
Abstract
Background
Global developmental delay or intellectual disability usually accompanies various genetic disorders as a part of the syndrome, which may include seizures, autism spectrum disorder and multiple congenital abnormalities. Next-generation sequencing (NGS) techniques have improved the identification of pathogenic variants and genes related to developmental delay. This study aimed to evaluate the yield of whole exome sequencing (WES) and neurodevelopmental disorder gene panel sequencing in a pediatric cohort from Ukraine. Additionally, the study computationally predicted the effect of variants of uncertain significance (VUS) based on recently published genetic data from the country’s healthy population.
Methods
The study retrospectively analyzed WES or gene panel sequencing findings of 417 children with global developmental delay, intellectual disability, and/or other symptoms. Variants of uncertain significance were annotated using CADD-Phred and SIFT prediction scores, and their frequency in the healthy population of Ukraine was estimated.
Results
A definitive molecular diagnosis was established in 66 (15.8%) of the individuals. WES diagnosed 22 out of 37 cases (59.4%), while the neurodevelopmental gene panel identified 44 definitive diagnoses among the 380 tested patients (12.1%). Non-diagnostic findings (VUS and carrier) were reported in 350 (83.2%) individuals. The most frequently diagnosed conditions were developmental and epileptic encephalopathies associated with severe epilepsy and GDD/ID (associated genes ARX, CDKL5, STXBP1, KCNQ2, SCN2A, KCNT1, KCNA2). Additionally, we annotated 221 VUS classified as potentially damaging, AD or X-linked, potentially increasing the diagnostic yield by 30%, but 18 of these variants were present in the healthy population of Ukraine.
Conclusions
This is the first comprehensive study on genetic causes of GDD/ID conducted in Ukraine. This study provides the first comprehensive investigation of the genetic causes of GDD/ID in Ukraine. It presents a substantial dataset of diagnosed genetic conditions associated with GDD/ID. The results support the utilization of NGS gene panels and WES as first-line diagnostic tools for GDD/ID cases, particularly in resource-limited settings. A comprehensive approach to resolving VUS, including computational effect prediction, population frequency analysis, and phenotype assessment, can aid in further reclassification of deleterious VUS and guide further testing in families.
Similar content being viewed by others


Background
Global developmental delay (GDD) and intellectual disability (ID) are terms used to describe individuals with significant delays in various developmental domains, including gross and fine motor skills, language and communication, and personal and social conduct [1]. While the designation GDD is reserved to children under the age of five, ID is applied for older children and adults. Both conditions are diagnosed when the standardized neurological tests fall two standard deviations below the age-appropriate mean [2]. While 40% of all GDD/ID cases are attributable to genetic disorders, other factors such as perinatal trauma, intrauterine infections, and toxic exposure, as well as postnatal events can also contribute to the developmental delay [3]. GDD/ID can also be accompanied by autism spectrum disorder (ASD) as well as anatomical abnormalities of other organ systems.
Traditionally, chromosomal microarray (CMA) and fragile X syndrome testing have been the primary diagnostic approaches for GDD/ID. However, CMA can only detect chromosomal deletions or duplications in about 20% of genetic cases. The advent of next-generation sequencing (NGS), specifically gene panel testing and whole exome sequencing (WES), has revolutionized the search for causative variants in neurodevelopmental disorders, increasing diagnostic success by an additional 25%. NGS techniques, with improved testing methods and bioinformatic algorithms, can now detect large copy number variations (CNVs) and chromosomal aberrations previously undetectable by NGS alone. Even though CMA and WES are not interchangeable, with the current improvements in testing techniques and bioinformatic algorithms, NGS gene panels and WES can accurately find large CNVs and chromosomal aberrations previously doomed undetectable by NGS technique [4].
Syndromic genetic disorders are the leading cause of pediatric disability in Ukraine [5]. However, the diagnosis of developmental delay in Ukraine has been delayed due to limited newborn screening and only the recent adoption of NGS genetic testing by physicians [6, 7].
Apart of isolated case reports, there has not been a comprehensive study on genetic causes of GDD/ID conducted in Ukraine, in particular, on diagnostic yield of NGS panels and WES techniques. This report will help pave the way to detecting locally significant candidate pathogenic variants for future variant resolution and familial studies in Ukraine and across Eastern Europe [8].
Methods
This is a retrospective study of the cohort consisted of a mixed set of individuals diagnosed with GDD/ID only, as well as GDD/ID patients with ASD and/or multiple congenital anomalies or other functional symptoms. The diagnostic protocol was performed according to the Diagnostic and Statistical Manual of Mental Disorders (DSM-5, APA 2013) [9]. The patients were referred to a medical geneticist by either pediatrician or pediatric neurologist for consultation. All 416 children enrolled in the study underwent sequencing of whole exome sequencing (WES, Invitae Inc., San Francisco, CA) or custom broad neurodevelopmental disorder (NDD) gene panel sequencing (Invitae Inc., San Francisco, CA). The medical geneticist obtained consent for testing and signed standardized requisition forms with optional clinical and demographic information. Family follow-up has not yet been performed with these patients’ family members. The study of deidentified aggregated data was approved as “No Human Subject Research” by the Institutional Review Board of Oakland University (Rochester, MI, Study #RB-FY2023-120).
NGS neurodevelopmental disorders panel and whole exome sequencing
The neurodevelopmental disorders panel (NDD) included 1,813 genes (Supplementary Table 1, sequencing and report limitations specified in the Supplementary file 1). For the NDD panel, genomic DNA from the submitted samples was extracted and enriched for targeted regions using the hybridization-based protocol [10]. For the WES sequencing, DNA libraries were prepared using the PCR-free method. The WES panel included a panel of more than 18,000 genes (Invitae Inc., San Francisco, CA).
All blood and saliva samples underwent double-step verification by visual identifiers (ID, Sex) and sex determined by sequencing, according to the company protocol (Invitae Inc., San Francisco, CA). All targeted regions were sequenced on Illumina NovaSeq 6000 platform (Illumina, San Diego, CA, USA). The average coverage for NDD panel testing was 50x, while for WES it was 35X across the entire exome.
Read mapping was performed to the reference GRCh37 human genome. To categorize the variants according to the laboratory, several pieces of evidence were considered, such as variant frequency and type, clinical findings, experimental research, and indirect and computational approaches.
Before being reported, clinically important variation that failed to meet strict NGS quality parameters had its accuracy verified by alternative methods [4], Sherloc [11], a points-based framework based on the joint consensus recommendations from the American College of Medical Genetics and Genomics and the Association for Molecular Pathology [12], was used to analyze the variations discovered by the bioinformatics pipeline. According to Invitae protocols, CNVs were confirmed through the application of either MLPA or Droplet Digital PCR (ddPCR). In cases where MLPA or ddPCR is unavailable, aCGH, was employed which involves a custom-designed microarray focused on exons.
Genomic data analysis.
Initially, a dataset was created in Ukraine containing reported genomic variants for each individual. This dataset also included information such as sex, age, and phenotypical descriptions. Subsequently, the data were transmitted to Oakland University, MI (USA). Due to the heterogeneous nature of data provided in the reports, the alleles with missing explicit rs-code were cross-referenced by their respective allele and protein notation according to Sequence Variant Nomenclature specifications, using the ENSEMBL Variant Recoder [13], resulting in the genomic positions of each reported variant for GRCh38 genomic reference and their relevant rs-codes. After the validation, we performed a detailed search on reported pathogenic (P) or likely pathogenic (LP) variants using ClinVar [14] and OMIM [15] databases.
Variants of uncertain significance (VUS) or heterozygous variants related to the autosomal recessive condition were considered non-diagnostic. Using genome data from 97 individuals from the “Genome Diversity in Ukraine” database [16, 17], as well as from the 150 whole genomes from the database of the cross-border cooperation project “Partnership for Genomic research in Ukraine and Romania” [18], we performed an additional annotation for the effect prediction among the VUS using CADD and SIFT scores and estimated their frequency in the general population of Ukraine.
Results
Demographic and clinical characteristics of the cohort
The cohort included 416 exclusively pediatric patients under 18 years old (age ranged between 1 and 18 years), with 60.9% males. Both sexes, males and females, had a similar mean age of around 7 yo (Table 1). Genetic information either from the NDD gene panel or from WES results as a first- or second-line test after inconclusive CMA was available for analysis in all individuals in this study. Diagnostic data for karyotypes, chromosomal microarrays, or FMR1 CGG-repeat expansion tests for the Fragile X syndrome, was not included in this study. Demographic and clinical information included in the analysis is summarized in Table 1.
Yield of definitive the molecular diagnosis
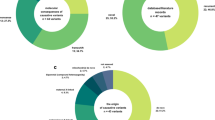
We identified a definitive molecular diagnosis in 66 or 16.3% of all individuals (Fig. 1). In general, WES positively diagnosed 22 out of 37 ordered cases (59.4%), while the NGS testing panel yielded 44 definitive diagnoses among the 379 tested patients (12.1%). Non-diagnostic variants (VUS and carrier) were identified in 348 (83.4%) individuals (details in Fig. 1).
Most of the known diagnosed conditions followed the AD mode of inheritance (41, or 62.11%), four with AR, nine with XLD, and two with XLR modes (Table 2). Compound heterozygosity was confirmed by segregation analysis. Chromosome 15 was most affected by these types of variants. The most commonly diagnosed same-gene condition was Rett syndrome: five cases were caused by single nucleotide variants (SNVs) or small indels in the MECP2 gene. Among the other diagnoses, 12 different conditions were observed in two individuals each, all the rest diseases were isolated cases only (Table 2). Out of 66 diagnosed cases, the rest 10 patients harbored large copy number variations (CNVs) encompassing multiple genes (15.1% of diagnosed cases) (Table 3).

The prevailing group of diseases diagnosed were classified as developmental and epileptic encephalopathies (type 1, 2, 4, 7, 11, 14, 32 and 43) characterized by severe epilepsy and GDD/ID (14 cases). The specific variants reported for each definitive diagnosis are reported in the Table 2.
Annotation and analysis of variants of uncertain significance
A total of 3,317 heterozygous variants of uncertain significance (or VUS) were identified in our cohort of 417 patients. In this VUS dataset, a CADD-Phred score between 10 and 20 were associated with 245 variants (considered 10% most deleterious substitutions in the human genome), while for 723 variants it was above 20 (the top 1% most deleterious variants) [19]. A deleterious SIFT-prediction score [20] for least one alternative transcript was calculated for 527 variants (Fig. 2; see details in Supplementary Table 2).

Among these deleterious variants identified in the study,165 were in the genes associated with AD conditions and 56 associated with X-linked dominant or recessive conditions according to OMIM (a total of 221). Here we reported these variants as potentially diagnostic and suggested a segregation analysys in 138 undiagnosed cases (Supplementary Table 2). However, 18 of these 221 variants had allele count 1 or 2 in 247 among the unaffected individuals from Ukraine (Supplementary Table 2). The rest of the variants (203) were absent in healthy individuals. Gene PACS2 (associated with AD developmental and epileptic encephalopathy 66, OMIM 618,067) was most frequently altered (seven individuals harboring rare highly deleterious heterozygous SNV) (Supplementary Table 2).
Discussion
Global developmental delay and intellectual disability (GDD/ID) is usually diagnosed for the patients with developmental delay before the exact genetic diagnosis is established [21].
GDD/ID is a complex set of symptoms with a wide range of genetic causes, including single nucleotide variants, large chromosomal indels, and copy number variants. However, in Ukraine a comprehensive study on the genetic causes of GDD/ID has not been conducted yet. This is mainly due to only the recent availability of NGS-based diagnostic tests. Also, the whole-genome data on the general genetic composition of the population has just been published recently [8, 16, 17], there was a need to use genome data available to evaluate the diagnostic yield of WES and NGS gene panel.
In this study, we report the largest to-date descriptive dataset of diagnosed genetic conditions which present with GDD/ID as a part of the clinical picture. Also, we report a combined diagnostic yield of the NDD gene panel of 1813 genes and WES at 16.3% on previously undiagnosed cases. Expectedly, individually WES had a much higher diagnostic yield compared to the NDD gene panel (59.4% and 12.1% respectively). Similarly, the reported diagnostic yield of target exome sequencing in patients with ID ranges from 21 to 55.7%. Pekeles et al. [22]. used four distinct panels in a trial with a sample of 48 patients and achieved a 21% rate of definitive diagnoses. With a sample of 133 patients, Yamamoto et al. [23] achieved a diagnostic rate of 29.3%. A similar rate of 34% was reported by Gieldon and colleagues in a study using 4 813 gene panels in 106 patients [24]. It is possible that the significant difference in genetic yield between individuals who underwent exome sequencing and those who received targeted sequencing could be attributed to the bias due limited sample size of 37 WES administered might have led to some variability in the results. In such a small sample, the observed difference could have occurred randomly without any underlying phenotypic differences between the groups. Also, the NDD panels were prescribed in many cases due to their significantly lower cost. However, it is also important to consider other factors that could contribute to the observed discrepancy. Exome sequencing is a more comprehensive approach compared to targeted sequencing, as it examines a larger portion of the genome. This broader coverage increases the likelihood of identifying disease-causing genetic variants, leading to a higher genetic yield. Additionally, exome sequencing may capture variants in genes that are not initially suspected based on the clinical presentation but still contribute to the observed phenotype.
Our study showed that both the NGS gene panel and WES can be diagnostic of large CNVs associated with the clinical picture of known syndromes in the absence of CMA testing. In these 10 cases, both the NDD gene panel and WES reported multiple whole genes deleted or multiplied, which was indicative of the cytogenic location to determine large aberration from the set of genes mutated.
Most of the reported variants in our cohort were variants of uncertain significance (VUS). Both patients and medical geneticists face challenges as a result of the discovery of a VUS [25]. A VUS may ultimately be reclassified as pathogenic or benign, but this process often takes several years and may never be completed for rare VUS, particularly if the condition is uncommon to find enough cases or too expensive to test relatives [26]. The clinical relevance of a VUS has been increasingly determined by a phenotypically driven in-silico approach [27]. Furthermore, variant interpretation can be enhanced by quantitative analysis of consortium disease cohorts and population controls [28].
The absence of family members’ genetic data was a major limiting factor to fully classifying or resolving the effect of the variants of uncertain significance in our study. Also, this prevented us from identifying de novo variants. Thus, we performed their annotation using CADD and SIFT predictive scores and found that as many as 527 variants were classified as deleterious by both scores (CADD-Phred > 10 and SIFT prediction “Deleterious”) with MAF < 0.01 and should be resolved for disease causation by family testing or phenotype confirmation tests. Out of these, 221 variants were associated with AD or X-linked conditions making them potentially diagnostic. This number of variants resolved could potentially increase the diagnostic yield in 138 undiagnosed case (by 33%). Interestingly, having WGS and phenotype data of 249 Ukrainians, we found that 18 of 221 potentially diagnostic variants are present at very low frequency in unaffected individuals (Supplementary Table 2), implying they might not be disease causative even with high prediction scores. Other 203 variants were absent in the sample of healthy individuals. Importantly, some of the VUS with high prediction scores and associated with AD or X-linked conditions were found in diagnosed individuals, potentially making their condition associated with multiple genetic aberrations.
Conclusions
This is the first comprehensive study on genetic causes of GDD/ID conducted in Ukraine, including diagnostic yield of NDD gene panel and WES techniques, comparing data from the cohort of pediatric patients and general population in the country. We report largest to date descriptive dataset of diagnosed genetic conditions which present with GDD/ID as a part of the clinical picture. Our results support the important role of NGS gene panel and WES in the diagnostic approach to GDD/ID cases as a first line choice in the scenario of scarce financial resources and logistic difficulty to perform multiple genetic tests in a family. A comprehensive approach to VUS resolution including computational effect prediction, comparative analysis of allele frequencies in population controls and phenotype assessment can be of extreme help to fully classify deleterious variants and narrow down the list for the further family follow-up testing.
Data availability
The data are available from the corresponding author on reasonable request.
Abbreviations
- GDD:
-
Global Developmental Delay
- ID:
-
Intellectual Disability
- WES:
-
Whole Exome Sequencing
- NGS:
-
Next Generation Sequencing
- AD:
-
Autosomal Dominant
- AR:
-
Autosomal Recessive
- XLD:
-
X-Linked Dominant
- XLR:
-
X-Linked Recessive
- CNVs:
-
Copy Number Variants
- NDD:
-
Neurodevelopmental Disorders
References
Shevell MI, Ashwal S, Donley D, Flint J, Gingold M, Hirtz D, et al. Practice parameter: evaluation of the child with global developmental delay [RETIRED]: report of the Quality standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurol AAN Enterprises. 2003;60:367–80.
Srour M, Shevell M. Genetics and the investigation of developmental delay/intellectual disability. Arch Dis Child BMJ Publishing Group Ltd. 2014;99:386–9.
Miclea D, Peca L, Cuzmici Z, Pop IV. Genetic testing in patients with global developmental delay/intellectual disabilities. A review. Clujul Medical. Volume 88. Romania: Universty of Medicine and Pharmacy of Cluj-Napoca; 2015. p. 288.
Lincoln SE, Truty R, Lin C-F, Zook JM, Paul J, Ramey VH, et al. A rigorous interlaboratory examination of the need to confirm next-generation sequencing–detected variants with an orthogonal method in clinical genetic testing. J Mol Diagnostics Elsevier. 2019;21:318–29.
Annual reports. [Internet]. Annual reports.Center of Medical statistics of Ministry of Health Care. 2021. Available from: http://medstat.gov.ua/ukr/reports.html.
Vrijenhoek T, Middelburg EM, Monroe GR, van Gassen KLI, Geenen JW, Hövels AM, et al. Whole-exome sequencing in intellectual disability; cost before and after a diagnosis. Eur J Hum Genet Nat Publishing Group. 2018;26:1566–71.
Знаменська ТК, Дубініна ТЮ, Воробйова. О. В. (2017). Стратегічні напрямки реконструкції системи охорони здоров’я новонароджених та дітей України.
Oleksyk TK, Wolfsberger WW, Schubelka K, Mangul S, O’Brien SJ. The Pioneer advantage: filling the blank spots on the map of genome diversity in Europe. Gigascience. 2022;11.
Division APA. Highlights of changes from dsm-iv to dsm-5: somatic symptom and related disorders. Focus (Madison). Am Psychiatric Assoc. 2013;11:525–7.
Kurian AW, Hare EE, Mills MA, Kingham KE, McPherson L, Whittemore AS, et al. Clinical evaluation of a multiple-gene sequencing panel for hereditary cancer risk assessment. J Clin Oncol Am Soc Clin Oncol. 2014;32:2001.
Nykamp K, Anderson M, Powers M, Garcia J, Herrera B, Ho Y-Y, et al. Sherloc: a comprehensive refinement of the ACMG–AMP variant classification criteria. Genetics in Medicine. Nat Publishing Group. 2017;19:1105–17.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in medicine. Nat Publishing Group. 2015;17:405–23.
Cunningham F, Allen JE, Allen J, Alvarez-Jarreta J, Amode MR, Armean IM, et al. Ensembl 2022. Nucleic acids res. Volume 50. Oxford University Press; 2022. pp. D988–95.
Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. Oxford University Press; 2016;44:D862–D868.
Hamosh A, Scott AF, Amberger J, Valle D, McKusick VA. Online mendelian inheritance in man (OMIM). Hum Mutat. Wiley Online Libr. 2000;15:57–61.
Oleksyk TK, Wolfsberger WW, Weber AM, Shchubelka K, Oleksyk OT, Levchuk O, et al. Genome diversity in Ukraine. Gigascience. Volume 10. Oxford University Press; 2021. p. giaa159.
Oleksyk TK, Wolfsberger WW. Dataset Genome Diversity in Ukraine [Internet]. 2020 [cited 2021 Sep 26]. Available from: http://gigadb.org/dataset/100835#.
Partnership for genomic research in. Ukraine and Romania [Internet]. Available from: https://genomes.uzhnu.edu.ua.
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47(D1):D886–94.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–81.
Schalock RL, Luckasson R. (2021). Intellectual disability, developmental disabilities, and the field of intellectual and developmental disabilities.
Pekeles H, Accogli A, Boudrahem-Addour N, Russell L, Parente F, Srour M. Diagnostic yield of intellectual disability gene panels. Pediatr Neurol Elsevier. 2019;92:32–6.
Yamamoto T, Imaizumi T, Yamamoto-Shimojima K, Lu Y, Yanagishita T, Shimada S, et al. Genomic backgrounds of Japanese patients with undiagnosed neurodevelopmental disorders. Brain Dev Elsevier. 2019;41:776–82.
Gieldon L, Mackenroth L, Kahlert A-K, Lemke JR, Porrmann J, Schallner J et al. Diagnostic value of partial exome sequencing in developmental disorders. PLoS One. Public Library of Science San Francisco, CA USA; 2018;13:e0201041.
Peyser A, Onel K, Hershlag A. Conflict resolution: discordant reporting of cancer gene variants: vus vs pathogenic. Fertil Steril Elsevier. 2022;118:e356.
Mohammadi L, Vreeswijk MP, Oldenburg R, van den Ouweland A, Oosterwijk JC, van der Hout AH, et al. A simple method for co-segregation analysis to evaluate the pathogenicity of unclassified variants; BRCA1 and BRCA2 as an example. BMC Cancer Springer. 2009;9:1–11.
Marinakis NM, Svingou M, Veltra D, Kekou K, Sofocleous C, Tilemis F-N, et al. Phenotype-driven variant filtration strategy in exome sequencing toward a high diagnostic yield and identification of 85 novel variants in 400 patients with rare mendelian disorders. Am J Med Genet Wiley Online Libr. 2021;185:2561–71.
Walsh R, Lahrouchi N, Tadros R, Kyndt F, Glinge C, Postema PG, et al. Enhancing rare variant interpretation in inherited arrhythmias through quantitative analysis of consortium disease cohorts and population controls. Genetics in medicine. Nat Publishing Group. 2021;23:47–58.
Acknowledgements
We would like to thank the Armed Forces of Ukraine for providing security to patients and scientists in Ukraine during the hard time this work was prepared.
Funding
KS, WW, YH, VM, MV and TKO were supported in part by 2SOFT/1.2/48 project “Partnership for Genomic Research in Ukraine and Romania” from Joint Operational Programme Romania-Ukraine 2014–2020 financed by the European Union through the European Neighborhood Instrument (ENI).
Author information
Authors and Affiliations
Contributions
Conceptualization: K.S., L.T., W.W.W., T.K.O.
Patient database establishment, phenotyping and patients records curation: L.T.O.K., A.M.
Methodology: K.S.
Data analysis: K.S., W.W., Y.H,V.M, M.V., K.K., K.W.
Project supervision: T.K.O.
Visualization: K.S., W.W.
Writing: K.S., L.T, W.W., K.K., KW., (original draft), and Y.H, V.M, M.V and T.K.O. (review and editing).
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by IRB of Oakland University with the waiver of consent as it is a retrospective anonymous study with the minimal possible risk to participants.
Consent for publication
Waiver of consent by IRB decision.
Competing interests
The authors declare that they have no competing interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shchubelka, K., Turova, L., Wolfsberger, W. et al. Genetic determinants of global developmental delay and intellectual disability in Ukrainian children. J Neurodevelop Disord 16, 13 (2024). https://doi.org/10.1186/s11689-024-09528-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s11689-024-09528-x