
Abstract
Background
Rare denovo variants represent a significant cause of neurodevelopmental delay and intellectual disability (ID).
Methods
Exome sequencing was performed on 4351 patients with global developmental delay, seizures, microcephaly, macrocephaly, motor delay, delayed speech and language development, or ID according to Human Phenotype Ontology (HPO) terms. All patients had previously undergone whole exome sequencing as part of diagnostic genetic testing with a focus on variants in genes implicated in neurodevelopmental disorders up to January 2017. This resulted in a genetic diagnosis in 1336 of the patients. In this study, we specifically searched for variants in 14 recently implicated novel neurodevelopmental disorder (NDD) genes.
Results
We identified 65 rare, protein-changing variants in 11 of these 14 novel candidate genes. Fourteen variants in CDK13, CHD4, KCNQ3, KMT5B, TCF20, and ZBTB18 were scored pathogenic or likely pathogenic. Of note, two of these patients had a previously identified cause of their disease, and thus, multiple molecular diagnoses were made including pathogenic/likely pathogenic variants in FOXG1 and CDK13 or in TMEM237 and KMT5B.
Conclusions
Looking for pathogenic variants in newly identified NDD genes enabled us to provide a molecular diagnosis to 14 patients and their close relatives and caregivers. This underlines the relevance of re-evaluation of existing exome data on a regular basis to improve the diagnostic yield and serve the needs of our patients.
Similar content being viewed by others

Background
Major congenital malformations, which include neurodevelopmental disorders (NDDs), are present in ~ 2–5% of children [1]. Children with NDD have variable severity of phenotypic features and different behavioral abnormalities. Often times, NDD arises from de-novo variants in genes important for central nervous system (CNS) development [2]. Whole exome sequencing has been critical and effective in diagnosing patients with NDD. Thus, treatment for NDD has become more refined through molecular genetic diagnosis rather than phenotype-driven management of symptoms [3]. Herein, we find novel pathogenic or likely pathogenic variants in six recently identified NDD genes, namely CDK13, CHD4, KCNQ3, KMT5B, TCF20, and ZBTB18.
Methods
Patients
From a total of 26,119 in-house exome data set, we included 4351 unrelated NDD patients in this study. Human Phenotype Ontology (HPO) nomenclature [4] was applied based on the clinical data provided by referring physician. In the context of this manuscript, NDD was defined by HPO terms described in Additional file 1: Figure S1. Patients had an average age of 7.75 (STD 8.04) years (Additional file 1: Table S1). All patients had previously undergone whole exome sequencing as part of their clinical genetic testing, following previously reported procedures [5]. These tests focused on NDD genes established before January 2017. Parents were available from 2030 patients to test for de novo occurrence of variants. Written informed consent was obtained from participants, and this study was approved by the Ethical Commission of the University of Rostock (registry no. A2015-0102). All samples were processed in Centogene’s laboratory, which is CAP and CLIA certified, adhering to the American College of Medical Genetics and Genomics (ACMG) guidelines [6].
Genetic testing
Patient DNA was extracted from EDTA blood or from dry blood spots in filter cards. WES was performed on the IonProton (n = 911 samples, enrichment with Ion AmpliSeq Exome RDY Kit (Life Technologies, Carlsbad, CA, USA)) or Illumina (n = 3440 samples, enrichment with Illumina’s NexteraRapid Capture Exome Kit (Illumina, Inc., San Diego, CA, USA)). Sequencing and bioinformatics were done as previously described [5, 7, 8]. We focused on genes of interest (fourteen recently nominated genes by the DDD study [9]; Additional file 1: Figure S1), filtered for rare variants (MAF < 0.0001), and an effect on the encoded protein sequence. Sanger validations were performed for all indels and variants with quality Phred score below 300 to rule out false-positive variants as previously described [5]. Further, we applied the ACMG criteria to score the pathogenicity of candidate variants [6].
Results
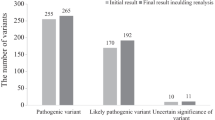
Among all 4351 NDD patients, we identified 65 heterozygous variant carriers (1.5%), for 65 different rare, protein sequence-changing variants in 11 out of 14 genes recently nominated by the DDD study [9] (Additional file 1: Figure S1 and Table S2). In 11 of 12 carriers for whom parents were available, the variant was shown to be de novo, and in one case (KCNQ3:p.Arg364Cys) inherited from the father whereby his affection status is unknown. The variant CDK13: p.His675Arg was found in two affected siblings. For all other patients, no relatives were available for testing. The 65 variants were either not present or at very low frequency (< 2.76 × 10− 4 frequency) in unaffected “in-house” exomes or in public databases (ExAC, GnomAD). Using ACMG recommendations, six of these 65 variants were scored as pathogenic (CDK13:p.Tyr351fs, CDK13:p.Gln544*, CDK13:p.Asn842Ser, KMT5B:p.Pro106fs, KMT5B:p.Ser116fs, and KCNQ3:p.Arg230Cys) and eight as likely pathogenic (CDK13:p.Thr500Met, CDK13:p.Asn843Ile, CDK13:p.Gly712Arg, CDK13:p.Tyr716Cys, CHD4:p.Lys634Arg, KMT5B:p.Ter394fs, ZBTB18:p.Arg436His, and TCF20:p.Pro1147Leu) (Table 1). The remaining 51 variants (78%) were categorized as variants of uncertain significance (VUS) (Additional file 1: Table S2 and S5; Fig. 1). This included a de novo splice region variant in KMT5B (c.-140+4T>G) which was predicted in silico (using HumanSplicingFinder and MaxEntScan) to results in alternative splicing for transcript NM_001300907.1. However, a fresh sample from this patient was not available to test for alterations in splicing. Patients’ clinical characteristics were compared across CDK13 and KMT5B variant carriers (Additional file 1: Figure S2 and S3).

a-f Composite figures of genes with pathogenic or likely pathogenic variants identified in this study: CDK13, CHD4, KCNQ3, KMT5B, TCF20, and ZBTB18 (adapted from the “Prevalence and architecture of de novo mutations in developmental disorders” study [9]). Boxes: pink highlighted variants are VUS and red highlighted variants are pathogenic or likely pathogenic changes. Functional domains of the encoded protein are indicated by blue boxes. Variants that have already been identifeid in the previous study are shown with red branching
There were two patients who had previously received a genetic diagnosis and thus carried an additional pathogenic variant in a previously established NDD gene (Additional file 1: Table S3). Thus, these two patients each carried multiple molecular diagnoses. This included a patient with a frameshift variant in FOXG1 (OMIM number 613454) and a missense change in CDK13 (OMIM number 603309) who had a complex phenotype beyond typical Rett-like syndrome presentation including MRI abnormalities and visual impairment. This patient also had delayed motor and language development, intellectual disability, muscular hypotonia, microcephaly, ventricular septal defect, failure to thrive and squint which aligns with the OMIM phenotype of congenital heart defects, dysmorphic facial features, and intellectual developmental disorder (CHDFIDD). The onset was at birth, and her parents were non-consanguineous, and there were no other affected siblings.
Another patient carried a homozygous c.869+1G>A variant in TMEM237 (OMIM number 614424) and a frameshift variant c.1180_*1delTAAG (p.Ter394fs) in KMT5B (OMIM number 617788). This male patient has been suspected to be affected with Joubert syndrome which is known to be linked to biallelic TMEM237 variants, and had defective vision and global developmental delay. Whether there is an additional contribution of the likely pathogenic KMT5B variant to the phenotype is difficult to determine, although some features overlap with the OMIM phenotype of mental retardation.
Discussion
In this study, we identified pathogenic/likely pathogenic variants in 14 NDD patients in six different, recently identified genes. Our findings highlight the importance of reanalyzing and revisiting exome sequencing data to reclassify variants of uncertain significance by taking into account novel observations published in the scientific literature. Since the initial study [9], 13 of the 14 investigated genes, with the exception of MSL3, have independently been replicated [10,11,12,13,14,15,16,17,18,19,20,21,22,23] including CDK13, CHD4, KCNQ3, KMT5B, TCF20, and ZBTB18.
In our sample, CDK13 (cyclin-dependent kinase 13) and KMT5B (lysine-specific methyltransferase 5B) harbored the most pathogenic/likely pathogenic variants while the most VUS were detected in TCF20. Of note, we found two unrelated patients with a change of the amino acid residue asparagine at position 842 in CDK13 (p.Asn842Ser and p.Asn842Ile). These patients had delayed speech and language development, motor delay, and abnormal facial shape (Additional file 1: Figure S3 and Table S4). The p.Asn842Ser has also been previously described in the DDD study [9], suggesting that position 842 could be a mutational hot spot.
Notably, there were two patients who carried two pathogenic/likely pathogenic variants in two different genes (n = 2/65, 3%) each. Of note, this is in the same range as a recent large-scale study (4.9%) [24], further underlining the importance to search for genetic causes with an exome-wide approach not to overlook relevant genetic diagnoses and also the importance of revisiting and reanalyzing exomes over time as more and more new genetic publications surface, even if one genetic cause has already been identified.
The genetic heterogeneity of NDD with hundreds of genes in which variants lead to NDD reflects the complex process of proper brain development. Many of the gene products function in multiple biological pathways but may result in strikingly different phenotypes. For example, patients with de novo variants in CDK13 and CHD4 may present with overlapping neurodevelopmental features and heart defects; the function of both genes is different [9, 25, 26]. CHD4 is part of the SNF2/RAD54 helicase family and is a core component of the nucleosome remodeling and histone deacetylase repressor complex which is important for epigenetic regulation of gene transcription. In contrast, CDK13 forms a complex with cyclin K and is predicted to have a role in regulating cell cycle but also transcription. On the other hand, a distinct phenotype can be seen for variants within the same gene. CHD4 somatic variants are also involved in uterine serous carcinoma, an aggressive endometrial cancer [27]. This illustrates the high time and spatial sensitivity of the developing brain/body to genetic variations.
Many novel NDD genes are involved in epigenetic mechanisms such as chromatin remodeling, histone modification, RNA splicing, transcription, and DNA binding including the two most relevant genes from our study, i.e., CDK13 and KMT5B. CDK13 forms a complex with cyclin K and is predicted to have a role in regulating cell cycle and transcription. Mutations can alter complex activity. KMT5B functions as a histone methyltransferases and trimethylate nucleosomal histone 5 [28]. KMT5B also trimethylates the oncogene ERK (extracellular signal-regulated kinases), and overexpression of KMT5B activates the ERK signaling pathway [29]. These kinases are important for brain development, proliferation of cells, and neuronal migration, and ERK1/2 deficits in mice have shown impaired neurogenesis [30]. Histone deacetylase inhibitors (HDACis) and DNA demethylating drugs (DNMTis) have been used in cancer therapy trials [31, 32] and may be emerging drugs in NDD [33].
Conclusions
Our study underlines the relevance of six additional NDD genes and highlights the significance of multiple genetic diagnoses in several patients. Our study accentuates the importance of re-evaluating whole exome sequencing data in light of new publications enabling reclassification of previously categorized variants of uncertain significance.
Availability of data and materials
All data on variants will be available on HGMD.
Abbreviations
- CDK13:
-
Cyclin-dependent kinase 13
- CHD4:
-
Chromodomain-helicase-DNA-binding protein 4
- DDD:
-
Deciphering Developmental Disorders
- DNA:
-
Deoxyribonucleic acid
- EDTA:
-
Ethylenediaminetetraacetic acid
- FOXG1:
-
Forkhead box G1
- KCNQ3:
-
Potassium voltage-gated channel subfamily Q member 3
- KMT5B:
-
Lysine methyltransferase 5B
- NDD:
-
Neurodevelopmental disorder
- TCF20:
-
Transcription factor 20
- TMEM237:
-
Transmembrane protein 237
- ZBTB18:
-
Zinc finger and BTB domain containing 18
References
Sheridan E, Wright J, Small N, Corry PC, Oddie S, Whibley C, Petherick ES, Malik T, Pawson N, McKinney PA, Parslow RC. Risk factors for congenital anomaly in a multiethnic birth cohort: an analysis of the Born in Bradford study. Lancet. 2013;382:1350–9.
Ku CS, Polychronakos C, Tan EK, Naidoo N, Pawitan Y, Roukos DH, Mort M, Cooper DN. A new paradigm emerges from the study of de novo mutations in the context of neurodevelopmental disease. Mol Psychiatry. 2013;18:141–53.
Aronson SJ, Rehm HL. Building the foundation for genomics in precision medicine. Nature. 2015;526:336–42.
Groza T, Kohler S, Moldenhauer D, Vasilevsky N, Baynam G, Zemojtel T, Schriml LM, Kibbe WA, Schofield PN, Beck T, et al. The human phenotype ontology: semantic unification of common and rare disease. Am J Hum Genet. 2015;97:111–24.
Trujillano D, Bertoli-Avella AM, Kumar Kandaswamy K, Weiss ME, Koster J, Marais A, Paknia O, Schroder R, Garcia-Aznar JM, Werber M, et al. Clinical exome sequencing: results from 2819 samples reflecting 1000 families. Eur J Hum Genet. 2017;25:176–82.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012;6:80–92.
study DDD. Prevalence and architecture of de novo mutations in developmental disorders. Nature. 2017;542:433–8.
van den Akker WMR, Brummelman I, Martis LM, Timmermans RN, Pfundt R, Kleefstra T, Willemsen MH, Gerkes EH, Herkert JC, van Essen AJ, et al. De novo variants in CDK13 associated with syndromic ID/DD: molecular and clinical delineation of 15 individuals and a further review. Clin Genet. 2018;93:1000–7.
Weiss K, Terhal PA, Cohen L, Bruccoleri M, Irving M, Martinez AF, Rosenfeld JA, Machol K, Yang Y, Liu P, et al. De novo mutations in CHD4, an ATP-dependent chromatin remodeler gene, cause an intellectual disability syndrome with distinctive Dysmorphisms. Am J Hum Genet. 2016;99:934–41.
Wang T, Guo H, Xiong B, Stessman HA, Wu H, Coe BP, Turner TN, Liu Y, Zhao W, Hoekzema K, et al. De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat Commun. 2016;7:13316.
Ambrosino P, Freri E, Castellotti B, Soldovieri MV, Mosca I, Manocchio L, Gellera C, Canafoglia L, Franceschetti S, Salis B, et al. Kv7.3 compound heterozygous variants in early onset encephalopathy reveal additive contribution of C-terminal residues to PIP2-dependent K(+) channel gating. Mol Neurobiol. 2018;55:7009–24.
Bowling KM, Thompson ML, Amaral MD, Finnila CR, Hiatt SM, Engel KL, Cochran JN, Brothers KB, East KM, Gray DE, et al. Genomic diagnosis for children with intellectual disability and/or developmental delay. Genome Med. 2017;9:43.
Moccia A, Srivastava A, Skidmore JM, Bernat JA, Wheeler M, Chong JX, Nickerson D, Bamshad M, Hefner MA, Martin DM, Bielas SL. Genetic analysis of CHARGE syndrome identifies overlapping molecular biology. Genet Med. 2018;20(9):1022–9.
Zhao JJ, Halvardson J, Zander CS, Zaghlool A, Georgii-Hemming P, Mansson E, Brandberg G, Savmarker HE, Frykholm C, Kuchinskaya E, et al. Exome sequencing reveals NAA15 and PUF60 as candidate genes associated with intellectual disability. Am J Med Genet B Neuropsychiatr Genet. 2018;177:10–20.
Ververi A, Splitt M, Dean JCS, Brady AF. Phenotypic spectrum associated with de novo mutations in QRICH1 gene. Clin Genet. 2018;93:286–92.
Strauss KA, Gonzaga-Jauregui C, Brigatti KW, Williams KB, King AK, Van Hout C, Robinson DL, Young M, Praveen K, Heaps AD, et al. Genomic diagnostics within a medically underserved population: efficacy and implications. Genet Med. 2018;20:31–41.
Stevens SJC, van der Schoot V, Leduc MS, Rinne T, Lalani SR, Weiss MM, van Hagen JM, Lachmeijer AMA, Stockler-Ipsiroglu SG, Lehman A, Brunner HG. De novo mutations in the SET nuclear proto-oncogene, encoding a component of the inhibitor of histone acetyltransferases (INHAT) complex in patients with nonsyndromic intellectual disability. Hum Mutat. 2018;39:1014–23.
Depienne C, Nava C, Keren B, Heide S, Rastetter A, Passemard S, Chantot-Bastaraud S, Moutard ML, Agrawal PB, VanNoy G, et al. Genetic and phenotypic dissection of 1q43q44 microdeletion syndrome and neurodevelopmental phenotypes associated with mutations in ZBTB18 and HNRNPU. Hum Genet. 2017;136:463–79.
Trinh J, Huning I, Budler N, Hingst V, Lohmann K, Gillessen-Kaesbach G. A novel de novo mutation in CSNK2A1: reinforcing the link to neurodevelopmental abnormalities and dysmorphic features. J Hum Genet. 2017;62:1005–6.
Jansen S, Geuer S, Pfundt R, Brough R, Ghongane P, Herkert JC, Marco EJ, Willemsen MH, Kleefstra T, Hannibal M, et al. De novo truncating mutations in the last and penultimate exons of PPM1D cause an intellectual disability syndrome. Am J Hum Genet. 2017;100:650–8.
Turner TN, Hormozdiari F, Duyzend MH, McClymont SA, Hook PW, Iossifov I, Raja A, Baker C, Hoekzema K, Stessman HA, et al. Genome sequencing of autism-affected families reveals disruption of putative noncoding regulatory DNA. Am J Hum Genet. 2016;98:58–74.
Posey JE, Harel T, Liu P, Rosenfeld JA, James RA, Coban Akdemir ZH, Walkiewicz M, Bi W, Xiao R, Ding Y, et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med. 2017;376:21–31.
Sifrim A, Hitz MP, Wilsdon A, Breckpot J, Turki SH, Thienpont B, McRae J, Fitzgerald TW, Singh T, Swaminathan GJ, et al. Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat Genet. 2016;48:1060–5.
Bostwick BL, McLean S, Posey JE, Streff HE, Gripp KW, Blesson A, Powell-Hamilton N, Tusi J, Stevenson DA, Farrelly E, et al. Phenotypic and molecular characterisation of CDK13-related congenital heart defects, dysmorphic facial features and intellectual developmental disorders. Genome Med. 2017;9:73.
Zhao S, Choi M, Overton JD, Bellone S, Roque DM, Cocco E, Guzzo F, English DP, Varughese J, Gasparrini S, et al. Landscape of somatic single-nucleotide and copy-number mutations in uterine serous carcinoma. Proc Natl Acad Sci U S A. 2013;110:2916–21.
Schotta G, Lachner M, Sarma K, Ebert A, Sengupta R, Reuter G, Reinberg D, Jenuwein T. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 2004;18:1251–62.
Vougiouklakis T, Sone K, Saloura V, Cho HS, Suzuki T, Dohmae N, Alachkar H, Nakamura Y, Hamamoto R. SUV420H1 enhances the phosphorylation and transcription of ERK1 in cancer cells. Oncotarget. 2015;6:43162–71.
Uriu-Adams JY, Keen CL. Zinc and reproduction: effects of zinc deficiency on prenatal and early postnatal development. Birth Defects Res B Dev Reprod Toxicol. 2010;89:313–25.
Sidhu H, Capalash N. UHRF1: the key regulator of epigenetics and molecular target for cancer therapeutics. Tumour Biol. 2017;39:1010428317692205.
Wahid B, Ali A, Rafique S, Idrees M. New insights into the epigenetics of hepatocellular carcinoma. Biomed Res Int. 2017;2017:1609575.
Hauser RM, Henshall DC, Lubin FD. The epigenetics of epilepsy and its progression. Neuroscientist. 2018;24(2):186–200. https://doi.org/10.1177/1073858417705840. Epub 2017 May 4.
Funding
JT acknowledges funding from Alexander Von Humboldt, Canadian Institutes of Health Research, and the Joachim Herz Stiftung. We acknowledge financial support by Land Schleswig-Holstein within the funding programme Open Access Publikations fonds.
Author information
Authors and Affiliations
Contributions
JT contributed to the execution of the research project; designed, executed, reviewed, and critiqued the statistical analysis; and wrote the first draft and reviewed and critiqued the manuscript preparation. KKK contributed to the execution of the research project; executed, reviewed, and critiqued the statistical analysis; and wrote the first draft and reviewed and critiqued the manuscript preparation. MW, MERW, GO, and SK contributed to the execution of the research project; executed, reviewed, and critiqued the statistical analysis; and reviewed and critiqued the manuscript preparation. KL and AR contributed to the conception, organization, and execution of the research project; designed, reviewed, and critiqued the statistical analysis; and wrote the first draft and reviewed and critiqued the manuscript preparation. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Written informed consent was obtained from participants, and this study was approved by the Ethical Commission of the University of Rostock (registry no. A2015-0102). All samples were processed in Centogene’s laboratory, which is CAP and CLIA certified, adhering to the ACMG guidelines.
Consent for publication
All authors consent for the publication of this work.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Table S1. Patient demographics for developmental disorders. Table S2. Variants of unknown significance identified in this study and pathogenicity scoring. Table S3. Individuals with dual molecular diagnoses. Table S4. HPO terms listed for all (likely) pathogenic mutation carriers. Table S5. Number of rare, protein-changing variants found in the NDD patients. Figure S1. Overview of study: workflow of identification of 14 (likely) pathogenic variants (6 of 14 candidate genes) in 14 of 4351 patients. Figure S2. HPO terms composite for CDK13 pathogenic/likely pathogenic carriers. HPO terms that overlap in different mutation carriers are highlighted in red. Figure S3. HPO terms composite for KMT5B pathogenic/likely pathogenic variant carriers. HPO terms that overlap in different mutation carriers are highlighted in red. (DOCX 96 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Trinh, J., Kandaswamy, K.K., Werber, M. et al. Novel pathogenic variants and multiple molecular diagnoses in neurodevelopmental disorders. J Neurodevelop Disord 11, 11 (2019). https://doi.org/10.1186/s11689-019-9270-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s11689-019-9270-4