhibitor treatment were fractionated and produced 39% more detections per protein than a single shot analysis [46]. The increased burden of LC–MS time requirements can be reduced by strategic concatenation of non-sequential fractions. Additionally, this strategy comes with increased sample handling and loss of peptides; an important consideration when clinical samples may be limited in protein input and availability [46]. In the gas phase, peptides can be further separated by ion mobility to achieve greater proteome coverage. These technologies include high-field asymmetric ion mobility spectrometry (FAIMS) and trapped ion mobility spectrometry (TIMS) aim to reduce MS1 complexity and thus MS2 contamination from co-eluted and co-isolated peptides [47, 48]. These technologies have demonstrated a 30% increase in peptide detections from routine analysis of cancer cell lines [49] and this improvement translates similarly to clinically-relevant specimens. Overall, the improvement of front-end peptide separation techniques is ongoing to meet the goal of increased proteome coverage.
MS scanning modes
The majority of discovery-based clinical proteomic studies continue to depend on data-dependent acquisition (DDA) to identify potential biomarkers or gain biological insights. This approach has some benefits including well established instrument operation, data analysis and processing pipelines. Additionally, DDA includes the option of label-dependent quantitation and associated multiplexing. On the downside, DDA is hampered by low inter-sample reproducibility of peptide detection due to random sampling, thus creating a “missing value” problem. While the premise of DDA has remained largely unchanged, improvements in the instrument’s efficiency have stemmed from the development of the BoxCar data-acquisition method [50]. Unlike past DDA advancements that focus on the MS2-level, BoxCar sequentially fills narrow m/z windows to increase the ion injection time more than ten-fold. The increase in ion collection significantly increases the signal-to-noise and overcomes issues of abundant peptides dominating the MS1 spectrum, while less abundant, co-eluting peptides were less likely to be selected for MS2. As a result, 90% of a cancer cell line proteome was detected in a 1-h analysis as opposed to 24 fractions. Reproducibility was also high, as the majority of proteins were quantified in all ten replicates. BoxCar will undoubtedly be applied to cancer-relevant studies in the near-future [51]. Another recent advancement in DDA is the MaxQuant.Live software that combines aspects of global and targeted MS. It applies on-the-fly mass, retention time and intensity calibration and controls the Orbitrap mass analyzer to predict the detection of significantly more precursors in real-time [52].
Due to the shortcomings of DDA, the field of clinical proteomics is observing a shift towards data-independent acquisition (DIA), a method that was originally described by Purvine et al. [53] and further reported by Venable et al. [54]. In DDA, the N most intense peptide precursors in a survey MS1 scan are selected for sequential fragmentation and MS2 detection. Whereas in Sequential Window Acquisition of All Theoretical Mass Spectra (SWATH-MS), a variation of classic DIA, all theoretical peptides in a sample are sequentially fragmented in narrow m/z windows to yield more complex MS2 spectra [55, 56]. These spectra are then matched to a pre-defined, empirically determined, spectral library of peptides with the goal of the library having achieved maximum proteome depth through extensive peptide fractionation. Additionally, peptide sequencing and quantitation are more robust since stochastic peptide detections as in DDA are no longer a concern. Current data analysis platforms such as Spectronaut Pulsar [57], OpenSWATH [58], and Scaffold EncyclopeDIA [59] (among others) are sufficient in de-convoluting the MS2 spectra to peptide sequences, at a rate comparable to DDA. Further improvements will allow less abundant peptides to be confidently matched. Clinical samples with greater proteomic diversity, such as tissues, make this MS2 complexity issue more troublesome. On the other hand, clinical samples of less complexity are well-suited for DIA analysis, such as urine. DIA’s reproducibility was tested across 11 laboratories using a benchmarked sample of stable heavy isotope labelled peptides spiked into a cell lysate digest with 91% of proteins detected across all centers [60]. Furthermore, public databases such as SWATHAtlas will continue to increase spectral library availability and increase DIA’s ascension in clinical proteomics [61, 62].
Fragmentation and MS detection techniques
MS instruments continue to show versatility with various peptide fragmentation and detection configurations. Collision-induced dissociation (CID) was first introduced in 1981 and is one the most fundamental fragmentation techniques in proteomics [63]. The ionized peptides are passed through a vacuum chamber where they collide with a neutral gas such as nitrogen, helium or argon. The vibrational energy cleaves the C–N (peptide bonds) to generate b and y ions series, followed by mass analyzer detection. Higher-energy collision dissociation (HCD) which is essentially a vendor-specific term for CID is often used in Orbitraps and hybrid LTQ-Orbitrap mass spectrometers which combine the cycle speed and sensitivity of the linear ion trap with the mass accuracy and resolution of the Orbitrap. Precursor ions are shuttled from the C-trap to the collision cell where the ions are similarly fragmented by a neutral gas [64]. The fragments are then transmitted to the Orbitrap analyzer resulting in improved MS2 spectrum quality, particularly with lower molecular mass [65].
Electron-transfer dissociation (ETD) is an ion–ion collision fragmentation-based method where cations (peptides or proteins) collide with charged radical reagent anions [66]. ETD is particularly useful in the study of diverse modifications because PTM integrity is preserved while still achieving the backbone fragmentation necessary for peptide detection. Hybrid ETD fragmentation methods like ETD-HCD have been reported where precursors sequentially undergo both types of fragmentation to yield b/y and c/z type fragment ions in a single spectrum [67]. Additionally, ETD is suitable for the study of top-down proteomics due their high-cationic nature including intact proteins, their PTMs, and protein–protein interactions. One such application is the study of engineered antibodies which have shown potential to be used as cancer therapies [68]. ETD-based MS methods have been reported in the literature to characterize monoclonal antibodies (mAb) and antibody drug conjugates (ADC) [69,70,71,72,73,74].
Protein quantitation
There is a large number of global and targeted protein quantitation approaches available, each with their unique sets of advantages and disadvantages. These can be categorized as relative or absolute quantitation, of which the former can be further divided into label-dependent and label-free techniques.
Relative quantitation often depends on the use of stable-isotopic labels that result in covalently derivatized peptides. Past methods include metabolic and chemical labelling, such as dimethylation and Stable Isotope Labeling with Amino Acids in Cell Culture (SILAC) which rely on MS1-level quantitation, but they are most applicable to cell line-based studies [75, 76]. Additionally, SILAC-based methods have also been adopted to in vivo studies, termed Super-SILAC, where heavy labelled cell line lysates are spiked into tumor tissue lysates as a global control [77]. This approach was used for histone PTM quantitation in breast cancer FFPE samples [78].
More recent labelling strategies, such as Tandem mass tag (TMT) and isobaric tag for relative and absolute quantitation (iTRAQ), have gained popularity as these techniques apply isobaric tags providing MS2-level relative quantitation while boosting the MS1 signal intensity to improve chances of peptide detection [79, 80]. A potential problem with these approaches is the co-isolation of precursor peptides that results in ratio compression. Improvements in instrument resolution with narrower isolation widths, and the combination of isobaric tags with ion mobility to reduce co-isolation have significantly improved the quality of these experiments. One notable advantage of using TMT in a clinical proteomic setting is the high degree of multiplexing, up to 16-plex, to significantly reduce the LC–MS time requirements of analyzing increasingly large patient cohorts. Potential caveats of TMT -based approaches are that extensive peptide-based fractionation is required to obtain deep proteome profiles and that 1–2 TMT channels are usually dedicated to a global control (i.e. a combined lysate of all clinical lysates analyzed). This reduces the ability of individual projects to be effectively compared against each other. More recently, the ratio compression of TMT labels and thus limited dynamic range of quantitative proteomic data can be overcome with further fragmentation. MS3-level quantitation further removes co-isolating and co-eluting peptides with the use of SPS (synchronous precursor selection) with only a marginal decrease in ion signal intensity [81].
The use of stable-isotopic labels does create added expenses in clinical proteomic studies. As an alternative, recent advances in data computation have given rise to powerful label-free proteomic techniques, with sample peptides often separated over long LC gradients. Relative protein abundances are derived from MS1-level peptide peak integration using software like MaxQuant [82], Proteome Discoverer and Skyline [83]. This allows for wider dynamic ranges spanning several orders of magnitude that are difficult to achieve with label-based techniques, albeit at a loss of precision (i.e. reproducibility). Label-free quantitation is more applicable to clinical proteomics due to the inter-patient and intra-patient variability of protein expression. Label-free strategies are applied to both DDA and DIA scanning modes. Latosinka et al. [84] compared label-free and isobaric tag (iTRAQ) strategies in muscle-invasive and non-muscle-invasive bladder cancer tissues. It was concluded that both methods provide comparable proteome coverages and proportional quantitative data, but label-free DDA identified a greater number of differentially expressed proteins.
In general, relative quantitation strategies are used for discovery-based clinical proteomics. Once proteins of interest have been identified they require further validation. While antibody-based techniques such as ELISA fulfill this role, MS-based targeted assays are well suited for validation, especially if no suitable antibodies are available. The most commonly used method is called multiple-reaction monitoring mass spectrometry (MRM-MS). This assay is well established for small molecules and peptides and is carried out using triple quadrupole mass analyzers. MRM-MS assays require prior knowledge of the parent ion (MS1) and fragment ion (MS/MS) mass-to-charge ratios. The combination of a parent ion and 3–5 associated fragment ions, termed transitions, are selected by the quadrupole. Quantitative data is obtained by measuring the area under the curve of the transitions extracted ion chromatograms. More recently, targeted assays have also been developed using quadrupole Orbitrap mass analyzers—termed parallel reaction monitoring mass spectrometry (PRM-MS), which leverage the high-resolution, accurate-mass (HRAM) Orbitrap for increased specificity [85, 86]. Similar to MRM-MS assays, prior knowledge of parent ion mass-to-charge ratios is required to develop PRM-MS assays. Since all fragment ions are generated and recorded in PRM-MS assays, prior knowledge of individual transitions (fragment ion mass-to-charge ratios) is not required. Rather the best fragment ions can be selected from the full MS/MS spectrum afterwards. Extracted fragment ion chromatograms, similar to described above, are used for quantitation. If appropriate controls such as stable isotope labelled peptide standards are used, targeted proteomics assays can provide absolute quantitation. Importantly, each targeted assay must be carefully optimized to achieve optimal performance. This includes optimized chromatographic separation, collision energies to achieve maximal fragment ion signal-to-noise and dwell times (MRM) or ion fill times (PRM), as recently described [87]. Optimization of these parameters also depends on the total number of peptides targeted. Ideally, for each peptide the assay linearity, level of detection (LOD) and lowest level of quantification (LLOQ) are experimentally determined using synthetic peptides [88]. Furthermore, the mass spectrometer’s detection efforts can be further optimized by using retention time information to schedule MRM/PRM target detection. These strategies require robust chromatography schemes that can be monitored using standard index retention time (iRT) peptides This allows for a substantial increase in the number of targets an assay can accurately monitor. Advances in instrument detection speed now allow for hundreds of targets to be monitored in a single injection [87, 89]. As the door opens for routine monitoring of proteomic signatures in the oncology clinic, targeted PRM approaches show great promise due their ability to monitor progressively more targets in a dependable fashion.
Tissue biopsy studies
Discovery-based studies
In the context of clinical proteomics, tissue analysis provides the most accurate reflection of the tumour’s physiological state. As mentioned, recent advancements in LC–MS technology have continuously enabled increased proteome coverage with reliable quantitation. These studies now enable routine proteomics analyses of patient tumor tissues for biomarker discovery, discovery of biological pathways and integrations with available genomics/transcriptomics profiles.
Tissue-based proteomic strategies have been applied to the study of many cancer types, including prostate [90, 91], breast [77, 92, 93], melanoma [94, 95], lung [96,97,98], ovarian [99, 100], and oropharyngeal carcinoma [101]. The studies above can be summarized by common themes. Clinical proteomic studies often compare cancerous tissue samples with “healthy” adjacent controls from the same patient for potential diagnostic biomarkers. Meanwhile, comparisons between patients with varying stages of cancer are compared for prognostic information. Once a smaller number of candidate proteins have been identified, pathway analyses give insight into how these proteins are associated with tumorigenesis, proliferation, metastasis and other cancer-driving processes. This is often followed by antibody-based techniques that complement and validate the differential expression findings in a larger independent cohort. The studies detailed below follow these guidelines and are summarized in Table 1.
A major application of discovery-based proteomics in cancer tissues is the development of risk stratification and cancer subtyping systems. This has been demonstrated by label-free strategies with several recent examples of DIA applications. Bouchal et al. [102] performed SWATH-MS on frozen breast cancer tumours. An extensive spectral library contained reference spectra for 28,233 proteotypic peptides and their modified variants, attributing to more than 4400 proteins. The discovery cohort consisted of 96 tissues belonging to five well-established clinical subtypes. Proteomic analysis resulted in the consistent quantitation of 2842 protein groups, the majority of which were involved in well-established breast cancer pathways. For the most part, proteomic analysis confirmed the conventional subtype classifications, but highlighted intra-subtype heterogeneity to further create a protein-based classification system. Namely, proteins INPP4B, CDK1 and ERBB2 were found to be linked to ER and HER2 status, patient outcome, and their expression was further validated in cell line models. A renal cell proteomic study was performed by Guo et al. [103] who analyzed tumor and healthy control tissue biopsies from nine different renal cell carcinoma patients (18 samples). The SWATH-MS approach resulted in the quantitation of more than 2000 proteins with high reproducibility. As above, several detected proteins were differentially expressed in the tumor region, with 296 proteins upregulated in tumor samples compared to controls. SWATH-MS was also used in a biomarker discovery study on hepatocellular carcinoma (HCC) performed by Zhu et al. [104]. In this study 38 biopsies from 19 HCC patients with punches from paired tumor and benign regions. The data was analysed by OpenSWATH using a pan-human SWATH assay library which led to the detection of 2570 proteins. Once again, several proteins were differentially expressed in tumor samples compared to benign tumors. In particular, a putative biomarker, MCM7 which is believed to be involved in liver cancer progression was further confirmed through IHC.
Aforementioned label-dependent protein quantitation techniques have also been applied to tissue proteomic for improved risk stratification. Iglesias-Gato et al. [31] performed a proteome-wide study of PCa progression with Super-SILAC quantitation. With an aim of finding prognostic biomarkers. Twenty-eight FFPE prostate tumor samples from with a range of Gleason scores 6–9, and eight adjacent non-malignant samples, were prepared for MS analysis. The peptides were fractionated, and the MS runs resulted in the quantitative detection of over 9000 proteins, with an elevated expression of CPT2, COPA and MSK1/2 in tumour tissues compared to non-malignant region. These elevated proteins were reported to play a role in the regulation of cell proliferation. Comparisons between Gleason groups highlighted pro-neuropeptide Y (Pro-NPY) to be associated with poor patient outcome in intermediate and high-risk patients (Gleason score ≥ 7), which was confirmed by IHC in an independent cohort.
Aside from improving risk stratification, and thus patient survival, clinical proteomics can be used to identify new targets for improved treatment efficiency. Recently, a label-free, global proteomics approach was used to investigate the proteomes of 25 FFPE ovarian cancer tissues, stratified by chemo-resistance. More than 9000 proteins were detected and the differential expression of CT45 was related to a positive chemotherapy response. Little was known about CT45’s molecular function, thus phosphoproteomic analysis of cell line models was pursued and suggested that CT45 increases the repair response to DNA damage. Additionally, CT45 produces immunogenic peptides that recruit cytotoxic T cells and promote tumour killing. Therefore, clinical proteomics was a powerful tool in identifying a target for future immunotherapies [105].
As demonstrated in the study above, tissue global proteomic studies can be further extended to the investigation of PTMs. Yang et al. [106] studied the global and glycoproteome of non-small cell lung carcinoma subtypes. In total 18 patient samples consisting of three squamous cell carcinoma (SqCC) tumor samples with matched benign tissues, six adenocarcinoma (ADC) samples with five matched benign samples and one normal healthy tissue. The digested samples were iTRAQ labelled and enriched for N-glycopeptides followed by reversed-phase LC fractionation prior to shotgun MS analysis. Different protein and glycoprotein signatures were found in ADC and SqCC samples, with pathways distinguishing between tumour types.
Protein kinases play an important role in signal transduction pathways, and the dysregulation of these pathways is an intriguing area of study in cancer [107]. Modern MS instruments and recent phosphopeptide enrichment methods have allowed for the large-scale detection and quantification of thousands of phosphorylation sites. Phosphoproteomic studies in clinical samples allow the identification of aberrantly activated kinases and their downstream substrates, which would otherwise be undetectable by traditional shotgun proteomics, serving as potential therapeutic targets. Many studies have shown important roles in understanding the molecular mechanism of cancers governed by phosphorylation-mediated pathways [108, 109]. Zagorac et al. [110] performed label-free quantitative analysis of the triple negative breast cancer (TNBC) phosphoproteome and compared relapsed and non-relapsed patients. In total 34 patient samples were lysed, digested and enriched for phosphopeptides. Label free single-shot runs resulted in the detection of more than 10,000 phosphosites, corresponding to 2643 phosphoproteins. The analysis showed 159 phosphosites to have increased phosphorylation status. Several different kinases were found to be hyperactivated in relapsed samples compared to the non-relapsed ones through pathway enrichment analysis. Six kinases, PNKP, CDK6, PRKFCE, c-Kit, P70S6K were considered to play a role in the relapse of cancer and showed potential as prognostic markers. Inhibitors against these six kinases were studied in TNBC PDX models and cell lines which showed antitumor activities suggesting potential therapeutic targets in TNBC.
Targeted and multi-site validation
Tissue proteomic studies to date have focused on discovery-based studies that highlight countless potential markers of cancer. We need to move towards rigorous validation of these targets if the field is to evolve towards routine analysis in the oncology clinic. This bridge is being formed by targeted MS methods. PRM of tissue lysates, or immuno-affinity pulldowns of specific proteins from lysates, will become more mainstream [111]. For example, a panel of 54 proteins involved in tumour suppression, drug metabolism and chemoresistance were monitored in FFPE tissue lysates from 50 metastatic CRC patients. This identified differentially expressed proteins that can be used to help guide eligibility of patients for clinical trials [112]. In a breast cancer tissue study, PRM assays were developed for proteins implicated in treatment sensitivity such as HER2, EGFR and PTEN with sub-femtomolar limits of quantitation. This was applied to monitor treatment effects in cell lines, PDX models, and extended to 46 frozen tissue lysates. Interestingly, a subset of the clinically annotated HER-2 positive tissues showed only minimal levels of HER2 by PRM [113]. Targeted LC–MS provides improved levels of detection and quantitation by only focussing on a subset of the proteome, but on occasion, particularly from complex tissue samples, target concentrations are still below reproducible levels of quantitation. Immuno-affinity enrichment of targets is then required to overcome this obstacle. Such a strategy was applied to the measurement of osteopontin in FFPE breast cancer tissues. Two rabbit IgGs were used to isolate two peptide targets from the lysates of normal tissue and breast cancer tumours. It was reproducibly demonstrated that the tumours contained 30× more osteopontin than normal healthy tissues using the first peptide and supported by the second peptide showing a 28× increase [114]. It is important to note that a tissue’s preservation method has an impact on targeted MS detection. Sprung et al. [115] targeted 114 peptides from FFPE and frozen tissues and reported that while the reproducibility of measurements was consistent between sample types, MRM signal intensities were reduced by 34% in FFPE samples.
When using the same sample preparation methods, LC–MS configuration and data analysis pipelines, considerable inter-lab reproducibility can be achieved. TMT-10 was used to multiplex PDX tissues derived from two breast cancer subtypes and compared across three independent laboratories. Overall, 7700 human-originating proteins were distinguishable from the 3100 mouse-derived stromal proteins with a maximum deviation across laboratories of 7%. Additionally, the TMT ratio between the two subtypes produced a minimum R2 correlation of 0.88. Phosphoproteomic results, with an average of 37,000 phosphosites quantified per sample, were less reproducible. The maximum deviation in phosphoproteome coverage was 24% with a R2 correlation of 0.72 [109]. Similar studies to assess inter-lab reproducibility in cancer cell lysates have been performed with similar results. Thirty stable-isotope labeled peptides were spiked into 1 µg digest of cell lysate and detected by SWATH-MS across 11 laboratories worldwide over a 1-week period. The inter-lab median CV of the standard peptide intensities was 47.3% in comparison to the inter-day and intra-day median CV’s at each site of 8.9% and 5.5%, respectively. 4000 proteins were detected in more than 80% of LC–MS runs. Of these proteins, the inter-lab median CV of protein intensity across sites was 22% [60]. Another study compared independently generated datasets. For example, one CPTAC CRC proteomic dataset generated by 2D LC–MS was compared to the proteome of another 40 CRC tissues prepared by GeLC–MS in a separate laboratory. The proteomes shared an 80% overlap in protein detections, of which quantitative measurements were strongly correlated (R2 = 0.8) [116]. Of course, any conclusions from this meta-analysis would require rigorous validation, but it does demonstrate that comparisons of independently-generated tissue proteomic datasets will be possible in the future [92]. Cross-referencing proteomic data findings with other public datasets such as The Cancer Genome Atlas (TCGA) and Human Protein Atlas [117] instills further confidence in biomarker identifications.
Spatial resolution in discovery proteomics
One factor to consider in quantifying changes in the tissue proteome is tumour heterogeneity. This was recently shown by sampling the proteome of frozen prostatectomy specimens by SWATH-MS from benign prostatic hyperplasia and adenocarcinoma patients. To account for inter-patient, intra-tissue and inter-tissue variability, tissue biopsy sections were investigated from different areas of the same sample, different samples and different patients. It was noticed that proteins involved in DNA repair pathways differ significantly between different areas of the same tumor tissue, owing to tumour heterogeneity. Thus, it was suggested to account for this variability in future studies when identifying protein biomarkers from tissues [90]. Efforts are being made to distinguish changes in protein expression originating from disease progression from those originating from tissue heterogeneity and secondary biology pathways. To overcome this, the development of laser-capture microdissection (LCM) provides a powerful tool to isolate specific regions of a tumour cross-section. LCM was used to isolate neoplastic islands and stroma from the tumour front and inner tumour of oral cancer tissues. Discovery-based proteomics revealed the lower expression of cystatin-B in the islands which was further verified by IHC. The study was extended to targeted methods to further distinguish patients with increased chances of lymph node metastasis [118]. Increasing the throughput of LCM to match that of discovery-based proteomic experiments with increasing cohort size is imperative for this technology to transfer to the clinic.
Another MS-based technique that provides spatial information and shows potential in the clinic is Mass Spectrometry Imaging (MSI). MSI has already been established in the detection of metabolomics-based markers, as is making strides in the proteomics sphere. A sectioned tissue biopsy is surveyed by MALDI-MS at ever-increasing spatial resolutions, as low as a few micrometers [119], and routine analysis at 100 µm. As the laser moves over the tissue, a protein marker’s intensity is correlated to a colour-coded heat map. Clinical MSI experiments are most commonly reserved for the detection of small molecules [120], glycans [121] and lipids [122] due to caveats in proteomics studies related to spectral complexity, difficult characterization of distinguishing peaks, and poor ionization of in-tact proteins. More recently, peptide ionization can be facilitated by the brief digestion of surface proteins prior to MSI. One applicable area of interest that has caught the attention of cancer research is the ECM proteome, as it relates to tumour metastasis and treatment resistance. It has been shown that pre-digestion of liver and colon cancer sections with matrix-metalloproteinase prior to MSI gives further insights into how these enzymes modulate ECM [123]. Alternatively, MSI can determine the distribution of cancerous and healthy cell types in heterogeneous tissue cross-sections [124].
This tumour heterogeneity creates the need for single-cell resolution in proteomics. Fluorescence-activated cell sorting (FACS) is an antibody-based technique used to sort a heterogeneous mixture of cells based on the expression of predetermined protein markers but comes with low multiplexability of markers. An MS-based alternative has emerged, called mass cytometry, and allows for dozens of protein markers to be monitored in individual cells. This is accomplished by coupling antibody probes to unique stable heavy metal isotopes rather than fluorophores [125]. The cells are nebulized by inductively-coupled plasma (ICP) and the metal ions provide the mass spectrometer a quantitative readout of a marker’s distribution within a sample, such as immune cell infiltration [126]. For example, single-cell mass cytometry was used to quantify a panel of 73 proteins related to tumour-immune cell signalling in 144 breast tumours and 50 healthy tissue samples. Higher grade tumours were found that have exhausted T cell counts and higher frequencies of PDL1+ tumor-associated macrophages [127]. The mass cytometry quantitative strategy can also be extended to intact tissue sections like MSI. Imaging mass cytometry was used to visualise the tumor microenvironment in FFPE CRC tissues with a focus on immune cell infiltration [128] or more recently to evaluate the single-cell pathology landscape of a large cohort of 352 breast cancer tissues [129].
Proteogenomics
Potentially the most powerful application of tissue proteomics to cancer research is using it in a concerted effort to complement genomics. This rapidly growing field is termed proteogenomics [130, 131]. Genomic research has significantly contributed to our understanding of cancer biology, through the identification of various cancer driver genes. It will be interesting to evaluate how some of these genomic aberrations modulate the cancer proteome. However, discovery proteomic analysis is dependent on reference databases of known peptides. Next-generation sequencing technologies can now provide comprehensive human genomes and transcriptomes that can detect single nucleotide polymorphisms and translocations. These variants are translated to unique proteoforms that would otherwise go undetected by traditional canonical sequence databases. Currently, MS-based detection of genomic aberrations is limited to only few cancer-specific variants, based on genomic estimations, due to the relatively low sequence coverage for each detected protein in a typical shotgun proteomic experiment. Future improvements in proteomics and computational approaches, such as open search algorithms like MSFragger [132], are expected to improve these detections. Alfaro et al. was able to combine publicly available databases with sample-specific genomic and transcriptomic data to interrogate proteomic data across 59 NCI cell lines. This resulted in the detection of 4771 mutations in 2200 gene products that would have otherwise escaped detection. This highlights the need for public availability of MS proteomics data so that expanded variant databases can be used for re-interrogation. In another example, Dimitrakopoulos et al. compared the DNA exome variants in 21 ER-positive breast cancer tissues to the proteomic data from the same samples [133]. This study demonstrated the limitations that still exist in the field as only 0.4% of these variants were detected at the proteome level. It was noteworthy that these detected variants belonged to the 6.3% most abundant mRNA transcripts which translated to many of the most abundant proteins. Nonetheless, the small subset of detectable variant peptides provides optimism that this class of potential biomarkers are well worth investigating in the future. Adding these variants to existing databases will make them more complete.
While the cancer genome and transcriptome of many cancers have been well elucidated, the cancer proteome and its relation to up-stream genomic alterations are poorly documented. In recent years, a growing number of studies have begun to integrate all levels of omics data to describe a comprehensive multi-omic assessment of tumours. These projects often require large collaborations between laboratories with unique skill sets, such as The Clinical Proteomic Tumor Analysis Consortium (CPTAC). This network was created by the National Cancer Institute to promote collaboration in an effort to accelerate our understanding of the molecular basis of cancer, with an initial focus on ovarian, breast and CRC (additional cancers have since been added). The consortium has produced increasingly complex multi-omic datasets, using standardized methods and applying them to growing cohort sizes of diverse surgical tissues. Stemming from the consortium, the Applied Proteogenomics Organizational Learning and Outcomes (APOLLO) network was created to bridge the CPTAC findings so that proteogenomic analysis becomes a routine component of personalized medicine. The International Cancer Proteogenome Consortium (ICPC) also works to bring cancer researchers together to share and compare data across 10 countries. The benefit of these networks is the validation of results in multiple centres. Irreproducibility in proteomics arises from differences in peptide digestion, pre-fractionation, chromatography, MS configuration and bioinformatics [134]. These aforementioned collaborations work to limit these factors with standard protocols and publicly available databases as detailed in the studies below, and summarized in Table 2.
Proteogenomic studies on breast cancer have recently demonstrated the ability to narrow down candidates for driver genes and to identify therapeutic targets. Mertins et al. [3] quantified the proteome and phosphoproteome of 105 breast cancer tissues which had previously been genomically annotated in a TCGA study. Trans-omic connections were made between cancer-centric pathways including CETN3 leading to elevated EGFR levels, or SKP1 loss leading to increased SRC expression. In another study, Johansson quantified nearly 10,000 proteins across all 45 breast cancer tumours prepared by FASP. Encouragingly, breast cancer subtypes were recapitulated by the proteomic data and among the subtypes with poor prognosis, and further classification was possible based on proteins related to immune infiltration. This study also included the mapping of protein products to non-coding genes which opens the door to new, tumour-specific, immunotherapeutic targets [135].
Human hepatocellular carcinoma has been investigated by a couple of proteogenomic studies. In collaboration with CPTAC, Gao et al. [136] applied TMT 11-plex to assess the proteome expression profiles from 165 patients with hepatitis-B virus-related hepatocellular carcinoma. Pathways related to tumour microenvironment regulation, cell proliferation and metabolic reprogramming, were studied in greater detail, and PYCR2 and ADH1A1 were identified as prognostic indicators of further patient subtyping. This study provides a valuable resource moving forward as the field acts to better understand liver cancer biology [136]. A group from the Chinese Human Proteome Project (CNHPP) consortium used proteomic and phosphoproteomics to stratify a cohort of 110 paired hepatocellular carcinoma and non-tumour tissues into subtypes with different clinical outcomes [137].
Sinha et al. [6] investigated the proteogenomic landscape of PCa through quantitation of the genome, epigenome, transcriptome and proteome. Label-free analysis of 76 localized, intermediate risk prostate tumours led to the quantitation of 7000 protein groups. This study led to several interesting observations. First, that established genomic subtypes of PCa converge on five proteomic subtypes, which are themselves associated with clinical outcomes. That ETS fusion genes, the most common mutation in prostate tumors, perturb the proteome and transcriptome in dramatically divergent ways, particularly influencing metabolic pathways. Similar to studies from CPTAC, that RNA abundance explains only ~ 10% of variability in protein levels in PCa, but there is a broad network of trans effects that converge on specific functional pathways and unique to this paper that biomarkers comprising genomic and proteomic features significantly out-perform those comprised of either molecular feature alone. Latonen et al. [138] also performed integrative proteomics in fresh frozen PCa tissues using a SWATH-MS strategy and also reported aberrations in the proteome cannot be reliably predicted by other omics datasets including gene copy number, DNA methylation and RNA expression. Similarly, in a 110 clear cell renal cell carcinoma study, it was reported that a handful of genes demonstrated an expected decrease in protein expression when the gene was increasingly methylated such as IQSEC1. On the other hand, this was not the case at the transcriptomic-level (mRNA), suggesting post-translational regulatory mechanisms were at play [136, 139].
Other notable cancer proteogenomic studies include the study of medulloblastoma [140], early-onset gastric cancer [141], lymphoblastic leukemia [142], and ovarian cancer [5]. Additionally, CPTAC has released proteogenomic datasets for colon and rectal cancers [4, 79]. While understanding cancer biology is critical, other studies have focussed on observing proteogenomic aberrations that may affect anti-cancer treatment responses. For example, CRC patients often receive treatment with anti-EGFR monoclonal antibodies. Yet, a study mined transcriptomic and proteomic data to validate that wild-type KRAS is necessary in tumours for effective treatment due to variant peptides [143]. Moving forward, the integration of proteomic datasets with genomic-level data will become increasingly common in future research of oncology and personalised medicine.
Proteomics of human body fluids
Blood-based proteomics
The promise of liquid biopsies is that they are thought to provide proteomic information representative of a given tumour or tissue type but can be collected in a less-invasive and longitudinal manner. In clinical laboratory assays, blood is the most widely used human body fluid in disease diagnosis, prognosis and treatment outcomes. Blood consists of cellular components (i.e. erythrocytes, thrombocytes, and lymphocytes) and a liquid component called plasma [144]. Blood is tested for various plasma proteins via enzymatic assays or antibody-based immunoassays. Plasma has a wide dynamic range of more than ten orders of magnitude in protein abundances, with only 22 proteins constituting 99% of the protein content [145]. These protein concentrations range from serum albumin (50 mg/mL), immunoglobulins and coagulation factors down to small protein hormones and cytokines (pg/mL) [144]. This large dynamic range has made it difficult to study the plasma proteome due to the masking of often low abundant potential disease biomarkers by the few very highly abundant proteins. Advances in MS-based proteomic detection technology and sample preparation have helped to partially overcome these issues.
Immunodepletion and fractionation has expanded the number of detected plasma proteins into the thousands [146]. Immunodepletion of high abundance proteins can be achieved through immunoaffinity-based [147,148,149] or dye-based depletion [150, 151]. Depletion methods have many limitations including off-target capture of other proteins or the depletion of proteins bound to abundant proteins like albumin [151]. More recently, Geyer et al. [152] performed a single-shot label free proteomic strategy from 1 µL of blood plasma through a single finger prick, without immunodepletion that resulted in detection of over 300 proteins. These included more than 40 FDA-approved biomarkers, inflammatory markers and gender-related proteins with high reproducibility.
After establishing the human proteome atlas project (HUPO) in 2001 [153], the Human plasma proteome project (HPPP) was initiated in 2002. The collaboration of 32 labs across 13 countries aims to generate an open source data repository of the human plasma and serum proteome via MS [154]. Additionally, the project evaluates various sample preparation workflows, MS instrumentations and analysis platforms across different laboratories. The multi-centre data was integrated and resulted in 9000 proteins detected by one or more peptides, or 3020 proteins detected more stringently by two or more peptides [154]. HPPP provided the first initiative for characterizing the human plasma proteome. More recently, Geyer et al. [155] acquired deep proteomic data of whole blood, platelet-enriched plasma and erythrocytes from 20 individual samples and compared it with the established plasma and serum proteomes. This resulted in the detection of more than 6000 proteins, and insights into the proteome of each blood compartment. The aim of the project was achieved in establishing a reference proteome which could identify the proteins from contaminating cell types and plasma.
Various studies have investigated the blood (plasma and serum) proteome from patients with various types of cancer, as summarized in Table 3. In one of these studies, Pan et al. [156] analysed the blood from healthy control and pancreatic cancer samples. The serum and plasma were isolated, immunodepleted and labelled with light and heavy-labelled acrylamide. The samples were pooled, fractionated by reversed-phase LC, tryptic digested and further fractionated by strong-cation-exchange (SCX) chromatography. This detected 1300 different proteins with several proteins differentially expressed in cancer compared to controls. Several of these differentially expressed proteins were confirmed using ELISA in an independent sample cohort with strong correlation to MS quantitation [157]. Proteins like TIMP1, ICAM1, AZGP1, APOA2 and LTF showed better predictive power than CA19-9 in differentiating the pancreatic samples from healthy controls, which is a gold standard blood biomarker for pancreatic cancer.
In a squamous cell lung carcinoma study, blood samples coming from the pulmonary artery and vein of noncancerous and cancerous lung regions were obtained during surgery. The plasma was immunodepleted and fractionated and peptides were quantified by iTRAQ. The results showed 50 proteins to be abundant in the vein draining part of the cancerous regions compared to the noncancerous sectors [158]. In another study done by Ahn et al. [159] one hundred plasma samples from different stages of CRC and healthy controls were pooled to create a SWATH library. Similarly, the abundant proteins were depleted and peptides were further fractionated. In total 37 proteins were differentially expressed in higher stages of CRC, and seven of these proteins were further validated by ELISA and western blot analysis.
Circulating tumor cells (CTC) are tumor cells that shed from the primary tumor into the circulatory system and can lead to metastasis in different organs, but provide a unique source of potential biomarkers [160]. The occurrence of CTCs in blood is quite rare, which is estimated to be one CTC per millilitre [161]. Different approaches have been reported for the isolation and detection of epithelial based CTCs through antibody-based EpCAM-coated ferromagnetic beads. This method has been FDA-approved for advanced PCa [162], breast cancer [163] and CRC [164] metastasis studies. The other CTC isolation methods include chip-based isolation [165, 166] and MagSweeper [167]. Alternative methods like micro-fluidic immunofluorescence [168] and microfluidic western blotting [169] detect a limited number of proteins in the CTC samples. Advances in MS-based analysis, especially recent single cell proteomics approaches [170] could potentially provide proteome-wide insights of these CTCs to identify novel protein markers for their detection and insights into tumour heterogeneity, cancer progression and treatment outcomes. Isolating minute amounts of these cells is a challenging task and all the methods mentioned above come with limitations such as leukocyte crosslinking [161], which leads to contamination of the target CTC proteome. Recent advancements in moving toward single-cell proteomics will improve coverage of the CTC proteome in future studies. A recent study by Li et al. [171] spiked varying amounts of MCF-7 cells in blood to mimic CTC amounts and were isolated with anti-EpCAM microbeads primed with antibodies specific to MCF-7 surface proteins. These captured MCF-7 cells were further prepared for LC–MS where 1327 proteins were detected from 50 spiked cells and 2026 proteins from 100 cells.
The plasma glycoproteome from various carcinomas were studied by Sajic et al. [172]. In this study, blood samples from 284 subjects from four different types of carcinomas (CRC, lung, PCa and pancreatic) and their own control groups were compared by SWATH-MS. This identified 1151 plasma glycoproteins from 4347 N-glycopeptides. Among the various expression similarities and differences that were revealed, an increased THBS1 expression was found to be common between all carcinomas. Cima et al. [173] analysed the effect of PTEN inactivation on the N-glycoproteome during PCa progression. The N-glycopeptides from serum and prostate tissue of wild-type and KO (PTEN) mice models were enriched and analysed by LC–MS. This generated a total of 757 N-Glycoproteins. The comparison between WT vs KO sera and tissue shortlisted 49 biomarker candidates. These candidate proteins were selected for SRM-based targeted assay in 143 patient serum samples (disease and control). In the end, 33 proteins were quantified by SRM in 80–105 patient samples. An additional nine proteins were validated by ELISA, totalling 39 proteins as potential PCa biomarkers.
In a recent study by Sinha et al. [87], potential biomarker discovery for high-grade serous ovarian carcinoma (HGSC) recurrence was performed using PDX models. Briefly, HGSC recurrent tumor tissues were engrafted into immune-compromised mice. Serum from unengrafted animals served as controls. N-glycopeptides were enriched from serum and tumor tissues using hydrazide chemistry and analyzed by label-free proteomics. This resulted in 3675 N-glycopeptides corresponding to 2200 proteins containing the Asn-X-Ser/Thr N-glycosylation sequon. Following species-assignment and bioinformatic prioritization the authors systematically developed targeted proteomics assays and applied them to two longitudinal cohorts of HGSC serum samples. This study reports four putative biomarkers for the early detection of HGSC recurrence. The study reports on a novel strategy for the discovery of tumor-derived proteins using a combination of N-glycoproteomics and PDX models. A similar study was performed by Hüttenhain et al. [174]. Genetically engineered ovarian cancer mouse models and control mice samples were used for the selection of N-glycoproteomic biomarker candidates which were quantified by SRM in 124 patient sera with epithelial ovarian cancer and 110 healthy controls. A protein signature consisting of IGHG2, L1CAM, THBS, DSG2 and LGALS3BP outperformed CA125, a known marker, in the detection of ovarian cancer.
Urine-based proteomics
Urine is another commonly sampled human body fluid because it is produced in large volumes and can be easily collected in a non-invasive manner. From a proteome perspective, urine is less complex than blood, with a narrower dynamic range, and is less prone to proteolytic degradation allowing for more stable storage over longer periods of time [175]. Improvements to LC–MS instrumentation have led to a series of studies reporting an increasing number of proteins that constitute the healthy human urine proteome, with Zhao et al. [176] reporting more than 6000 proteins. This has caught the attention of organizations seeking non-invasive biomarkers, including HUPO which has dedicated the Human Kidney and Urine Proteome Project (HKUPP) specifically to the analysis of urinary biomarkers [177]. The challenges associated with urinary proteomics studies include inter-patient variability since urine protein concentrations depend on kidney filtration and reabsorption performance which greatly fluctuates within a population. Secondly, intra-patient variability needs further characterization because urinary protein concentrations are affected by time of day, exercise, diet and age. For example, a study used capillary electrophoresis (CE-MS) to profile the urinary peptidomes and found that the expression of 112 urinary peptides strongly correlated with age in both healthy and diseased groups (mostly originating from collagen, uromodulin and fibrinogen) [178]. As such, the planning and selection of patient cohorts is an important component of urine proteomics studies in the future. Lastly, targeted approaches are highly applicable to liquid biopsy proteomic studies but do come with some caveats. Fu et al. [179] highlighted that target selection is crucial for accurate quantitation when they quantified twelve uromodulin (the most abundant protein in healthy urine) peptides by SRM in urine samples. However, only four were robustly correlated with ELISA protein concentrations due to the unpredictable confounding factors of clinical samples such as proteoform complexity. The urine-based proteomic studies detailed below are also summarized in Table 3.
It is estimated that 70% of urine proteins originate from the kidney and urinary tracts. As such, this proximity makes urine a valuable resource for monitoring urinary tract cancers [180]. Currently, bladder cancer relies on the detection of urinary NMP22 [181] by ELISA but this test suffers from low sensitivity. A second ELISA test for BTA [182] lacks specificity since the protein is also detected at high concentrations in blood that would yield a false-positive result in patients with poor filtration performance. CE-MS was used by Frantzi et al. [45] to develop a panel of peptide markers that distinguish primary from recurrent urothelial bladder cancer. The multi-centre discovery cohort was followed by a validation cohort combining 1357 patients. Intensities were normalized by 29 internal standard peptides. The predictive power of the current standard, cytology data, was augmented when combined with this peptide panel. Ortiz et al. [183] applied a shotgun approach to the study of urines from 115 kidney cancer patients. While distinctions could be made between healthy and cancer patient samples, the analysis demonstrated further diagnostic power by revealing EHD4 expression is elevated in clear-cell renal cell carcinoma relative to a benign oncocytoma. These techniques have also been applied to childhood cancers. Wilms tumour is the most common form of childhood kidney cancer and gel electrophoresis was used to fractionate urinary proteins from 49 patients followed by LC–MS. After validation in a larger cohort by ELISA, it was determined that prohibitin can be used for early disease detection, non-invasive monitoring of disease progression and as a target to block chemoresistance [184].
The remaining 30% of urinary proteins are from the glomerular filtration of blood suggesting that urine can also provide insight into cancers of distant organs [180]. A multi-centre, multi-disease study analyzed the proteomes of 231 patients by label-free proteomics. Prior to tryptic digestion, the urine samples were denatured to further release proteins from the uromodulin network. From this study, a protein signature was developed that not only distinguished lung cancer patients from healthy controls and other benign lung conditions such as pneumonia, but it was also from the urinary proteomes of other cancer types including: lung, bladder, cervical, CRC, esophageal, and gastric cancers [185].
Since the urethra passes through the prostate, urine is a valuable source of PCa biomarkers. A desirable trait of PCa markers is that they originate from the prostate rather than from other organs in the male urogenital tract. The Early Detection Research Network (EDRN) has studied post-DRE (digital rectal exam) urines which has been shown to contain a trove of potential biomarkers indicative of PCa status, including a non-coding RNA transcript, PCA3 [186]. However, these proteins are often lower abundance in urine creating the need for targeted MS quantitation. This approach was used by Shi et al. [187] and demonstrated that it is feasible to accurately multiplex the quantitation of 10 low-abundance, PCa-associated proteins in clinical urines. While detection of these proteins is not as feasible for shotgun proteomics, urine can also be collected after a digital rectal exam (DRE), a standard diagnostic test for patients with suspected PCa (termed post-DRE urine). The idea is that a DRE expels a small amount of prostatic secretions, a fluid often referred to as expressed prostatic secretions [14] that can then be collected within urine. Post-DRE urine is usually collected as the first flow, first catch urine (~ 50 ml) following a DRE. Kim et al. [188] used a shotgun approach to identify 232 proteotypic peptides that were differentially expressed between organ-confined and extracapsular PCa in expressed prostatic secretions. These peptides were then used to systematically develop targeted proteomics assays for evaluation in post-DRE urines. Briefly, SRM-MS assays were developed using synthetic, stable-isotope labelled peptides and subsequently applied to two independent cohorts of post-DRE urines. Statistical approaches were applied to develop clinical predictive models for PCa diagnosis (PCa patients vs. controls) and prognosis (patients with organ-confined compared to men with extracapsular disease). This study provided evidence that computationally guided proteomics in combination with richly annotated urine cohorts can discover highly accurate non-invasive biomarkers [13, 188].
Urinary extracellular vesicles (EVs) have attracted significant interest in recent years [189]. Although many different types of EVs (i.e. exosomes, microvesicles, etc.) are naturally released from healthy cells, their rate of release and cargo expression are reprogrammed in cancer to promote proliferation and metastasis while modulating the tumour microenvironment and immune response. These nanovesicles can be collected from urine through differential centrifugation, sucrose gradient density ultracentrifugation or filtration techniques [190]. iTRAQ was used to quantify 3500 proteins from the EV’s in post-DRE urine of PCa patients and identified FABP5 as a potential high-risk PCa marker. This differential expression was further validated by MS in cell line models, MRM quantitation and IHC staining [191]. A similar study was performed by Sequeiros et al. [192] who used a targeted MS approach to quantify 64 EV proteins by SRM from a larger cohort of 107 post-DRE urines composed of healthy men and those suffering from low-risk and high-risk PCa. A combination of two proteins (ADSV and TGM4) distinguished PCa patients from healthy controls, while a panel of five proteins (CD63, GLPK5, SPHMPSA, and PAPP) accurately classified patients by risk group that would help guide further treatment. The diagnostic panel was further verified by tissue microarray in prostate tissues.
Alternative liquid biopsies sources
Aside from blood and urine, there are a variety of alternative human body fluids that could potentially be used for biomarker discovery. Some of these non-conventional fluid samples are rich sources of organ-specific proteins due to their close proximity, but often come at a cost of invasive sample collection. Thus, they may not be as applicable to routine clinical practices such as early cancer detection or longitudinal monitoring of cancer progression. The aforementioned prostatic secretions (often referred to as expressed prostatic secretion—EPS) is a fluid naturally produced by the prostate. The EPS proteome is hence a rich source of prostate-derived proteins and this fluid’s proteome has been directly profiled in the absence of a urinary background [14, 193,194,195]. One limitation of EPS is that it is in general only collected prior to radical prostatectomy, making it not suitable for routine clinical assays. It nevertheless provides an opportunity to discover prostate-derived biomarkers that can be further evaluated in post-DRE urines by targeted proteomics assays. Similarly, cerebrospinal fluid (CSF) surrounds the brain and spinal cord providing mechanical and immunological protection but is not sampled as often as blood in brain cancer treatments due to its invasive collection. Nonetheless, CSF cannot be overlooked as a valuable biomarker source since it was recently demonstrated to contain more than 3300 total proteins, with an enrichment in brain-specific proteins [196]. Spreafico et al. [17] analyzed the CSF proteome of 40 patients and identified a panel of six proteins that distinguished metastatic pediatric brain cancer from healthy controls. Likewise, ascites fluid collection from the peritoneal cavity is invasive but it has been sampled to detect biomarkers indicative of malignant gastric cancer [197], ovarian cancer [18, 19], hepatocellular carcinoma [198], changes in the N-glycoproteome related to epithelial ovarian cancer [199], and the proteomes of tumour cells derived from post-chemotherapy ovarian cancers [200].
Meanwhile, there exists a subset of alternative bodily fluid types that can be collected more readily but their proteomes have not been characterized as thoroughly as more conventional body fluids. Tears provide an intriguing biomarker source and only recently have there been efforts to characterize its proteome [16]. Tears from both eyes of eight healthy controls were analyzed and unsurprisingly, a significant proportion of the identified proteins were enzymes [201]. Similarly to urine, a subset of tear protein expressions have been reported to correlate with age [202]. While tear proteins are sampled more commonly for ocular-related diseases, some efforts have been made to associate tear proteins with primary breast cancer using MALDI [203]. Multiple studies report characterization of the saliva proteome since it is a good proxy for oral cancers. These studies often focus on the most common oral cancer, oral squamous cell carcinoma, with more than 1000 proteins regularly detected in a sample of saliva [204]. Label-free quantitation identified 22 overexpressed proteins in oral cancer patient saliva with resistin correlating with advanced stage and metastasis [15]. Finally, proteomic studies of stool samples have shown that feces are another potential source of biomarkers. Most notably, CRC escapes early detection since existing IHC tests demonstrate limited sensitivity. Komor et al. [205] performed LC–MS/MS on a cohort of nearly 300 fecal samples from healthy controls, adenoma patients, and CRC patients. A panel of proteins included haptoglobin, LAMP1, SYNE2, LRG1, RBP4, FN1 and ANXA6 and was able to distinguish the cancerous patients from controls with a high degree of specificity and sensitivity. Haptoglobin was further verified as a biomarker in a large (n = 795) validation cohort by antibody-based assays. Similarly, Bosch et al. [206] used MS to detect 834 proteins in feces, of which 29 were statistically enriched in CRC patient samples. Combinations of these new potential biomarker candidates even outperformed the current IHC standard hemoglobin.
Moving forward, supplementary inter- and intra-patient variability studies will be needed to confirm proper sample collection protocols and patient selection. As clinical fluid sample preparation methods become more established and standardized, these alternative body fluid proteomes will be characterized in further detail as part of the search for robust non-invasive cancer biomarkers.
Conclusions and future directions
In summary, recent advances in biobanking and proteomics technologies now enable the robust profiling of clinical samples to unprecedented depth. To become a mainstream technology in clinical laboratories, similar to well-established genomics assays, proteomics technologies and investigators must embark on large-scale, possibly multi-institutional validations studies. Targeted proteomics assays are likely to play a central role for the validation of tissue or fluid-based biomarkers. Technologies such as MRM-MS (or more recently PRM-MS) are already firmly established in clinical biochemistry laboratories around the world for the detection and quantification of small molecules. The establishment of protein assays is the next logical step, but requires rigorous assay development metrics and large richly annotated validation cohorts.
Recent proteogenomics studies have also demonstrated the complementarity of proteomics and genomics technologies. Interrogation of biomolecules along the central dogma are expected to provide novel biological insights, multi-omics biomarkers and possible novel drug targets. While a handful of impressive studies have been published in recent years these have mainly focused on technical aspects of proteogenomics. What is missing is proteogenomics studies with a clear clinical question and large tissue cohorts to arrive at statistically powered conclusions. Another significant bottleneck of proteogenomics is the current lack of appropriate analyses strategies. While the field is capable of generating these large datasets, computational analysis strategies will require further improvements. This will require close collaboration between genomics, proteomics and data science/statistics investigators.
A unique feature of proteomics technologies is the ability to detect subcellular localizations [207], protein complexes [208] and post-translational modifications [209]. These so-called proteoforms [8] must be directly detected at the protein level and cannot be simply predicted from upstream genomics/transcriptomics data. For example, proteogenomics technologies have recently demonstrated great utility in the area of immune-oncology, in particular for the detection of druggable tumor-specific antigens [210]. A major hurdle for the development of cancer vaccines and T cell-based immunotherapies is the direct detection of MHC-associated neoantigens. Proteomics technologies have been developed for their isolation and MS-based detection. As these technologies further mature, including the development of more advanced analysis pipelines, direct clinical impact is to be expected.
Moving forward, the field of clinical proteomics is likely to see a rapid expansion of clinical cohort sizes as a result of standardized, high-throughput sample preparation techniques. This will minimize the frequency of studies that suffer from statistical underpowering and improve efficiency of translating biomarker candidates and drug targets to clinical application. Proteomics will increasingly become a critical part of cancer systems biology that integrate multi-omics data from genomics, epigenomics, transcriptomics and PTMs. This will create demands for superior computing power to handle and analyze increasingly large amounts of data. Further improvements in MS instrument sensitivity and speed will make deep proteome coverage more regularly attainable, especially without the need for extensive pre-fractionation. Improvements in detection/quantitation levels will also allow clinical proteomics to expand towards minimal input material and single-cell proteomics. Finally, data analysis pipeline will continue to enable the detection of protein panels and signatures that provide more diagnostics and prognostic accuracy relative to singular markers. These advancements will all be required for MS-based clinical proteomics to reach its full potential in translating research discoveries to improvements to clinical practice.
Availability of data and materials
Not applicable.
Abbreviations
- ACN:
-
Acetonitrile
- ADC:
-
Adenocarcinoma
- APOLLO:
-
Applied Proteogenomics Organizational Learning and Outcomes
- CDX:
-
Cell derived Xenografts
- CE-MS:
-
Capillary electrophoresis
- CID:
-
Collision-induced dissociation
- CNHPP:
-
Chinese Human Proteome Project
- CPTAC:
-
Clinical Proteomic Tumor Analysis Consortium
- CSF:
-
Cerebrospinal fluid
- CRC:
-
Colorectal cancer
- CTC:
-
Circulating tumor cell
- DDA:
-
Data dependent acquisition
- DIA:
-
Data independent acquisition
- DRE:
-
Digital rectal exam
- ECM:
-
Extracellular matrix
- EDRN:
-
Early Detection Research Network
- ELISA:
-
Enzyme-linked immunosorbent assay
- EPI:
-
Epigenetic
- EPS:
-
Expressed prostatic secretion
- ESI:
-
Electrospray ionization
- ETD:
-
Electron-transfer dissociation
- EVs:
-
Extracellular vesicles
- FACS:
-
Fluorescence assisted cell sorting
- FAIMS:
-
High-field asymmetric ion mobility spectrometry
- FASP:
-
Filter-aided sample preparation
- FF:
-
Fresh frozen
- FFPE:
-
Formalin-fixed, Paraffin-embedded
- GEN:
-
Genomic
- GLY:
-
Glycoproteomics
- HCD:
-
Higher-energy collisional dissociation
- HGSC:
-
High-grade serous ovarian carcinoma
- HKUPP:
-
Human Kidney and Urine Proteome Project
- HPPP:
-
Human plasma proteome project
- HRAM:
-
High-resolution, accurate-mass
- HUPO:
-
Human Proteome Atlas Project
- ICPC:
-
International Cancer Proteogenome Consortium
- IEF:
-
Isoelectric focussing
- IHC:
-
Immunohistochemistry
- iRT:
-
Indexed retention time
- iTRAQ:
-
Isobaric tag for relative and absolute quantitation
- LC:
-
Liquid chromatography
- LCM:
-
Laser capture-microdissection
- LLOQ:
-
Lowest limit of quantification
- LOD:
-
Limit of detection
- mAb:
-
Monoclonal antibody
- MALDI:
-
Matrix-assisted laser desorption ionization
- MRM:
-
Multiple reaction monitoring
- MS:
-
Mass spectrometry
- MSI:
-
Mass spectrometry imaging
- MW:
-
Molecular weight
- OCT:
-
Optimal cutting temperature
- OF:
-
Orbitrap Fusion
- OV:
-
Orbitrap Velos
- PDX:
-
Patient-derived xenografts
- PHO:
-
Phosphoproteomic
- PRM:
-
Parallel reaction monitoring
- PSA:
-
Prostate-specific antigen
- PCa:
-
Prostate cancer
- PTMs:
-
Post-translational modifications
- PVDF:
-
Polyvinylidene difluoride
- QE:
-
Q-Exactive
- RPF:
-
Reversed-phase fractionation
- SAX:
-
Strong-anion-exchange
- SCX:
-
Strong-cation-exchange
- SDC:
-
Sodium deoxycholate
- SDS:
-
Sodium dodecyl sulfate
- SILAC:
-
Stable isotope labeling with amino acids in cell culture
- SPS:
-
Synchronous precursor selection
- sqCC:
-
Squamous cell carcinoma
- S-trap:
-
Suspension trapping
- SWATH-MS:
-
Sequential Window Acquisition of All Theoretical Mass Spectra
- TCGA:
-
The Cancer Genome Atlas
- TFE:
-
2,2,2-Trifluoroethanol
- TIMS:
-
Trapped ion mobility spectrometry
- TMT:
-
Tandem mass tag
- TNBC:
-
Triple negative breast cancer
- TOF:
-
Time-of-flight
- TRA:
-
Transcriptomic
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30.
Yaffe MB. Why geneticists stole cancer research even though cancer is primarily a signaling disease. Sci Signal. 2019;12(565):1–3.
Mertins P, Mani DR, Ruggles KV, Gillette MA, Clauser KR, Wang P, et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. 2016;534(7605):55–62.
Zhang B, Wang J, Wang X, Zhu J, Liu Q, Shi Z, et al. Proteogenomic characterization of human colon and rectal cancer. Nature. 2014;513(7518):382–7.
Zhang H, Liu T, Zhang Z, Payne SH, Zhang B, McDermott JE, et al. Integrated proteogenomic characterization of human high-grade serous ovarian cancer. Cell. 2016;166(3):755–65.
Sinha A, Huang V, Livingstone J, Wang J, Fox NS, Kurganovs N, et al. The Proteogenomic Landscape of Curable Prostate Cancer. Cancer Cell. 2019;35(3):414–27.
Aebersold R, Agar JN, Amster IJ, Baker MS, Bertozzi CR, Boja ES, et al. How many human proteoforms are there? Nat Chem Biol. 2018;14(3):206–14.
Smith LM, Kelleher NL. Proteoform: a single term describing protein complexity. Nat Methods. 2013;10(3):186–7.
Łupicka-Słowik A, Grzywa R, Leporowska E, Procyk D, Oleksyszyn J, Sieńczyk M. Development and evaluation of an immunoglobulin Y-based ELISA for measuring prostate specific antigen in human serum. Ann Lab Med. 2019;39(4):373–80.
Vyberg M, Nielsen S, Røge R, Sheppard B, Ranger-Moore J, Walk E, et al. Immunohistochemical expression of HER2 in breast cancer: socioeconomic impact of inaccurate tests. BMC Health Serv Res. 2015;15(1):1–9.
Kim JI, Choi KU, Lee IS, Choi YJ, Kim WT, Shin DH, et al. Expression of hypoxic markers and their prognostic significance in soft tissue sarcoma. Oncol Lett. 2015;9(4):1699–706.
Hillen F, Baeten CIM, Van De Winkel A, Creytens D, Van Der Schaft DWJ, Winnepenninckx V, et al. Leukocyte infiltration and tumor cell plasticity are parameters of aggressiveness in primary cutaneous melanoma. Cancer Immunol Immunother. 2008;57(1):97–106.
Principe S, Kim Y, Fontana S, Ignatchenko V, Nyalwidhe JO, Lance RS, et al. Identification of prostate-enriched proteins by in-depth proteomic analyses of expressed prostatic secretions in urine. J Proteome Res. 2012;11(4):2386–96.
Drake RR, Elschenbroich S, Lopez-Perez O, Kim Y, Ignatchenko V, Ignatchenko A, et al. In-depth proteomic analyses of direct expressed prostatic secretions. J Proteome Res. 2010;9(5):2109–16.
Wu CC, Chu HW, Hsu CW, Chang KP, Liu HP. Saliva proteome profiling reveals potential salivary biomarkers for detection of oral cavity squamous cell carcinoma. Proteomics. 2015;15(19):3394–404.
de Souza GA, Godoy LMF, Mann M. Identification of 491 proteins in the tear fluid proteome reveals a large number of proteases and protease inhibitors. Genome Biol. 2006;7(8):1–11.
Spreafico F, Bongarzone I, Pizzamiglio S, Magni R, Taverna E, De Bortoli M, et al. Proteomic analysis of cerebrospinal fluid from children with central nervous system tumors identifies candidate proteins relating to tumor metastatic spread. Oncotarget. 2017;8(28):46177–90.
Elschenbroich S, Ignatchenko V, Clarke B, Kalloger SE, Boutros PC, Gramolini AO, et al. In-depth proteomics of ovarian cancer ascites: combining shotgun proteomics and selected reaction monitoring mass spectrometry. J Proteome Res. 2011;10(5):2286–99.
Gortzak-Uzan L, Ignatchenko A, Evangelou AI, Agochiya M, Brown KA, St. Onge P, et al. A proteome resource of ovarian cancer ascites: integrated proteomic and bioinformatic analyses to identify putative biomarkers. J Proteome Res. 2008;7(1):339–51.
Georges LMC, De Wever O, Galván JA, Dawson H, Lugli A, Demetter P, et al. Cell line derived xenograft mouse models are a suitable in vivo model for studying tumor budding in colorectal cancer. Front Med. 2019;6:1–7.
Guo S, Jiang X, Mao B, Li QX. The design, analysis and application of mouse clinical trials in oncology drug development. BMC Cancer. 2019;19(1):1–14.
Collins AT, Lang SH. A systematic review of the validity of patient derived xenograft (PDX) models: the implications for translational research and personalised medicine. PeerJ. 2018;2018(11):1–22.
Wang H, Qian WJ, Mottaz HM, Clauss TRW, Anderson DJ, Moore RJ, et al. Development and evaluation of a micro- and nanoscale proteomic sample preparation method. J Proteome Res. 2005;4(6):2397–403.
Eckert MA, Coscia F, Chryplewicz A, Chang JW, Hernandez KM, Pan S, et al. Proteomics reveals NNMT as a master metabolic regulator of cancer-associated fibroblasts. Nature. 2019;569(7758):723–8.
Manza LL, Stamer SL, Ham AJL, Codreanu SG, Liebler DC. Sample preparation and digestion for proteomic analyses using spin filters. Proteomics. 2005;5(7):1742–5.
Wiśniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6(5):359–62.
Hughes CS, Moggridge S, Müller T, Sorensen PH, Morin GB, Krijgsveld J. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat Protoc. 2019;14(1):68–85.
Distler U, Kuharev J, Navarro P, Tenzer S. Label-free quantification in ion mobility-enhanced data-independent acquisition proteomics. Nat Protoc. 2016;11(4):795–812.
Wiśniewski JR, Ostasiewicz P, Mann M. High recovery FASP applied to the proteomic analysis of microdissected formalin fixed paraffin embedded cancer tissues retrieves known colon cancer markers. J Proteome Res. 2011;10(7):3040–9.
Wis̈niewski JR, Dus̈-Szachniewicz K, Ostasiewicz P, Ziólkowski P, Rakus D, Mann M. Absolute proteome analysis of colorectal mucosa, adenoma, and cancer reveals drastic changes in fatty acid metabolism and Plasma membrane transporters. J Proteome Res. 2015;14(9):4005–18.
Iglesias-Gato D, Wikström P, Tyanova S, Lavallee C, Thysell E, Carlsson J, et al. The Proteome of Primary Prostate Cancer. Eur Urol. 2016;69(5):942–52.
Iglesias-Gato D, Thysell E, Tyanova S, Crnalic S, Santos A, Lima TS, et al. The proteome of prostate cancer bone metastasis reveals heterogeneity with prognostic implications. Clin Cancer Res. 2018;24(21):5433–44.
Berger ST, Ahmed S, Muntel J, Polo NC, Bachur R, Kentsis A, et al. MStern blotting-high throughput polyvinylidene fluoride (PVDF) membrane-based proteomic sample preparation for 96-well plates. Mol Cell Proteomics. 2015;14(10):2814–23.
Zougman A, Selby PJ, Banks RE. Suspension trapping (STrap) sample preparation method for bottom-up proteomics analysis. Proteomics. 2014;14(9):1000–6.
Hailemariam M, Eguez RV, Singh H, Bekele S, Ameni G, Pieper R, et al. S-trap, an ultrafast sample-preparation approach for shotgun proteomics. J Proteome Res. 2018;17(9):2917–24.
Ludwig KR, Schroll MM, Hummon AB. Comparison of in-solution, FASP, and S-trap based digestion methods for bottom-up proteomic studies. J Proteome Res. 2018;17(7):2480–90.
Hughes CS, Foehr S, Garfield DA, Furlong EE, Steinmetz LM, Krijgsveld J. Ultrasensitive proteome analysis using paramagnetic bead technology. Mol Syst Biol. 2014;10(10):757.
Davis S, Scott C, Ansorge O, Fischer R. Development of a Sensitive, Scalable Method for Spatial, Cell-Type-Resolved Proteomics of the Human Brain. J Proteome Res. 2019;18(4):1787–95.
Sielaff M, Kuharev J, Bohn T, Hahlbrock J, Bopp T, Tenzer S, et al. Evaluation of FASP, SP3, and iST protocols for proteomic sample preparation in the low microgram range. J Proteome Res. 2017;16(11):4060–72.
Moggridge S, Sorensen PH, Morin GB, Hughes CS. Extending the compatibility of the SP3 paramagnetic bead processing approach for proteomics. J Proteome Res. 2018;17(4):1730–40.
Leutert M, Rodríguez-Mias RA, Fukuda NK, Villén J. R2–P2 rapid-robotic phosphoproteomics enables multidimensional cell signaling studies. Mol Syst Biol. 2019;15(12):1–20.
Hughes CS, Mcconechy MK, Cochrane DR, Nazeran T, Karnezis AN, Huntsman DG, et al. Quantitative profiling of single formalin fixed tumour sections: proteomics for translational research. Sci Rep. 2016;6:1–14.
Owen DR, Wong HL, Bonakdar M, Jones M, Hughes CS, Morin GB, et al. Molecular characterization of ERBB2- amplified colorectal cancer identifies potential mechanisms of resistance to targeted therapies: a report of two instructive cases. Cold Spring Harb Mol Case Stud. 2018;4(2):1–18.
Kulak NA, Pichler G, Paron I, Nagaraj N, Mann M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat Methods. 2014;11(3):319–24.
Frantzi M, Van Kessel KE, Zwarthoff EC, Marquez M, Rava M, Malats N, et al. Development and validation of urine-based peptide biomarker panels for detecting bladder cancer in a multi-center study. Clin Cancer Res. 2016;22(16):4077–86.
Lee HJ, Kim HJ, Liebler DC. Efficient microscale basic reverse phase peptide fractionation for global and targeted proteomics. J Proteome Res. 2016;15(7):2346–54.
Cooper HJ. To what extent is FAIMS beneficial in the analysis of proteins? J Am Soc Mass Spectrom. 2016;27(4):566–77.
Michelmann K, Silveira JA, Ridgeway ME, Park MA. Fundamentals of trapped ion mobility spectrometry. J Am Soc Mass Spectrom. 2014;26(1):14–24.
Hebert AS, Prasad S, Belford MW, Bailey DJ, Mcalister GC, Abbatiello SE, et al. Comprehensive single-shot proteomics with FAIMS on a hybrid orbitrap mass spectrometer. Anal Chem. 2018;90:9529–37.
Meier F, Geyer PE, Virreira Winter S, Cox J, Mann M. BoxCar acquisition method enables single-shot proteomics at a depth of 10,000 proteins in 100 minutes. Nat Methods. 2018;15(6):440–8.
Wewer Albrechtsen NJ, Geyer PE, Doll S, Treit PV, Bojsen-Møller KN, Martinussen C, et al. Plasma proteome profiling reveals dynamics of inflammatory and lipid homeostasis markers after Roux-En-Y gastric bypass surgery. Cell Syst. 2018;7(6):601–12.
Wichmann C, Meier F, Winter SV, Brunner AD, Cox J, Mann M. MaxQuant.live enables global targeting of more than 25,000 peptides. Mol Cell Proteomics. 2019;18(5):982–94.
Purvine S, Eppel JT, Yi EC, Goodlett DR. Shotgun collision-induced dissociation of peptides using a time of flight mass analyzer. Proteomics. 2003;3(6):847–50.
Venable JD, Dong MQ, Wohlschlegel J, Dillin A, Yates JR. Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat Methods. 2004;1(1):39–45.
Meyer JG, Schilling B. Clinical applications of quantitative proteomics using targeted and untargeted data-independent acquisition techniques. Expert Rev Proteomics. 2017;14(5):419–29.
Gillet LC, Navarro P, Tate S, Röst H, Selevsek N, Reiter L, et al. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics. 2012;11(6):1–17.
Kelstrup CD, Bekker-Jensen DB, Arrey TN, Hogrebe A, Harder A, Olsen JV. Performance evaluation of the Q exactive HF-X for shotgun proteomics. J Proteome Res. 2018;17(1):727–38.
Röst HL, Rosenberger G, Navarro P, Gillet L, Miladinoviä SM, Schubert OT, et al. OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat Biotechnol. 2014;32(3):219–23.
Searle BC, Pino LK, Egertson JD, Ting YS, Lawrence RT, MacLean BX, et al. Chromatogram libraries improve peptide detection and quantification by data independent acquisition mass spectrometry. Nat Commun. 2018;9(1):1–12.
Collins BC, Hunter CL, Liu Y, Schilling B, Rosenberger G, Bader SL, et al. Multi-laboratory assessment of reproducibility, qualitative and quantitative performance of SWATH-mass spectrometry. Nat Commun. 2017;8(1):1–11.
Rosenberger G, Koh CC, Guo T, Röst HL, Kouvonen P, Collins BC, et al. A repository of assays to quantify 10,000 human proteins by SWATH-MS. Sci Data. 2014;1:1–15.
Song Y, Zhong L, Zhou J, Lu M, Xing T, Ma L, et al. Data-independent acquisition-based quantitative proteomic analysis reveals potential biomarkers of kidney cancer. Proteomics Clin Appl. 2017;11(11–12):1–10.
Porter CJ, Proctor CJ, Beynon JH. The dependence of collision induced fragmentation pattern on the internal energy of the precursor ion. Org Mass Spectrom. 1981;16(2):62–7.
Olsen JV, Macek B, Lange O, Makarov A, Horning S, Mann M. Higher-energy C-trap dissociation for peptide modification analysis. Nat Methods. 2007;4(9):709–12.
Jedrychowski MP, Huttlin EL, Haas W, Sowa ME, Rad R, Gygi SP. Evaluation of HCD- and CID-type fragmentation within their respective detection platforms for murine phosphoproteomics. Mol Cell Proteomics. 2011;10(12):1–20.
Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci USA. 2004;101(26):9528–33.
Frese CK, Altelaar AFM, Van Den Toorn H, Nolting D, Griep-Raming J, Heck AJR, et al. Toward full peptide sequence coverage by dual fragmentation combining electron-transfer and higher-energy collision dissociation tandem mass spectrometry. Anal Chem. 2012;84(22):9668–73.
Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov. 2010;9(10):767–74.
Hernandez-Alba O, Houel S, Hessmann S, Erb S, Rabuka D, Huguet R, et al. A case study to identify the drug conjugation site of a site-specific antibody-drug-conjugate using middle-down mass spectrometry. J Am Soc Mass Spectrom. 2019;30(11):2419–29.
Fornelli L, Ayoub D, Aizikov K, Liu X, Damoc E, Pevzner PA, et al. Top-down analysis of immunoglobulin G isotypes 1 and 2 with electron transfer dissociation on a high-field Orbitrap mass spectrometer. J Proteomics. 2017;159:67–76.
Tran BQ, Barton C, Feng J, Sandjong A, Yoon SH, Awasthi S, et al. Comprehensive glycosylation profiling of IgG and IgG-fusion proteins by top-down MS with multiple fragmentation techniques. J Proteomics. 2016;134:93–101.
Chen B, Lin Z, Zhu Y, Jin Y, Larson E, Xu Q, et al. Middle-down multi-attribute analysis of antibody-drug conjugates with electron transfer dissociation. Anal Chem. 2019;91(18):11661–9.
Skinner OS, Catherman AD, Early BP, Thomas PM, Compton PD, Kelleher NL. Fragmentation of integral membrane proteins in the gas phase. Anal Chem. 2014;86(9):4627–34.
Catherman AD, Durbin KR, Ahl DR, Early BP, Fellers RT, Tran JC, et al. Large-scale top-down proteomics of the human proteome: membrane proteins, mitochondria, and senescence. Mol Cell Proteomics. 2013;12(12):3465–73.
Wang X, He Y, Ye Y, Zhao X, Deng S, He G, et al. SILAC-based quantitative MS approach for real-time recording protein-mediated cell-cell interactions. Sci Rep. 2018;8(1):1–9.
Kotowski U, Erović BM, Schnöll J, Stanek V, Janik S, Steurer M, et al. Quantitative proteome analysis of Merkel cell carcinoma cell lines using SILAC. Clin Proteomics. 2019;16:1–13.
Pozniak Y, Balint-Lahat N, Rudolph JD, Lindskog C, Katzir R, Avivi C, et al. System-wide clinical proteomics of breast cancer reveals global remodeling of tissue homeostasis. Cell Syst. 2016;2(3):172–84.
Noberini R, Osti D, Miccolo C, Richichi C, Lupia M, Corleone G, et al. Extensive and systematic rewiring of histone post-translational modifications in cancer model systems. Nucleic Acids Res. 2018;46(8):3817–32.
Vasaikar S, Huang C, Wang X, Petyuk VA, Savage SR, Wen B, et al. Proteogenomic analysis of human colon cancer reveals new therapeutic opportunities. Cell. 2019;177(4):1035–1049.e19.
Yan B, Chen B, Min S, Gao Y, Zhang Y, Xu P, et al. iTRAQ-based comparative serum proteomic analysis of prostate cancer patients with or without bone metastasis. J Cancer. 2019;10(18):4165–77.
Erickson BK, Rose CM, Braun CR, Erickson AR, Knott J, McAlister GC, et al. A strategy to combine sample multiplexing with targeted proteomics assays for high-throughput protein signature characterization. Mol Cell. 2017;65(2):361–70.
Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26(12):1367–72.
Schilling B, Rardin MJ, Maclean BX, Zawadzka AM, Frewen BE, Cusack MP, et al. Quantitation of proteomic data using MS1 extracted ion chromatograms in skyline. Mol Cell Proteomics. 2012;11(5):202–14.
Latosinska A, Vougas K, Makridakis M, Klein J, Mullen W, Abbas M, et al. Comparative analysis of label-free and 8-plex iTRAQ approach for quantitative tissue proteomic analysis. PLoS ONE. 2015;10(9):1–25.
Gallien S, Duriez E, Crone C, Kellmann M, Moehring T, Domon B. Targeted proteomic quantification on quadrupole-orbitrap mass spectrometer. Mol Cell Proteomics. 2012;11(12):1709–23.
Peterson AC, Russell JD, Bailey DJ, Westphall MS, Coon JJ. Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol Cell Proteomics. 2012;11(11):1475–88.
Sinha A, Hussain A, Ignatchenko V, Ignatchenko A, Tang KH, Ho VWH, et al. N-Glycoproteomics of patient-derived xenografts: a strategy to discover tumor-associated proteins in high-grade serous ovarian cancer. Cell Syst. 2019;8(4):345–51.
Song E, Gao Y, Wu C, Shi T, Nie S, Fillmore TL, et al. Targeted proteomic assays for quantitation of proteins identified by proteogenomic analysis of ovarian cancer. Sci Data. 2016;2017(4):1–13.
You J, Kao A, Dillon R, Croner LJ, Benz R, Blume JE, et al. A large-scale and robust dynamic MRM study of colorectal cancer biomarkers. J Proteomics. 2017;2018(187):80–92.
Guo T, Li L, Zhong Q, Rupp NJ, Charmpi K, Wong CE, et al. Multi-region proteome analysis quantifies spatial heterogeneity of prostate tissue biomarkers. Life Sci Alliance. 2018;1(2):1–15.
Shao W, Guo T, Toussaint NC, Xue P, Wagner U, Li L, et al. Comparative analysis of mRNA and protein degradation in prostate tissues indicates high stability of proteins. Nat Commun. 2019;10(1):1–8.
Yanovich G, Agmon H, Harel M, Sonnenblick A, Peretz T, Geiger T. Clinical proteomics of breast cancer reveals a novel layer of breast cancer classification. Cancer Res. 2018;78(20):6001–10.
Tyanova S, Albrechtsen R, Kronqvist P, Cox J, Mann M, Geiger T. Proteomic maps of breast cancer subtypes. Nat Commun. 2016;7:1–11.
Bassani-Sternberg M, Bräunlein E, Klar R, Engleitner T, Sinitcyn P, Audehm S, et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat Commun. 2016;7:1–16.
Harel M, Ortenberg R, Varanasi SK, Mangalhara KC, Mardamshina M, Markovits E, et al. Proteomics of melanoma response to immunotherapy reveals mitochondrial dependence. Cell. 2019;179(1):236–50.
Doll S, Kriegmair MC, Santos A, Wierer M, Coscia F, Neil HM, et al. Rapid proteomic analysis for solid tumors reveals LSD1 as a drug target in an end-stage cancer patient. Mol Oncol. 2018;12(8):1296–307.
He W, Zhang H, Wang Y, Zhou Y, Luo Y, Cui Y, et al. CTHRC1 induces non-small cell lung cancer (NSCLC) invasion through upregulating MMP-7/MMP-9. BMC Cancer. 2018;18(1):1–14.
Li L, Wei Y, To C, Zhu CQ, Tong J, Pham NA, et al. Integrated Omic analysis of lung cancer reveals metabolism proteome signatures with prognostic impact. Nat Commun. 2014;5:1–12.
Li QK, Shah P, Tian Y, Hu Y, Roden RBS, Zhang H, et al. An integrated proteomic and glycoproteomic approach uncovers differences in glycosylation occupancy from benign and malignant epithelial ovarian tumors. Clin Proteomics. 2017;14(1):1–9.
Dieters-Castator DZ, Rambau PF, Kelemen LE, Siegers GM, Lajoie GA, Postovit LM, et al. Proteomics-derived biomarker panel improves diagnostic precision to classify endometrioid and high-grade serous ovarian carcinoma. Clin Cancer Res. 2019;25(14):4309–19.
Sepiashvili L, Waggott D, Hui A, Shi W, Su S, Ignatchenko A, et al. Integrated omic analysis of oropharyngeal carcinomas reveals human papillomavirus (HPV)—Dependent regulation of the activator protein 1 (AP-1) pathway. Mol Cell Proteomics. 2014;13(12):3572–84.
Bouchal P, Schubert OT, Faktor J, Capkova L, Imrichova H, Zoufalova K, et al. Breast cancer classification based on proteotypes obtained by SWATH mass spectrometry. Cell Rep. 2019;28(3):832–43.
Guo T, Kouvonen P, Koh CC, Gillet LC, Wolski WE, Röst HL, et al. Rapid mass spectrometric conversion of tissue biopsy samples into permanent quantitative digital proteome maps. Nat Med. 2015;21(4):407–13.
Zhu Y, Zhu J, Lu C, Zhang Q, Xie W, Sun P, et al. Identification of protein abundance changes in hepatocellular carcinoma tissues Using PCT–SWATH. Proteomics Clin Appl. 2019;13(1):1–13.
Coscia F, Lengyel E, Duraiswamy J, Ashcroft B, Bassani-Sternberg M, Wierer M, et al. Multi-level proteomics identifies CT45 as a chemosensitivity mediator and immunotherapy target in ovarian cancer. Cell. 2018;175(1):159-170.e16.
Yang S, Chen L, Chan DW, Li QK, Zhang H. Protein signatures of molecular pathways in non-small cell lung carcinoma (NSCLC): comparison of glycoproteomics and global proteomics. Clin Proteomics. 2017;14(1):1–15.
Brognard J, Hunter T. Protein kinase signaling networks in cancer. Curr Opin Genet Dev. 2011;21(1):4–11.
Wei W, Shin YS, Xue M, Matsutani T, Masui K, Yang H, et al. Single-cell phosphoproteomics resolves adaptive signaling dynamics and informs targeted combination therapy in glioblastoma. Cancer Cell. 2016;29(4):563–73.
Mertins P, Tang LC, Krug K, Clark DJ, Gritsenko MA, Chen L, et al. Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography-mass spectrometry. Nat Protoc. 2018;13(7):1632–61.
Zagorac I, Fernandez-Gaitero S, Penning R, Post H, Bueno MJ, Mouron S, et al. In vivo phosphoproteomics reveals kinase activity profiles that predict treatment outcome in triple-negative breast cancer. Nat Commun. 2018;9(1):1–15.
He J, Sun X, Shi T, Schepmoes AA, Fillmore TL, Petyuk VA, et al. Antibody-independent targeted quantification of TMPRSS2-ERG fusion protein products in prostate cancer. Mol Oncol. 2014;8(7):1169–80.
Serna G, Ruiz-Pace F, Cecchi F, Fasani R, Jimenez J, Thyparambil S, et al. Targeted multiplex proteomics for molecular prescreening and biomarker discovery in metastatic colorectal cancer. Sci Rep. 2019;9(1):1–10.
Guerin M, Gonçalves A, Toiron Y, Baudelet E, Pophillat M, Granjeaud S, et al. Development of parallel reaction monitoring (PRM)-based quantitative proteomics applied to HER2-Positive breast cancer. Oncotarget. 2018;9(73):33762–77.
Macur K, Hagen L, Ciesielski TM, Konieczna L, Skokowski J, Jenssen BM, et al. A targeted mass spectrometry immunoassay to quantify osteopontin in fresh-frozen breast tumors and adjacent normal breast tissues. J Proteomics. 2019;208:103469.
Sprung RW, Martinez MA, Carpenter KL, Ham AJL, Washington MK, Arteaga CL, et al. Precision of multiple reaction monitoring mass spectrometry analysis of formalin-fixed, paraffin-embedded tissue. J Proteome Res. 2012;11(6):3498–505.
Jimenez CR, Zhang H, Kinsinger CR, Nice EC. The cancer proteomic landscape and the HUPO cancer proteome project. Clin Proteomics. 2018;15(1):1–7.
Berglund L, Bjo E, Oksvold P, Fagerberg L, Asplund A, Szigyarto CA, et al. A genecentric human protein atlas for expression profiles based on antibodies. Mol Cell Proteomics. 2019;7(10):2019–27.
Carnielli CM, Macedo CCS, De Rossi T, Granato DC, Rivera C, Domingues RR, et al. Combining discovery and targeted proteomics reveals a prognostic signature in oral cancer. Nat Commun. 2018;9(1):1–17.
Zavalin A, Yang J, Hayden K, Vestal M, Caprioli RM. Tissue protein imaging at 1 µm laser spot diameter for high spatial resolution and high imaging speed using transmission geometry MALDI TOF MS. Anal Bioanal Chem. 2017;407(8):2337–42.
Liu X, Flinders C, Mumenthaler SM, Hummon AB. MALDI mass spectrometry imaging for evaluation of therapeutics in colorectal tumor organoids. J Am Soc Mass Spectrom. 2018;29(3):516–26.
Powers TW, Holst S, Wuhrer M, Mehta AS, Drake RR. Two-dimensional N-glycan distribution mapping of hepatocellular carcinoma tissues by MALDI-imaging mass spectrometry. Biomolecules. 2015;5(4):2554–72.
Jones EE, Powers TW, Neely BA, Cazares LH, Troyer DA, Parker AS, et al. MALDI imaging mass spectrometry profiling of proteins and lipids in clear cell renal cell carcinoma. Proteomics. 2014;14(7–8):924–35.
Angel PM, Comte-Walters S, Ball LE, Talbot K, Mehta A, Brockbank KGM, et al. Mapping Extracellular matrix proteins in formalin-fixed, paraffin-embedded tissues by MALDI imaging mass spectrometry. J Proteome Res. 2018;17(1):635–46.
Hoffmann F, Umbreit C, Krüger T, Pelzel D, Ernst G, Kniemeyer O, et al. Identification of proteomic markers in head and neck cancer using MALDI–MS imaging, LC–MS/MS, and immunohistochemistry. Proteomics Clin Appl. 2019;13(1):1–10.
Danova M, Torchio M, Comolli G, Sbrana A, Antonuzzo A, Mazzini G. The role of automated cytometry in the new era of cancer immunotherapy (Review). Mol Clin Oncol. 2018;9(4):355–61.
Zhang T, Lv J, Tan Z, Wang B, Warden AR, Li Y, et al. Immunocyte profiling using single-cell mass cytometry reveals EpCAM + CD4+ T cells abnormal in colon cancer. Front Immunol. 2019;10:1–12.
Wagner J, Rapsomaniki MA, Chevrier S, Anzeneder T, Langwieder C, Dykgers A, et al. A single-cell atlas of the tumor and immune ecosystem of human breast cancer. Cell. 2019;177(5):1330–1345.e18.
Ijsselsteijn ME, van der Breggen R, Sarasqueta AF, Koning F, de Miranda NFCC. A 40-marker panel for high dimensional characterization of cancer immune microenvironments by imaging mass cytometry. Front Immunol. 2019;10(OCT):1–8.
Jackson HW, Fischer JR, Zanotelli VRT, Ali HR, Mechera R, Soysal SD, et al. The single-cell pathology landscape of breast cancer. Nature. 2020;578(7796):615–20.
Nesvizhskii AI. Proteogenomics: concepts, applications and computational strategies. Nat Methods. 2014;11(11):1114–25.
Alfaro JA, Sinha A, Kislinger T, Boutros PC. Onco-proteogenomics: cancer proteomics joins forces with genomics. Nat Methods. 2014;11(11):1107–13.
Kong AT, Leprevost FV, Avtonomov DM, Mellacheruvu D, Nesvizhskii AI. MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat Methods. 2017;14(5):513–20.
Dimitrakopoulos L, Prassas I, Sieuwerts AM. Proteome-wide onco-proteogenomic somatic variant identi fi cation in ER- positive breast cancer. Clin Biochem. 2019;66:63–75.
Tabb DL, Wang X, Carr SA, Clauser KR, Mertins P, Chambers MC, et al. Reproducibility of differential proteomic technologies in CPTAC fractionated xenografts. J Proteome Res. 2016;15(3):691–706.
Johansson HJ, Socciarelli F, Vacanti NM, Haugen MH, Zhu Y, Siavelis I, et al. Breast cancer quantitative proteome and proteogenomic landscape. Nat Commun. 2019;10(1):1–14.
Gao Q, Zhu H, Dong L, Shi W, Chen R, Song Z, et al. Integrated proteogenomic characterization of HBV-related hepatocellular carcinoma. Cell. 2019;179(2):561–77.
Jiang Y, Sun A, Zhao Y, Ying W, Sun H, Yang X, et al. Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature. 2019;567(7747):257–61.
Latonen L, Afyounian E, Jylhä A, Nättinen J, Aapola U, Annala M, et al. Integrative proteomics in prostate cancer uncovers robustness against genomic and transcriptomic aberrations during disease progression. Nat Commun. 2018;9(1):1–13.
Clark DJ, Dhanasekaran SM, Petralia F, Pan J, Song X, Hu Y, et al. Integrated proteogenomic characterization of clear cell renal cell carcinoma. Cell. 2019;179(4):964–983.
Forget A, Martignetti L, Puget S, Calzone L, Brabetz S, Picard D, et al. Aberrant ERBB4-SRC signaling as a hallmark of group 4 medulloblastoma revealed by integrative phosphoproteomic profiling. Cancer Cell. 2018;34(3):379–395.e7.
Mun DG, Bhin J, Kim S, Kim H, Jung JH, Jung Y, et al. Proteogenomic characterization of human early-onset gastric cancer. Cancer Cell. 2019;35(1):111–124.e10.
Yang M, Vesterlund M, Siavelis I, Moura-Castro LH, Castor A, Fioretos T, et al. Proteogenomics and Hi-C reveal transcriptional dysregulation in high hyperdiploid childhood acute lymphoblastic leukemia. Nat Commun. 2019;10(1):1–15.
Woo S, Cha SW, Bonissone S, Na S, Tabb DL, Pevzner PA, et al. Advanced proteogenomic analysis reveals multiple peptide mutations and complex immunoglobulin peptides in colon cancer. J Proteome Res. 2015;14(9):3555–67.
Geyer PE, Holdt LM, Teupser D, Mann M. Revisiting biomarker discovery by plasma proteomics. Mol Syst Biol. 2017;13(9):942.
Tirumalai RS, Chan KC, Prieto DRA, Issaq HJ, Conrads TP, Veenstra TD. Characterization of the low molecular weight human serum proteome. Mol Cell Proteomics. 2003;2(10):1096–103.
Keshishian H, Burgess MW, Gillette MA, Mertins P, Clauser KR, Mani DR, et al. Multiplexed, quantitative workflow for sensitive biomarker discovery in plasma yields novel candidates for early myocardial injury. Mol Cell Proteomics. 2015;14(9):2375–93.
Steel LF, Trotter MG, Nakajima PB, Mattu TS, Gonye G, Block T. Efficient and specific removal of albumin from human serum samples. Mol Cell Proteomics. 2003;2(4):262–70.
Björhall K, Miliotis T, Davidsson P. Comparison of different depletion strategies for improved resolution in proteomic analysis of human serum samples. Proteomics. 2005;5(1):307–17.
Gundry RL, White MY, Nogee J, Tchernyshyov I, Van Eyk JE. Assessment of albumin removal from an immunoaffinity spin column: critical implications for proteomic examination of the albuminome and albumin-depleted samples. Proteomics. 2009;9(7):2021–8.
Ahmed N, Barker G, Oliva K, Garfin D, Talmadge K, Georgiou H, et al. An approach to remove albumin for the proteomic analysis of low abundance biomarkers in human serum. Proteomics. 2003;3(10):1980–7.
Bellei E, Bergamini S, Monari E, Fantoni LI, Cuoghi A, Ozben T, et al. High-abundance proteins depletion for serum proteomic analysis: concomitant removal of non-targeted proteins. Amino Acids. 2011;40(1):145–56.
Geyer PE, Kulak NA, Pichler G, Holdt LM, Teupser D, Mann M. Plasma proteome profiling to assess human health and disease. Cell Syst. 2016;2(3):185–95.
Hanash S, Celis JE. The Human Proteome Organization: a mission to advance proteome knowledge. Mol Cell Proteomics. 2002;1(6):413–4.
Omenn GS, States DJ, Adamski M, Blackwell TW, Menon R, Hermjakob H, et al. Overview of the HUPO Plasma Proteome Project: results from the pilot phase with 35 collaborating laboratories and multiple analytical groups, generating a core dataset of 3020 proteins and a publicly-available database. Proteomics. 2005;5(13):3226–45.
Geyer PE, Voytik E, Treit PV, Doll S, Kleinhempel A, Niu L, et al. Plasma Proteome Profiling to detect and avoid sample-related biases in biomarker studies. EMBO Mol Med. 2019;11(11):1–22.
Pan S, Chen R, Crispin DA, May D, Stevens T, McIntosh MW, et al. Protein alterations associated with pancreatic cancer and chronic pancreatitis found in human plasma using global quantitative proteomics Profiling. J Proteome Res. 2011;10(5):2359–76.
Pleskow DK, Berger HJ, Gyves J, Allen E, McLean A, Podolsky DK. Evaluation of a serologic marker, CA19-9, in the diagnosis of pancreatic cancer. Ann Intern Med. 1989;110(9):704–9.
Geary B, Walker MJ, Snow JT, Lee DCH, Pernemalm M, Maleki-Dizaji S, et al. Identification of a biomarker panel for early detection of lung cancer patients. J Proteome Res. 2019;18(9):3369–82.
Ahn SB, Sharma S, Mohamedali A, Mahboob S, Redmond WJ, Pascovici D, et al. Potential early clinical stage colorectal cancer diagnosis using a proteomics blood test panel. Clin Proteomics. 2019;16(1):1–20.
Kim MY, Oskarsson T, Acharyya S, Nguyen DX, Zhang XHF, Norton L, et al. Tumor self-seeding by circulating cancer cells. Cell. 2009;139(7):1315–26.
Krebs MG, Metcalf RL, Carter L, Brady G, Blackhall FH, Dive C. Molecular analysis of circulating tumour cells—biology and biomarkers. Nat Rev Clin Oncol. 2014;11(3):129–44.
De Bono JS, Scher HI, Montgomery RB, Parker C, Miller MC, Tissing H, et al. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin Cancer Res. 2008;14(19):6302–9.
Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC, et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med. 2004;351(8):781–91.
Cohen SJ, Punt CJA, Iannotti N, Saidman BH, Sabbath KD, Gabrail NY, et al. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26(19):3213–21.
Nagrath S, Sequist LV, Maheswaran S, Bell DW, Irimia D, Ulkus L, et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature. 2007;450(7173):1235–9.
Chen H. A triplet parallelizing spiral microfluidic chip for continuous separation of tumor cells. Sci Rep. 2018;8(1):1–8.
Talasaz AH, Powell AA, Huber DE, Berbee JG, Roh KH, Yu W, et al. Isolating highly enriched populations of circulating epithelial cells and other rare cells from blood using a magnetic sweeper device. Proc Natl Acad Sci USA. 2009;106(10):3970–5.
Lu Y, Xue Q, Eisele MR, Sulistijo ES, Brower K, Han L, et al. Highly multiplexed profiling of single-cell effector functions reveals deep functional heterogeneity in response to pathogenic ligands. Proc Natl Acad Sci U S A. 2015;112(7):E607–15.
Sinkala E, Sollier-Christen E, Renier C, Rosàs-Canyelles E, Che J, Heirich K, et al. Profiling protein expression in circulating tumour cells using microfluidic western blotting. Nat Commun. 2017;8:1–12.
Zhu Y, Scheibinger M, Ellwanger DC, Krey JF, Choi D, Kelly RT, et al. Single-cell proteomics reveals changes in expression during hair-cell development. Elife. 2019;8:1–26.
Li S, Plouffe BD, Belov AM, Ray S, Wang X, Murthy SK, et al. An integrated platform for isolation, processing, and mass spectrometry-based proteomic profiling of rare cells in whole blood. Mol Cell Proteomics. 2015;14(6):1672–83.
Sajic T, Liu Y, Arvaniti E, Surinova S, Williams EG, Schiess R, et al. Similarities and differences of blood N-glycoproteins in five solid carcinomas at localized clinical stage analyzed by SWATH-MS. Cell Rep. 2018;23(9):2819–2831.e5.
Cima I, Schiess R, Wild P, Kaelin M, Schüffler P, Lange V, et al. Cancer genetics-guided discovery of serum biomarker signatures for diagnosis and prognosis of prostate cancer. Proc Natl Acad Sci USA. 2011;108(8):3342–7.
Hüttenhain R, Choi M, de la Fuente LM, Oehl K, Chang CY, Zimmermann AK, et al. A targeted mass spectrometry strategy for developing proteomic biomarkers: a case study of epithelial ovarian cancer. Mol Cell Proteomics. 2019;18(9):1836–50.
Hüttenhain R, Soste M, Selevsek N, Röst H, Sethi A, Carapito C, et al. Reproducible quantification of cancer-associated proteins in body fluids using targeted proteomics. Sci Transl Med. 2012;4(142):142ra94.
Zhao M, Li M, Yang Y, Guo Z, Sun Y, Shao C, et al. A comprehensive analysis and annotation of human normal urinary proteome. Sci Rep. 2017;7(1):1–13.
Hirao Y, Saito S, Fujinaka H, Miyazaki S, Xu B, Quadery AF, et al. Proteome profiling of diabetic mellitus patient urine for discovery of biomarkers by comprehensive MS-based proteomics. Proteomes. 2018;6(1):9.
Nkuipou-Kenfack E, Bhat A, Klein J, Jankowski V, Mullen W, Vlahou A, et al. Identification of ageing-associated naturally occurring peptides in human urine. Oncotarget. 2015;6(33):34106–17.
Fu Q, Grote E, Zhu J, Jelinek C, Köttgen A, Coresh J, et al. An empirical approach to signature peptide choice for selected reaction monitoring: quantification of uromodulin in urine. Clin Chem. 2016;62(1):198–207.
Spahr C, Davis M, McGinley MD, Robinson JH, Bures EJ, Beierle J, et al. Towards defining the urinary proteome using liquid chromatography-tandem mass spectrometry I. Profiling an unfractionated tryptic digest. Proteomics. 2001;1(1):93–107.
Wang Z, Que H, Suo C, Han Z, Tao J, Huang Z, et al. Evaluation of the NMP22 BladderChek test for detecting bladder cancer: a systematic review and meta-analysis. Oncotarget. 2017;8(59):100648–56.
Guo A, Wang X, Gao L, Shi J, Sun C, Wan Z. Bladder tumour antigen (BTA stat) test compared to the urine cytology in the diagnosis of bladder cancer: a meta-analysis. Can Urol Assoc J. 2014;8(5–6):E347–52.
Di Meo A, Batruch I, Brown MD, Yang C, Finelli A, Jewett MA, et al. Searching for prognostic biomarkers for small renal masses in the urinary proteome. Int J Cancer. 2020;146(8):2315–25.
Ortiz MV, Ahmed S, Burns M, Henssen AG, Hollmann TJ, MacArthur I, et al. Prohibitin is a prognostic marker and therapeutic target to block chemotherapy resistance in Wilms’ tumor. JCI Insight. 2019;4(15):e127098.
Zhang C, Leng W, Sun C, Lu T, Chen Z, Men X, et al. Urine proteome profiling predicts lung cancer from control cases and other tumors. EBioMedicine. 2018;30:120–8.
Tomlins SA, Day JR, Lonigro RJ, Hovelson DH, Siddiqui J, Kunju LP, et al. Urine TMPRSS2:eRG Plus PCA3 for individualized prostate cancer risk assessment. Eur Urol. 2016;70(1):45–53.
Shi T, Quek SI, Gao Y, Nicora CD, Nie S, Fillmore TL, et al. Multiplexed targeted mass spectrometry assays for prostate cancer-associated urinary proteins. Oncotarget. 2017;8(60):101887–98.
Kim Y, Jeon J, Mejia S, Yao CQ, Ignatchenko V, Nyalwidhe JO, et al. Targeted proteomics identifies liquid-biopsy signatures for extracapsular prostate cancer. Nat Commun. 2016;7:1–10.
Principe S, Jones EE, Kim Y, Sinha A, Nyalwidhe JO, Brooks J, et al. In-depth proteomic analyses of exosomes isolated from expressed prostatic secretions in urine. Proteomics. 2013;13(10–11):1667–71.
Merchant ML, Rood IM, Deegens JKJ, Klein JB. Isolation and characterization of urinary extracellular vesicles: implications for biomarker discovery. Nat Rev Nephrol. 2017;13(12):731–49.
Fujita K, Kume H, Matsuzaki K, Kawashima A, Ujike T, Nagahara A, et al. Proteomic analysis of urinary extracellular vesicles from high Gleason score prostate cancer. Sci Rep. 2017;7:1–9.
Sequeiros T, Rigau M, Chiva C, Montes M, Garcia-Grau I, Garcia M, et al. Targeted proteomics in urinary extracellular vesicles identifies biomarkers for diagnosis and prognosis of prostate cancer. Oncotarget. 2017;8(3):4960–76.
Li R, Guo Y, Han BM, Yan X, Utleg AG, Li W, et al. Proteomics cataloging analysis of human expressed prostatic secretions reveals rich source of biomarker candidates. Proteomics Clin Appl. 2008;2(4):543–55.
Zhao T, Zeng X, Bateman NW, Sun M, Teng PN, Bigbee WL, et al. Relative quantitation of proteins in expressed prostatic secretion with a stable isotope labeled secretome standard. J Proteome Res. 2012;11(2):1089–99.
Kim Y, Ignatchenko V, Yao CQ, Kalatskaya I, Nyalwidhe JO, Lance RS, et al. Identification of differentially expressed proteins in direct expressed prostatic secretions of men with organ-confined versus extracapsular prostate cancer. Mol Cell Proteomics. 2012;11(12):1870–84.
Macron C, Lane L, Núnez Galindo A, Dayon L. Deep dive on the proteome of human cerebrospinal fluid: a valuable data resource for biomarker discovery and missing protein identification. J Proteome Res. 2018;17(12):4113–26.
Jin J, Son M, Kim H, Kim H, Kong SH, Kim HK, et al. Comparative proteomic analysis of human malignant ascitic fluids for the development of gastric cancer biomarkers. Clin Biochem. 2018;56:55–61.
Zhang J, Liang R, Wei J, Ye J, He Q, Chunlingyuan, et al. Identification of candidate biomarkers in malignant ascites from patients with hepatocellular carcinoma by iTRAQ-based quantitative proteomic analysis. Biomed Res Int. 2018;2018:5484976.
Biskup K, Braicu EI, Sehouli J, Tauber R, Blanchard V. The ascites N-glycome of epithelial ovarian cancer patients. J Proteomics. 2017;157:33–9.
Ahmed N, Greening D, Samardzija C, Escalona RM, Chen M, Findlay JK, et al. Unique proteome signature of post-chemotherapy ovarian cancer ascites-derived tumor cells. Sci Rep. 2016;6:1–13.
Dor M, Eperon S, Lalive PH, Guex-Crosier Y, Hamedani M, Salvisberg C, et al. Investigation of the global protein content from healthy human tears. Exp Eye Res. 2018;2019(179):64–74.
Nättinen J, Jylhä A, Aapola U, Mäkinen P, Beuerman R, Pietilä J, et al. Age-associated changes in human tear proteome. Clin Proteomics. 2019;16(1):1–11.
Böhm D, Keller K, Pieter J, Boehm N, Wolters D, Siggelkow W, et al. Comparison of tear protein levels in breast cancer patients and healthy controls using a de novo proteomic approach. Oncol Rep. 2012;28(2):429–38.
Sivadasan P, Kumar Gupta M, Sathe GJ, Balakrishnan L, Palit P, Gowda H, et al. Data from human salivary proteome—A resource of potential biomarkers for oral cancer. Data Br. 2015;4:374–8.
Komor MA, Bosch LJW, Coupé VMH, Rausch C, Pham TV, Piersma SR, et al. Proteins in stool as biomarkers for non-invasive detection of colorectal adenomas with high risk of progression. J Pathol. 2020;250(3):288–98.
Bosch LJW, De Wit M, Pham TV, Coupé VMH, Hiemstra AC, Piersma SR, et al. Novel stool-based protein biomarkers for improved colorectal cancer screening. Ann Intern Med. 2017;167(12):855–66.
Kislinger T, Cox B, Kannan A, Chung C, Hu P, Ignatchenko A, et al. Global survey of organ and organelle protein expression in mouse: combined proteomic and transcriptomic profiling. Cell. 2006;125(1):173–86.
Gingras AC, Abe KT, Raught B. Getting to know the neighborhood: using proximity-dependent biotinylation to characterize protein complexes and map organelles. Curr Opin Chem Biol. 2018;2019(48):44–54.
Doll S, Gnad F, Mann M. The case for proteomics and phospho-proteomics in personalized cancer medicine. Proteomics Clin Appl. 2019;13(2):1–10.
Laumont CM, Vincent K, Hesnard L, Audemard É, Bonneil É, Laverdure JP, et al. Noncoding regions are the main source of targetable tumor-specific antigens. Sci Transl Med. 2018;10(470):eaau5516.
Acknowledgements
The authors thank all members of the Kislinger labs for helpful suggestions.
Funding
This study was supported by an operating grant from the National Cancer Institute Early Detection Research Network (1U01CA214194-01). This research was funded in part by the Ontario Ministry of Health and Long-Term Care.
Author information
Authors and Affiliations
Contributions
AM and SK contributed equally. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Yes.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Macklin, A., Khan, S. & Kislinger, T. Recent advances in mass spectrometry based clinical proteomics: applications to cancer research. Clin Proteom 17, 17 (2020). https://doi.org/10.1186/s12014-020-09283-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12014-020-09283-w