
Abstract
Background
Migraine is a highly prevalent disorder with significant economical and personal burden. Despite the development of effective therapeutics, the causes which precipitate migraine attacks remain elusive. Clinical studies have highlighted altered metabolic flux and mitochondrial function in patients. In vivo animal experiments can allude to the metabolic mechanisms which may underlie migraine susceptibility. Understanding the translational relevance of these studies are important to identifying triggers, biomarkers and therapeutic targets in migraine.
Main body
Functional imaging studies have suggested that migraineurs feature metabolic syndrome, exhibiting hallmark features including upregulated oxidative phosphorylation yet depleted available free energy. Glucose hypometabolism is also evident in migraine patients and can lead to altered neuronal hyperexcitability such as the incidence of cortical spreading depression (CSD). The association between obesity and increased risk, frequency and worse prognosis of migraine also highlights lipid dysregulation in migraine pathology. Calcitonin gene related peptide (CGRP) has demonstrated an important role in sensitisation and nociception in headache, however its role in metabolic regulation in connection with migraine has not been thoroughly explored. Whether impaired metabolic function leads to increased release of peptides such as CGRP or excessive nociception leads to altered flux is yet unknown.
Conclusion
Migraine susceptibility may be underpinned by impaired metabolism resulting in depleted energy stores and altered neuronal function. This review discusses both clinical and in vivo studies which provide evidence of altered metabolic flux which contribute toward pathophysiology. It also reviews the translational relevance of animal studies in identifying targets of biomarker or therapeutic development.
Similar content being viewed by others

Background
Migraine is a highly prevalent and disabling disorder, affecting over 1 billion people worldwide [1] and ranking the leading cause of disability amongst neurological conditions [2]. In addition to economical loss, migraine significantly reduces quality of life, [3] and is often comorbid with stress, anxiety and depression [4]. In recent years there has been development of effective migraine therapeutics which target the release of the nociceptive neuropeptide, calcitonin gene related peptide (CGRP). Although these agents are effective at reducing migraine days and improving quality of life [5], the factors which contribute towards the precipitation of headache attack in those with migraine remain unclear.
Migraine is a prevalent feature of both mitochondrial and metabolic disorders (lifetime prevalence of 61% for mitochondrial [6] and 1-year prevalence of 11.9% in men and 22.5% in women with metabolic disorders [7]), which suggests migraine may be a common clinical manifestation of brain energy dysfunction [8]. Moreover, advancement in metabolic imaging and monitoring techniques has led to the hypothesis that the metabolic flux is altered in migraine patients, [9] leading to an energetic deficit which could underlie headache susceptibility. This review aims to evaluate animal and clinical studies which provide evidence of the perturbations in metabolic flux in migraine. Furthermore, it will discuss the translational relevance of these studies and identify altered energetic pathways which may serve as biomarkers or targets for therapeutic development.
Current understanding of migraine pathogenesis
Sensitization of the trigeminovascular system and cortical hyperexcitability are two mechanisms thought to be crucial to pathophysiology of migraine.
The trigeminovascular system is composed of trigeminal sensory neurons innervating the dura mater as well as cerebral and pial blood vessels. They synapse in the pars caudalis of the ipsilateral spinal trigeminal nucleus. The main projection of these nociceptive fibres is the ventral posteromedial thalamic nucleus, which then relays to the primary sensory cortex. This system is fundamental to nociception and migraine pathophysiology [10]. Trigeminal afferents express a range of receptors, such as transient receptor potential (TRP) channels, which are targets of nociceptive and vasoactive agents able to promote allodynia and hyperalgesia and, hence, induce headache attacks [11]. An important molecule released by afferents is calcitonin gene-related peptide (CGRP) [12], a 37-amino acid peptide implicated in migraine and a target of recent effective migraine therapeutics [13,14,15,16,17].
Hyperexcitability of the cerebral cortex is also thought to contribute towards migraine pathophysiology [18]. It has been hypothesised that hyperexcitability of trigeminovascular neurons may account for headache during migraine attacks in the absence of aura. Hyperresponsiveness of the visual cortex may result in photophobia or of the auditory cortex which corresponds to avoidance of noise [19]. Moreover, Familial Hemiplegic migraine (FHM), features mutations in calcium, ATP or sodium channels which result in a lowered threshold for neuronal activation. One mechanism of hyperexcitability which may contribute toward headache pathophysiology is cortical spreading depression (CSD): a wave of depolarisation across the cortical surface, leading to release in neuropeptides and alterations in cerebral blood flow [20, 21]. Functional imaging studies have associated this neurophysiological event with migraine aura: [22] a period of temporary visual disturbances and focal neurological symptoms that can precede a migraine attack [23]. Mechanistic insights from animal studies suggest that CSD can also activate meningeal nociceptors and, hence, may contribute towards headache pain [24]. Cortical spreading depression is of particular importance to this review as it is a metabolically stressful event and has been hypothesised to be triggered by or lead to, an energetic imbalance in migraine patients [25].
Dysfunctional glucose metabolism in migraine
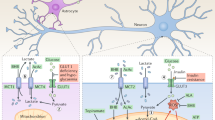
Glucose is the major energy substrate of the brain. It diffuses across the blood–brain-barrier via the glucose transporter GLUT1 and can be taken up by neurons via GLUT3 [26]. Its major metabolic pathways are: glycolysis (yielding ATP molecules as well as pyruvate or lactate), glycogenesis (providing glycogen stores which can be utilised during hypoglycaemia or ischaemia) and the pentose phosphate pathway (involved in ribose-5-phosphate and NADPH metabolism). Pyruvate can be metabolised in the tricarboxylic acid (TCA) cycle following its oxidative decarboxylation into Acetyl CoA via the highly regulated and irreversible pyruvate dehydrogenase reaction (Fig. 1).

Hypothesised alterations in metabolic flux which may contribute to migraine pathophysiology. The activity of numerous vital metabolic pathways is altered in migraine patients, which may lead to the upregulation of nociceptive peptides. Moreover, metabolic hormones including insulin and amylin act on receptors implicated in nociception in trigeminal nerves and vasodilation in endothelial cells, pathways which are both involved in migraine pathophysiology. α-KG; α-ketoglutarate, ADP; Adenosine di-phosphate, AMY1; Amylin type 1 receptor, ATP; Adenosine tri-phosphate, CGRP; Calcitonin gene related peptide, CoQ; CoenzymeQ10, Cyt c; Cytochrome C, FAD; Flavin adenine dinucleotide, GLUT; Glucose transporter, TCA; tricarboxylic acid cycle
Glucose metabolism is fine-tuned within and among cells to ensure maintenance of adequate concentrations of substrates. At rest, the metabolism of the brain is compartmentalised between neurons and astrocytes; neurons can rely on both glycolytic and oxidative metabolism, whereas astrocytes tend to be primarily glycolytic and can metabolically support neurons [27]. At times of increased energy demand, glycolysis is upregulated in neurons as the pathway can provide ATP at a faster rate than mitochondrial oxidative phosphorylation [28]. Hypoglycaemia leading to depleted glucose supply or dysfunctions in glucose uptake and glycolysis can have deleterious effects on cerebral function and have been demonstrated in migraine.
In vivo murine models have been useful in providing in-depth understanding of mechanisms linking abnormal glycolysis and migraine (Table 1). In rodents, hypoglycaemia induced by food deprivation or insulin was shown to reduce CSD threshold whilst increasing its duration [29, 30]. Spontaneous depolarisation events, which are analogous to anoxic depolarisation, have also been recorded in hypoglycaemic rats [31]. These studies propose the theory that inability to maintain ionic homeostasis, due to depleted ATP production, may increase excitability of cortical tissue and, therefore, susceptibility to CSD. In contrast, hyperglycaemia provided a protective effect, increasing the electrical threshold and reducing the frequency of potassium chloride-induced CSDs [29]. In other models however, both glucagon (increasing blood glucose) and insulin (decreasing blood glucose) inhibited dural-evoked neuronal firing in the trigeminocervical-complex [32]. These studies were very useful to determine that trigeminal nerves are glucose responsive, and suggest a role for hormones regulating metabolic homeostasis in pain (further detailed in sections below) [32].
In clinical practice, migraineurs often report that attacks are precipitated by fasting or skipping meals, [33] and indeed food deprivation was associated with headache in 58% of patients with frequent migraine or tension type headache [34]. In addition to being a migraine trigger, hypoglycaemia is thought to have a causative role in fasting headache, defined as occurring following 16 h of fasting and resolving within 72 h of food intake [23]. Another example is the GLUT1 deficiency syndrome: it results in impaired facilitated diffusion of glucose into the brain and can cause epileptic encephalopathy, however, there are milder phenotypes that demonstrate hemiplegic migraine [35, 36]. It is plausible that GLUT1 deficiency causes a reduction of the major energetic substrate of the brain and may trigger headache attacks via insufficient ATP production. Interestingly, hemiplegic migraines resolved with a modified Atkins diet which supplies ketone bodies as an alternative energy source [35]. This observation further supports the theory that metabolic imbalances are highly relevant to migraine and may be corrected by providing appropriate metabolic support.
The gold standard to assess cerebral glucose uptake in vivo in patients is 18F-Fluorodeoxyglucose PET (18F-FDG PET) which has provided further evidence for the role of downregulated glycolysis in migraine. The cerebral areas affected by glucose hypometabolism appear to be different in episodic migraineurs compared to chronic patients. In episodic migraineurs both with and without aura, hypometabolism has been detected in temporal [37], and fronto-temporal [38] regions involved in pain processing, whereas prefrontal cortex appears to be implicated in chronic patients [39]. Hypometabolism was also associated with disease duration in patients with and without aura, [37, 39] suggesting that repeated migraine attacks and activation of nociceptive regions may lead to abnormalities in glucose metabolism. Hypometabolism was improved in episodic migraine patients following external trigeminal nerve stimulation, which also decreased migraine attack frequency [38]. These studies suggest that improving glucose availability and utilisation may offer a prophylactic solution in some migraine patients and is an avenue to be explored further to expand available migraine treatment options.
Lactate
In the absence of sufficient oxygen, lactate is produced via anaerobic glycolysis of pyruvate (Fig. 1), hence, it has often been used as a pathological marker of hypoxia. However, lactate is routinely produced by specific cells and is an important signalling molecule involved in regulating neuronal plasticity, excitation, and homeostasis [40].
Results of animal and clinical studies suggest that lactate excess may be related to CSD. This hypothesis was corroborated in rat proton magnetic resonance spectroscopy (1H-MRS) studies during CSD, [41] and also supported by analyses of metabolites in brain tissue which demonstrated increased lactate, reduced glucose and pH. These results suggest the occurrence of lactic acidosis in neuronal tissue, therefore it is possible that vascular changes during CSD may lead to tissue hypoxia and the preferential use of anaerobic processes [42]. Not only is anaerobic glycolysis less efficient at ATP production, but it is plausible that it may also delay the recovery period, which is particularly energy intense. Interestingly, some studies have shown that during the metabolically challenging period following a CSD, lactate is produced in excess by astrocytes and utilised as an energy substrate by neurons [43]. Although healthy neuronal tissue demonstrates plasticity to changes in energetic flux in vivo, this process may become pathological during chronification of migraine and repeated CSD events, possibly via an impaired astrocytic-neuronal interaction.
Further evidence for the role of lactate in migraine comes from studies conducted in humans (Table 1). Upregulated lactate concentrations have been shown in migraine patients both with and without aura during interictal periods in direct measurements of serum and plasma [44,45,46]. Interestingly, exercise can be a trigger of migraine attacks in some migraineurs, and, although the exact pathophysiology remains unknown, excessive lactate production may contribute towards aetiology. For example, in a study including 20 chronic migraine patients and 20 controls, lactate was increased in migraine in comparison to controls during aerobic exercise (although significant results did not survive correction for multiple comparisons) [47]. 1H-MRS studies, which allows direct detection of lactate in vivo in brain, have also demonstrated increased lactate in familial hemiplegic migraine [48] and in migraine with aura, [49, 50] although not in migraine without aura. These observations strengthen the link between CSD (which is associated to aura) and lactate in migraine [51].
Lipid metabolism: cholesterol
Lipids can also be utilised as an energy source and are stored by astrocytes. Lipid droplets can be metabolised via fatty acid β-oxidation to produce acetyl-CoA which can then be oxidised in the TCA (Fig. 1). Fatty acids can also cross the blood–brain-barrier, and following oxidation, provide metabolic support to neurons in the form of ketones, NADH, acetyl CoA, and FADH2.
Dysregulation of lipid metabolism has become evident in migraine, since obesity has been associated with a higher risk, [52] increased frequency, [53] worse prognosis and chronification of migraine [54, 55]. CGRP has also demonstrated a key role in lipid metabolism and energy homeostasis, since high fat meals lead to increased plasma CGRP concentrations [56]. Moreover, plasma CGRP concentrations were increased in obese women compared to normal weight controls [56]. The expression of several neurotransmitters and oestrogen receptors in adipose tissue, [57] has led to the theory that the hypothalamic-pituitary-adipose axis may contribute towards headache pathology. It is possible that CGRP has a pivotal role in this axis. Whilst CGRP monoclonal antibodies are effective at treating migraine, their long-term effects on bodyweight are still unknown and would need further investigation.
Alterations in blood lipids has been identified in migraine patients (particularly cholesterol and low-density lipoprotein -LDL-), [58,59,60,61] with higher lipid content in serum being associated with elevated frequency and severity of attacks (Table 1) [59, 60, 62].
Importantly, such direct associations with cholesterol and LDL may provide an explanation for the increased cardiovascular and stroke risk in migraine [63, 64]. There is a significant association between stroke and migraine with aura, [64, 65] and although some studies identified stronger associations between elevated total cholesterol and triglycerides and migraine with aura, [60], others found that patients exhibited lipid alterations independent of aura symptoms [59]. Both total and LDL cholesterol were reduced in patients after therapy, [62] suggesting that perturbed lipid metabolism is reversible. It is not yet known if increased cholesterol in migraine is a cause, an effect or correlations are due to underlying common factors. However, metabolite measurements in blood from mice following CSD also demonstrate alterations in lipid metabolism including increased prostaglandin and anti-inflammatory lipid mediators [66]. Additional studies in murine model are needed to further elucidate links between migraine, aura subtypes and cholesterol metabolism as well as pathophysiology. They could translate into changes into clinical practice as lowering total cholesterol and LDL may improve migraine [62].
Secondary headache and lipid metabolism
An association between obesity and headache is also evident in secondary headache disorders including idiopathic intracranial hypertension (IIH). IIH is characterised by raised intracranial pressure and features headache with migraine-like characteristics [67]. Over 90% of IIH patients are obese, [68] and the incidence of the disease is increasing in parallel with rising obesity rates [69]. In newly diagnosed female IIH patients, higher BMI and moderate weight gain of 5% were associated with greater risk of IIH [70]. Moreover, adiposity was found to be associated with disease activity and insulin resistance in IIH, with adipocytes being transcriptional primed for gaining adipose mass [71]. Weight loss via surgical or diet interventions is therapeutic in IIH and was associated with improvement in headache measures [72, 73]. Investigating the metabolic pathways underpinning the pathogenesis of IIH would be beneficial to development of biomarkers and therapeutic targets.
Fatty acid metabolism
Prolonged activity neurons leads to upregulated β-oxidation and consequently increased reactive oxygen species (ROS) and peroxidated fatty acids [74, 75]. Using high throughput mass spectrometry methods, plasma fatty acids were found to be differentially metabolized in chronic migraine patients compared to controls, with levels correlating with depression [76]. Improving fatty acid metabolism may also provide therapeutic benefits. This has been suggested in preliminary studies testing the effects of supplementation with Palmitoylethanolamide (PEA), an endogenous fatty acid amide found within the central nervous system and able to stimulate fatty acid oxidation [77]. PEA supplementation reduced number of headache attacks per month and pain intensity in paediatric patients with migraine without aura, [78] whereas it reduced pain intensity (when given in combination with non-steroidal anti-inflammatory analgesics) in migraine with aura [79]. Moreover, diets rich in omega 3 fatty acids (such as containing 1.5 g of eicosapentaenoic acid and docosahexaenoic acid a day) were also found to reduce headache frequency compared with a normal intake of fatty acids (150 mg of eicosapentaenoic acid and docosahexaenoic acid a day) [80].
Fatty acids also play a role in modulating neuroinflammation, which is a function potentially of benefit in neurodegenerative diseases [81]. Hence, supplementing the diet with sources of fatty acids is a migraine treatment approach that needs exploring in further randomised trials which may show benefits in other conditions as well.
Mitochondrial function and oxidative phosphorylation
Healthy neurons rely primarily on oxidative phosphorylation for energy, a process hosted by mitochondria which are vital organelles.
For the investigation of mitochondrial function in vivo, most models utilise CSD as a mechanism of migraine. Use of two-photon fluorescent imaging to measure the ratio of autofluorescent reduced NADH to non-fluorescent NAD+ allows quantification of changes in mitochondrial redox potential [82]. Using this method, it has been possible to observe an initial surge in NADH oxidation and ATP production followed by a prolonged period of depleted oxidation [42, 83, 84]. It is plausible that this prolonged period of reduced NADH utilisation may be due to the limited supply of oxygen in neural tissue due to vascular changes during CSD [42]. Moreover, repetitive CSD events were able to gradually decrease baseline fluorescent changes, suggesting that chronic CSD events may lead to long-term alterations in the oxidative capacity of the tissue [84]. In addition to reduced oxidative capacity, CSD also resulted in a decreased mitochondrial membrane potential and therefore activity of the ATP synthase in rats [85]. Dural exposure to inflammatory soup resulted in a reduced spare respiratory capacity, specifically in the trigeminal nucleus caudalis in acute brain slices [86]. In these models, reduced mitochondrial function not only resulted in depleted ATP supply, but also in increased production of reactive oxygen species and calcium influx, which may all contribute towards the pathology of headache besides altering metabolic flux.
The modality of choice to investigate mitochondrial function in vivo in humans is 31-Phosphorus magnetic resonance spectroscopy (31P-MRS). Studies in migraine have consistently highlighted primary mitochondrial dysfunction in patients as evidenced by increased ADP in those with aura and FHM [87,88,89] and decreased phosphorylation potential (indicating reduced available free energy) in both patients with and without aura (Table 1) [90,91,92]. Together these results are consistent with those reported in 31P-MRS studies of mitochondrial cytopathies and some neurodegenerative conditions such as amyotrophic lateral sclerosis [93, 94]. Results suggest an imbalance between energy demand and production which may be the underlying reason for inability to maintain optimal intracellular ionic milieu and the reduced threshold for migraine attack. 31P-MRS data in patients may reciprocate findings in CSD animal models, since migraine with aura patients most frequently demonstrate alterations in mitochondrial function. Results regarding changes in ATP concentrations remain divided, with most studies either not reporting ATP levels, or concentrations remaining unchanged or similar to that of controls [95]. ATP may remain constant whilst free energy is reduced since depleted ATP is demonstrated mostly in necrotic tissue, [96, 97] which is not typically a feature of migraine. Some further indirect evidence was provided by analysis of the transcriptome of peripheral blood mononuclear cells from migraine patients, which also revealed alteration in pathways linked to oxidative phosphorylation, with the majority of altered genes being downregulated compared to controls [46].
Lastly, a strong argument for dysfunction in oxidative phosphorylation in migraine is provided by the therapeutic benefits of coenzyme Q10 (CoQ10) supplementation. CoQ10 is an electron acceptor in the electron transport chain which has been shown to improve mitochondrial respiration in mitochondrial cytopathies [98]. Several trials have demonstrated the ability of CoQ10 to reduce migraine attack duration, [99] frequency, [100] severity and migraine days per month [101]. In addition to acting in the electron transfer chain, CoQ10 also has an anti-inflammatory role which may be favourable in the treatment of migraine, and was able to reduce nitric oxide (NO), [102] TNF-α and CGRP in a placebo-controlled trial [103]. Reduction in attack frequency, [104] and severity [104, 105] was also accomplished with use of riboflavin further supporting the role of dysfunctional oxidative phosphorylation in migraine. Riboflavin is a precursor of both FAD flavin adenine dinucleotide (FAD) an electron donor involved in complex II and flavin mononucleotide (FMN), a component of complex I.
Insulin
Insulin has become a hormone of interest in migraine since numerous studies have identified insulin resistance in patients [106,107,108,109]. Resistance was also associated with migraine disease duration in a study which reported metabolic syndrome in 31.9% of chronic migraine patients [109]. Although there were no differences found between those with or without aura, identification of a single nucleotide polymorphism in the insulin receptor gene has been found to be associated with migraine aura, further implicating the role of insulin function in hyperexcitability involved in migraine aetiology [110]. However, until studies are conducted comparing insulin function in migraine patients with obesity or diabetes versus obese or diabetes controls, it remains difficult to attribute changes in insulin resistance solely to migraine.
Animal studies have revealed that insulin may potentially modulate the release of CGRP. In particular, insulin can induce the release of CGRP via sensitization of neuronal Transient Receptor Potential Cation Channel Subfamily V Member 1 (TRVP1—Fig. 1) [111]. Insulin is also able to sensitize vascular TRPV1 receptors to induce vasodilation, a similar effect shown in isolated mesenteric blood vessels of rats [112]. In addition to their activation, vascular TRPV1 receptors are also sensitized to its agonists following insulin interaction, inducing a more pronounced vasoconstrictive effect when activated with capsaicin [111].
CGRP is also able to regulate insulin secretion [113] via TRPV1 activation, [114] potentially suggesting a feedback loop between excessive nociception and altered insulin function. TRPV1 knockout mice demonstrated glucose intolerance, whereas agonism of TRVP1 in wild type mice induced insulin secretion [114]. Moreover, CGRP infusions in rat and dog models have exhibited multiple markers of insulin resistance including increased plasma glucose, [115] decreased glucose uptake, [116] impaired glycogen synthesis in muscle, [117] and increased hepatic glucose production [116]. Ex vivo investigation of pancreatic islets in a model of diabetes and diet-induced obesity exhibited CGRP’s ability to block glucose-stimulated insulin secretion, insulin-2 gene expression and reduce glycolytic capacity [118]. One may speculate as to whether insulin dysfunction may be a predisposing feature of metabolic abnormalities in migraine, or if it is a result of chronic CGRP exposure. Investigating the effects of disturbed insulin secretion such as in type-2 diabetes patients on CGRP concentrations and migraine prevalence would provide further evidence of a reciprocal relationship.
Although antagonism of CGRP signalling has demonstrated significant efficacy in migraine prophylaxis, the effects on insulin function have not yet been assessed in patients. CGRP receptor antagonism in mice has shown moderate improvements in oral glucose tolerance [119]. CGRP-α knockout mice exhibited improved insulin sensitivity and glucose handling in response to a high-fat diet [120]. In other models of diabetes and diet-induced obesity, the use of antibodies to block CGRP-α also improved glucose tolerance, insulin sensitivity and resulted in weight loss [118]. The α isoform of CGRP may have a more dominant role in glucose handling, since inhibition of both α and β isoforms upregulated insulin secretion but did not affect plasma glucose concentrations [121]. Since CGRP antagonism has shown promising results at improving glucose and insulin metabolism in vivo, it would be important to probe putative benefits in metabolism in patients with migraine.
Amylin
Amylin is a pancreatic hormone co-released with insulin in response to food intake [122]. It lowers serum glucose by reducing glycogen release and may have a direct role in trigeminal nerve sensitisation, since it shares a substantial amino acid similarity to CGRP [123]. Both amylin and CGRP can bind to theAMY1 receptor which is located throughout the trigeminovascular system [124]. It has also been shown to be present in elevated concentrations in migraine patients [125]. Moreover, a recent provocation study demonstrated that infusion of the amylin analogue Pramlintide, induced migraine attack in migraine without aura patients [126]. Similar studies in mice also indicated sensitisation following amylin infusion by decreasing Von Frey thresholds and increasing aversion of bright light, particularly in female mice [126]. Squint-detecting assays, used as an automated measure of grimacing and pain, also detected increased squinting in female mice following amylin injection [127].
Whilst CGRP has become a popular target for migraine therapeutics, the therapeutic potentials of amylin antagonism have not yet been fully explored. Pramlintide, an amylin receptor agonist, is currently licensed for diabetes in the United States [128]. It may be important to consider headache as a side effect in diabetic patients, since its off-target effects may include activation of the trigeminovascular system.
Conclusion
The pathophysiology of migraine is evolving and may also feature mitochondrial and metabolic deficits. Moreover, migraine is prevalent in those with mitochondrial disorders and structural and biochemical impairments in electron transfer chain have been identified in migraineurs [129]. Interesting, CGRP a major nociceptive peptide involved in migraine demonstrates a reciprocal relationship with multiple metabolic pathways including glucose and lipid utilisation. It is still uncertain as to whether metabolic deficits result in excessive CGRP-mediated nociception or vice versa. Attributing metabolic perturbations exclusively to migraine may be difficult, since common migraine comorbidities also feature some metabolic alterations such as obesity and depression [130]. However, both clinical and in vivo evidence suggests that an imbalance between energetic demand and supply may contribute towards migraine pathology. Therefore, perturbation of metabolic pathways which exacerbate this imbalance may be the basis for the metabolic component of migraine.
Availability of data and materials
Not applicable.
Abbreviations
- 31P-MRS:
-
Phosphorus magnetic resonance spectroscopy
- 18F:
-
Fluorodeoxyglucose PET (18F-FDG PET)
- ADP:
-
Adenosine diphosphate
- AMY1:
-
Amylin type 1
- ATP:
-
Adenosine triphosphate
- CGRP:
-
Calcitonin gene related peptide
- CoQ10:
-
Coenzyme Q10
- CSD:
-
Cortical spreading depression
- FADH2 :
-
Flavin adenine dinucleotide
- GLUT:
-
Glucose transporter
- LDL:
-
Low density lipoproteins
- MRS:
-
Magnetic resonance spectroscopy
- NADH:
-
Nicotinamide adenine dinucleotide
- TNF-α:
-
Tumour necrosis factor alpha
- TRP:
-
Transient receptor potential
- TRPV1:
-
Transient Receptor Potential Cation Channel Subfamily V Member 1
- TCA:
-
Tricarboxylic acid
References
GBD 2017 Disease and Injury Incidence and Prevalence Collaborators (2018) Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 392(10159):1789–858. https://doi.org/10.1016/s0140-6736(18)32279-7
Steiner TJ, Stovner LJ, Birbeck GL (2013) Migraine: the seventh disabler. J Headache Pain 14:1–1. https://doi.org/10.1186/1129-2377-14-1
Lipton RB, Hamelsky SW, Kolodner KB, Steiner TJ, Stewart WF (2000) Migraine, quality of life, and depression. Neurol 55(5):629. https://doi.org/10.1212/WNL.55.5.629
Hamelsky SW, Lipton RB (2006) Psychiatric comorbidity of migraine. Headache 46(9):1327–1333. https://doi.org/10.1111/j.1526-4610.2006.00576.x
Deng H, Li G-g, Nie H, Feng Y-y, Guo G-y, Guo W-l et al (2020) Efficacy and safety of calcitonin-gene-related peptide binding monoclonal antibodies for the preventive treatment of episodic migraine – an updated systematic review and meta-analysis. BMC Neurol 20(1):57. https://doi.org/10.1186/s12883-020-01633-3
Terrin A, Bello L, Valentino ML, Caporali L, Sorarù G, Carelli V et al (2022) The relevance of migraine in the clinical spectrum of mitochondrial disorders. Sci Rep 12(1):4222. https://doi.org/10.1038/s41598-022-08206-z
Guldiken B, Guldiken S, Taskiran B, Koc G, Turgut N, Kabayel L et al (2009) Migraine in metabolic syndrome. Neurologist 15(2):55–58. https://doi.org/10.1097/NRL.0b013e31817781b6
Sinclair AJ, Matharu M (2012) Migraine, cerebrovascular disease and the metabolic syndrome. Ann Indian Acad Neurol 15(Suppl 1):S72–S77. https://doi.org/10.4103/0972-2327.100015
Grech O, Mollan SP, Wakerley BR, Fulton D, Lavery GG, Sinclair AJ (2021) The Role of Metabolism in Migraine Pathophysiology and Susceptibility. Life. 11(5):415. https://doi.org/10.3390/life11050415
Goadsby PJ, Lipton RB, Ferrari MD (2002) Migraine–current understanding and treatment. N Engl J Med 346(4):257–270. https://doi.org/10.1056/NEJMra010917
Benemei S, De Cesaris F, Fusi C, Rossi E, Lupi C, Geppetti P (2013) TRPA1 and other TRP channels in migraine. J Headache Pain 14(1):71. https://doi.org/10.1186/1129-2377-14-71
Meng J, Ovsepian SV, Wang J, Pickering M, Sasse A, Aoki KR et al (2009) Activation of TRPV1 mediates calcitonin gene-related peptide release, which excites trigeminal sensory neurons and is attenuated by a retargeted botulinum toxin with anti-nociceptive potential. J Neurosci 29(15):4981–4992. https://doi.org/10.1523/jneurosci.5490-08.2009
Ho TW, Mannix LK, Fan X, Assaid C, Furtek C, Jones CJ et al (2008) Randomized controlled trial of an oral CGRP receptor antagonist, MK-0974, in acute treatment of migraine. Neurol 70(16):1304. https://doi.org/10.1212/01.WNL.0000286940.29755.61
Hewitt DJ, Aurora SK, Dodick DW, Goadsby PJ, Ge Y, Bachman R et al (2011) Randomized controlled trial of the CGRP receptor antagonist MK-3207 in the acute treatment of migraine. Cephalalgia 31(6):712–722. https://doi.org/10.1177/0333102411398399
Olesen J, Diener HC, Husstedt IW, Goadsby PJ, Hall D, Meier U et al (2004) Calcitonin gene-related peptide receptor antagonist BIBN 4096 BS for the acute treatment of migraine. N Engl J Med 350(11):1104–1110. https://doi.org/10.1056/NEJMoa030505
Dodick DW, Goadsby PJ, Silberstein SD, Lipton RB, Olesen J, Ashina M et al (2014) Safety and efficacy of ALD403, an antibody to calcitonin gene-related peptide, for the prevention of frequent episodic migraine: a randomised, double-blind, placebo-controlled, exploratory phase 2 trial. Lancet Neurol 13(11):1100–1107. https://doi.org/10.1016/S1474-4422(14)70209-1
Dodick DW, Goadsby PJ, Spierings ELH, Scherer JC, Sweeney SP, Grayzel DS (2014) Safety and efficacy of LY2951742, a monoclonal antibody to calcitonin gene-related peptide, for the prevention of migraine: a phase 2, randomised, double-blind, placebo-controlled study. Lancet Neurol 13(9):885–892. https://doi.org/10.1016/S1474-4422(14)70128-0
Vecchia D, Pietrobon D (2012) Migraine: a disorder of brain excitatory–inhibitory balance? Trends Neurosci 35(8):507–520
Goadsby PJ, Holland PR, Martins-Oliveira M, Hoffmann J, Schankin C, Akerman S (2017) Pathophysiology of migraine: a disorder of sensory processing. Physiol Rev 97(2):553–622. https://doi.org/10.1152/physrev.00034.2015
Hadjikhani N, Sanchez Del Rio M, Wu O, Schwartz D, Bakker D, Fischl B et al (2001) Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci USA 98(8):4687–4692. https://doi.org/10.1073/pnas.071582498
Leao AAP, (1944) SPREADING DEPRESSION OF ACTIVITY IN THE CEREBRAL CORTEX. J Neurophysiology, 7, 359–390. https://doi.org/10.1152/jn.1944.7.6.359.
Bowyer SM, Aurora KS, Moran JE, Tepley N, Welch KM (2001) Magnetoencephalographic fields from patients with spontaneous and induced migraine aura. Ann Neurol 50(5):582–587. https://doi.org/10.1002/ana.1293
Headache classification committee of the international headache S (2013) The international classification of headache disorders, 3rd edition (beta version). Cephalalgia 33(9):629–808. https://doi.org/10.1177/0333102413485658
Zhang X, Levy D, Noseda R, Kainz V, Jakubowski M, Burstein R (2010) Activation of meningeal nociceptors by cortical spreading depression: implications for migraine with aura. J Neurosci : J Soc Neurosci 30(26):8807–8814. https://doi.org/10.1523/JNEUROSCI.0511-10.2010
Borkum JM (2021) Brain energy deficit as a source of oxidative stress in migraine: a molecular basis for migraine susceptibility. Neurochem Res 46(8):1913–1932. https://doi.org/10.1007/s11064-021-03335-9
Gerhart DZ, LeVasseur RJ, Broderius MA, Drewes LR (1989) Glucose transporter localization in brain using light and electron immunocytochemistry. J Neurosci Res 22(4):464–472. https://doi.org/10.1002/jnr.490220413
Mongeon R, Venkatachalam V, Yellen G (2016) Cytosolic NADH-NAD+ redox visualized in brain slices by two-photon fluorescence lifetime biosensor imaging. Antioxid Redox Signal 25(10):553–563
Díaz-García CM, Yellen G (2019) Neurons rely on glucose rather than astrocytic lactate during stimulation. J Neurosci Res 97(8):883–889. https://doi.org/10.1002/jnr.24374
Hoffmann U, Sukhotinsky I, Eikermann-Haerter K, Ayata C (2013) Glucose modulation of spreading depression susceptibility. J Cereb Blood Flow Metab : J Int Soc Cereb Blood Flow Metab 33(2):191–195. https://doi.org/10.1038/jcbfm.2012.132
Bures J, Buresova O (1960) Activation of latent foci of spreading cortical depression in rats. J Neurophysiol 23:225–236. https://doi.org/10.1152/jn.1960.23.3.225
Astrup J, Norberg K (1976) Potassium activity in cerebral cortex in rats during progressive severe hypoglycemia. Brain Res 103(2):418–423. https://doi.org/10.1016/0006-8993(76)90817-9
Martins-Oliveira M, Akerman S, Holland PR, Hoffmann JR, Tavares I, Goadsby PJ (2017) Neuroendocrine signaling modulates specific neural networks relevant to migraine. Neurobiol Dis 101:16–26. https://doi.org/10.1016/j.nbd.2017.01.005
Giffin NJ, Ruggiero L, Lipton RB, Silberstein SD, Tvedskov JF, Olesen J et al (2003) Premonitory symptoms in migraine. Neurol 60(6):935. https://doi.org/10.1212/01.WNL.0000052998.58526.A9
Martin PR, Seneviratne HM (1997) Effects of food deprivation and a stressor on head pain. Health Psychol 16(4):310
Mohammad SS, Coman D, Calvert S (2014) Glucose transporter 1 deficiency syndrome and hemiplegic migraines as a dominant presenting clinical feature. J Paediatr Child Health 50(12):1025–1026. https://doi.org/10.1111/jpc.12613
Scoppola C, Magli G, Conti M, Fadda M, Luzzu GM, Simula DM et al (2021) CACNA1A-Linked hemiplegic migraine in GLUT 1 deficiency syndrome: a case report. Front Neurol 12:679354. https://doi.org/10.3389/fneur.2021.679354
Kim JH, Kim S, Suh SI, Koh SB, Park KW, Oh K (2010) Interictal metabolic changes in episodic migraine: a voxel-based FDG-PET study. Cephalalgia 30(1):53–61. https://doi.org/10.1111/j.1468-2982.2009.01890.x
Magis D, D’Ostilio K, Thibaut A, De Pasqua V, Gerard P, Hustinx R et al (2017) Cerebral metabolism before and after external trigeminal nerve stimulation in episodic migraine. Cephalalgia 37(9):881–891. https://doi.org/10.1177/0333102416656118
Torres-Ferrus M, Pareto D, Gallardo VJ, Cuberas-Borrós G, Alpuente A, Caronna E et al (2021) Cortical metabolic and structural differences in patients with chronic migraine. An exploratory 18FDG-PET and MRI study. J Headache Pain 22(1):75. https://doi.org/10.1186/s10194-021-01289-5
Magistretti PJ, Allaman I (2018) Lactate in the brain: from metabolic end-product to signalling molecule. Nat Rev Neurosci 19(4):235–249. https://doi.org/10.1038/nrn.2018.19
Scheller D, Kolb J, Tegtmeier F (1992) Lactate and pH change in close correlation in the extracellular space of the rat brain during cortical spreading depression. Neurosci Lett 135(1):83–86. https://doi.org/10.1016/0304-3940(92)90141-s
Takano T, Tian GF, Peng W, Lou N, Lovatt D, Hansen AJ et al (2007) Cortical spreading depression causes and coincides with tissue hypoxia. Nat Neurosci 10(6):754–762. https://doi.org/10.1038/nn1902
Feuerstein D, Backes H, Gramer M, Takagaki M, Gabel P, Kumagai T et al (2016) Regulation of cerebral metabolism during cortical spreading depression. J Cereb Blood Flow Metab 36(11):1965–1977. https://doi.org/10.1177/0271678x15612779
Proia P, Amato A, Contrò V, Monaco AL, Brusa J, Brighina F et al (2019) Relevance of lactate level detection in migrane and fibromyalgia. Eur J Trans Myol 29(2):8202. https://doi.org/10.4081/ejtm.2019.8202
Okada H, Araga S, Takeshima T, Nakashima K (1998) plasma lactic acid and pyruvic acid levels in migraine and tension-type headache. headache: J Head Face Pain 38(1):39–42. https://doi.org/10.1046/j.1526-4610.1998.3801039.x
Aczél T, Körtési T, Kun J, Urbán P, Bauer W, Herczeg R et al (2021) Identification of disease- and headache-specific mediators and pathways in migraine using blood transcriptomic and metabolomic analysis. J Headache Pain 22(1):117. https://doi.org/10.1186/s10194-021-01285-9
Ribeiro GACdS, Scola RH, Piovesan EJ, Wollmann Junior DR, Paiva EdS, Cunha CLPd et al (2015) A importância de ácido láctico na enxaqueca e na fibromialgia. Rev Bras Reumatol 55(6):471–6. https://doi.org/10.1016/j.rbr.2015.02.002
Grimaldi D, Tonon C, Cevoli S, Pierangeli G, Malucelli E, Rizzo G et al (2010) Clinical and neuroimaging evidence of interictal cerebellar dysfunction in FHM2. Cephalalgia 30(5):552–559. https://doi.org/10.1111/j.1468-2982.2009.01979.x
Shibata K, Yamane K, Otuka K, Iwata M (2008) Abnormal visual processing in migraine with aura: a study of steady-state visual evoked potentials. J Neurol Sci 271(1–2):119–126. https://doi.org/10.1016/j.jns.2008.04.004
Watanabe H, Kuwabara T, Ohkubo M, Tsuji S, Yuasa T (1996) Elevation of cerebral lactate detected by localized 1H-magnetic resonance spectroscopy in migraine during the interictal period. Neurol 47(4):1093–1095. https://doi.org/10.1212/wnl.47.4.1093
Reyngoudt H, Paemeleire K, Dierickx A, Descamps B, Vandemaele P, De Deene Y et al (2011) Does visual cortex lactate increase following photic stimulation in migraine without aura patients? A functional 1H-MRS study. J Headache Pain 12(3):295–302. https://doi.org/10.1007/s10194-011-0295-7
Peterlin BL, Rosso AL, Williams MA, Rosenberg JR, Haythornthwaite JA, Merikangas KR et al (2013) Episodic migraine and obesity and the influence of age, race, and sex. Neurol 81(15):1314. https://doi.org/10.1212/WNL.0b013e3182a824f7
Bigal ME, Tsang A, Loder E, Serrano D, Reed ML, Lipton RB et al (2007) Body mass index and episodic headaches: a population-based study. Arch Intern Med 167(18):1964–1970. https://doi.org/10.1001/archinte.167.18.1964
Scher IA, Stewart FW, Ricci AJ, Lipton BR (2003) Factors associated with the onset and remission of chronic daily headache in a population-based study. PAIN. 106(1):81
Bigal ME, Lipton RB (2006) Obesity is a risk factor for transformed migraine but not chronic tension-type headache. Neurol 67(2):252. https://doi.org/10.1212/01.wnl.0000225052.35019.f9
Zelissen PM, Koppeschaar HP, Lips CJ, Hackeng WH (1991) Calcitonin gene-related peptide in human obesity. Peptides 12(4):861–863
Nicolaysen A, Gammelsaeter R, Storm-Mathisen J, Gundersen V, Iversen PO (2007) The components required for amino acid neurotransmitter signaling are present in adipose tissues. J Lipid Res 48(10):2123–2132. https://doi.org/10.1194/jlr.M700021-JLR200
Hedman C, Winther K, Knudsen J (1988) Platelet function in classic migraine during attack-free periods. Acta Neurol Scand 78(4):271–277
Gruber HJ, Bernecker C, Pailer S, Lechner A, Horejsi R, Möller R et al (2010) Lipid profile in normal weight migraineurs–evidence for cardiovascular risk. Eur J Neurol 17(3):419–425
Rist PM, Tzourio C, Kurth T (2011) Associations between lipid levels and migraine: cross-sectional analysis in the epidemiology of vascular ageing study. Cephalalgia 31(14):1459–1465. https://doi.org/10.1177/0333102411421682
Onderwater GLJ, Ligthart L, Bot M, Demirkan A, Fu J, van der Kallen CJH et al (2019) Large-scale plasma metabolome analysis reveals alterations in HDL metabolism in migraine. Neurol 92(16):e1899. https://doi.org/10.1212/WNL.0000000000007313
Tana C, Santilli F, Martelletti P, di Vincenzo A, Cipollone F, Davì G et al (2015) Correlation between migraine severity and cholesterol levels. Pain Pract 15(7):662–670
Scher AI, Terwindt GM, Picavet HSJ, Verschuren WMM, Ferrari MD, Launer LJ (2005) Cardiovascular risk factors and migraine. Neurol 64(4):614. https://doi.org/10.1212/01.WNL.0000151857.43225.49
Hu X, Zhou Y, Zhao H, Peng C (2017) Migraine and the risk of stroke: an updated meta-analysis of prospective cohort studies. Neurol Sci 38(1):33–40
Mahmoud AN, Mentias A, Elgendy AY, Qazi A, Barakat AF, Saad M et al (2018) Migraine and the risk of cardiovascular and cerebrovascular events: a meta-analysis of 16 cohort studies including 1 152 407 subjects. BMJ Open 8(3):e020498. https://doi.org/10.1136/bmjopen-2017-020498
Loonen ICM, Kohler I, Ghorasaini M, Giera M, van den Maagdenberg A, Mayboroda OA et al (2022) Changes in plasma lipid levels following cortical spreading depolarization in a transgenic mouse model of familial hemiplegic migraine. Metabolites. 12(3):220. https://doi.org/10.3390/metabo12030220
Mollan SP, Grech O, Sinclair AJ (2021) Headache attributed to idiopathic intracranial hypertension and persistent post-idiopathic intracranial hypertension headache: A narrative review. Headache: J Head Face Pain 61(6):808–16. https://doi.org/10.1111/head.14125
Mollan SP, Ali F, Hassan-Smith G, Botfield H, Friedman DI, Sinclair AJ (2016) Evolving evidence in adult idiopathic intracranial hypertension: pathophysiology and management. J Neurol Neurosurg Psychiatr 87(9):982–992. https://doi.org/10.1136/jnnp-2015-311302
Kilgore KP, Lee MS, Leavitt JA, Mokri B, Hodge DO, Frank RD et al (2017) Re-evaluating the incidence of idiopathic intracranial hypertension in an era of increasing obesity. Ophthalmol 124(5):697–700. https://doi.org/10.1016/j.ophtha.2017.01.006
Daniels AB, Liu GT, Volpe NJ, Galetta SL, Moster ML, Newman NJ et al (2007) Profiles of obesity, weight gain, and quality of life in idiopathic intracranial hypertension (pseudotumor cerebri). Am J Ophthalmol 143(4):635–641. https://doi.org/10.1016/j.ajo.2006.12.040
Westgate CS, Botfield HF, Alimajstorovic Z, Yiangou A, Walsh M, Smith G et al (2021) Systemic and adipocyte transcriptional and metabolic dysregulation in idiopathic intracranial hypertension. JCI Insight. 6(10):e145346. https://doi.org/10.1172/jci.insight.145346
Sinclair AJ, Burdon MA, Nightingale PG, Ball AK, Good P, Matthews TD et al (2010) Low energy diet and intracranial pressure in women with idiopathic intracranial hypertension: prospective cohort study. BMJ 341:c2701. https://doi.org/10.1136/bmj.c2701
Mollan SP, Mitchell JL, Ottridge RS, Aguiar M, Yiangou A, Alimajstorovic Z et al (2021) Effectiveness of bariatric surgery vs community weight management intervention for the treatment of idiopathic intracranial hypertension: a randomized clinical trial. JAMA Neurol 78:678. https://doi.org/10.1001/jamaneurol.2021.0659
Barber CN, Raben DM (2019) Lipid metabolism crosstalk in the brain: glia and neurons. Front Cell Neurosci 13:212. https://doi.org/10.3389/fncel.2019.00212
Tracey TJ, Kirk SE, Steyn FJ, Ngo ST (2021) The role of lipids in the central nervous system and their pathological implications in amyotrophic lateral sclerosis. Semin Cell Dev Biol 112:69–81. https://doi.org/10.1016/j.semcdb.2020.08.012
Fonteh AN, Castor K, Arakaki X, Woldeamanuel Y, Cowan R, Harrington M (2020) Plasma lipid metabolism exemplifies the interoceptive nature of chronic migraine. FASEB J 34(S1):1. https://doi.org/10.1096/fasebj.2020.34.s1.08999
Minnich A, Tian N, Byan L, Bilder G (2001) A potent PPARα agonist stimulates mitochondrial fatty acid β-oxidation in liver and skeletal muscle. Ame J Physiol-Endocrinol Metab 280(2):E270–E279. https://doi.org/10.1152/ajpendo.2001.280.2.E270
Papetti L, Sforza G, Tullo G, Alaimo di Loro P, Moavero R, Ursitti F et al (2020) Tolerability of palmitoylethanolamide in a pediatric population suffering from migraine: a pilot study. Pain Res Manag 2020:3938640. https://doi.org/10.1155/2020/3938640
Chirchiglia D, Cione E, Caroleo MC, Wang M, Di Mizio G, Faedda N. et al. Effects of add-on ultramicronized n-palmitol ethanol amide in patients suffering of migraine with aura: a pilot study. Front Neurol. 2018;9 (674). https://doi.org/10.3389/fneur.2018.00674.
Ramsden CE, Zamora D, Faurot KR, MacIntosh B, Horowitz M, Keyes GS et al (2021) Dietary alteration of n-3 and n-6 fatty acids for headache reduction in adults with migraine: randomized controlled trial. BMJ 374:n1448. https://doi.org/10.1136/bmj.n1448
Calviello G, Su HM, Weylandt KH, Fasano E, Serini S, Cittadini A (2013) Experimental evidence of ω-3 polyunsaturated fatty acid modulation of inflammatory cytokines and bioactive lipid mediators: their potential role in inflammatory, neurodegenerative, and neoplastic diseases. Biomed Res Int 2013:743171. https://doi.org/10.1155/2013/743171
Vergen J, Hecht C, Zholudeva LV, Marquardt MM, Hallworth R, Nichols MG (2012) Metabolic imaging using two-photon excited NADH intensity and fluorescence lifetime imaging. Microsc Microanal : J Microsc Soc Ame, Microbeam Anal Soc, Microsc Soc Canada 18(4):761–770. https://doi.org/10.1017/S1431927612000529
Galeffi F, Somjen GG, Foster KA, Turner DA (2011) Simultaneous monitoring of tissue PO2 and NADH fluorescence during synaptic stimulation and spreading depression reveals a transient dissociation between oxygen utilization and mitochondrial redox state in rat hippocampal slices. J Cereb Blood Flow Metab 31(2):626–639. https://doi.org/10.1038/jcbfm.2010.136
Carlson AP, Carter RE, Shuttleworth CW (2012) Vascular, electrophysiological, and metabolic consequences of cortical spreading depression in a mouse model of simulated neurosurgical conditions. Neurol Res 34(3):223–231. https://doi.org/10.1179/1743132811Y.0000000077
Li F, Qiu E, Dong Z, Liu R, Wu S, Yu S (2011) Protection of flunarizine on cerebral mitochondria injury induced by cortical spreading depression under hypoxic conditions. J Headache Pain 12(1):47–53. https://doi.org/10.1007/s10194-011-0300-1
Fried NT, Moffat C, Seifert EL, Oshinsky ML (2014) Functional mitochondrial analysis in acute brain sections from adult rats reveals mitochondrial dysfunction in a rat model of migraine. Am J Physiol Cell Physiol 307(11):C1017–C1030. https://doi.org/10.1152/ajpcell.00332.2013
Uncini A, Lodi R, Di Muzio A, Silvestri G, Servidei S, Lugaresi A et al (1995) Abnormal brain and muscle energy metabolism shown by 31P-MRS in familial hemiplegic migraine. J Neurol Sci 129(2):214–222. https://doi.org/10.1016/0022-510x(94)00283-t
Barbiroli B, Montagna P, Cortelli P, Funicello R, Iotti S, Monari L et al (1992) Abnormal brain and muscle energy metabolism shown by 31P-magnetic resonance spectroscopy in patients affected by migraine with aura. Neurol 42(6):1209. https://doi.org/10.1212/WNL.42.6.1209
Sacquegna T, Lodi R, De Carolis P, Tinuper P, Cortelli P, Zaniol P et al (1992) Brain energy metabolism studied by 31P-MR spectroscopy in a case of migraine with prolonged aura. Acta Neurol Scand 86(4):376–380. https://doi.org/10.1111/j.1600-0404.1992.tb05104.x
Schulz UG, Blamire AM, Corkill RG, Davies P, Styles P, Rothwell PM (2007) Association between cortical metabolite levels and clinical manifestations of migrainous aura: an MR-spectroscopy study. Brain 130(Pt 12):3102–3110. https://doi.org/10.1093/brain/awm165
Montagna P, Cortelli P, Monari L, Pierangeli G, Parchi P, Lodi R et al (1994) 31P-magnetic resonance spectroscopy in migraine without aura. Neurol 44(4):666–669. https://doi.org/10.1212/wnl.44.4.666
Reyngoudt H, Paemeleire K, Descamps B, De Deene Y, Achten E (2011) 31P-MRS demonstrates a reduction in high-energy phosphates in the occipital lobe of migraine without aura patients. Cephalalgia 31(12):1243–1253. https://doi.org/10.1177/0333102410394675
Barbiroli B, Montagna P, Martinelli P, Lodi R, Iotti S, Cortelli P et al (1993) Defective brain energy metabolism shown by in vivo 31P MR spectroscopy in 28 patients with mitochondrial cytopathies. J Cereb Blood Flow Metab 13(3):469–474. https://doi.org/10.1038/jcbfm.1993.61
Sassani M, Alix JJ, McDermott CJ, Baster K, Hoggard N, Wild JM et al (2020) Magnetic resonance spectroscopy reveals mitochondrial dysfunction in amyotrophic lateral sclerosis. Brain 143(12):3603–3618. https://doi.org/10.1093/brain/awaa340
Bottomley PA, Hardy CJ (1989) Rapid, reliable in vivo assays of human phosphate metabolites by nuclear magnetic resonance. Clin Chem 35(3):392–395. https://doi.org/10.1093/clinchem/35.3.392
Grusch M, Polgar D, Gfatter S, Leuhuber K, Huettenbrenner S, Leisser C et al (2002) Maintenance of ATP favours apoptosis over necrosis triggered by benzamide riboside. Cell Death Differ 9(2):169–178. https://doi.org/10.1038/sj.cdd.4400937
Ekstrand M, Widell E, Hammar A, Akyürek LM, Johansson M, Fagerberg B et al (2017) Depletion of ATP and glucose in advanced human atherosclerotic plaques. PLoS One 12(6):e0178877-e. https://doi.org/10.1371/journal.pone.0178877
Barbiroli B, Iotti S, Cortelli P, Martinelli P, Lodi R, Carelli V et al (1999) Low brain intracellular free magnesium in mitochondrial cytopathies. J Cereb Blood Flow Metab 19(5):528–532. https://doi.org/10.1097/00004647-199905000-00007
Sazali S, Badrin S, Norhayati MN, Idris NS (2021) Coenzyme Q10 supplementation for prophylaxis in adult patients with migraine—a meta-analysis. BMJ Open 11(1):e039358. https://doi.org/10.1136/bmjopen-2020-039358
Sándor PS, Di Clemente L, Coppola G, Saenger U, Fumal A, Magis D et al (2005) Efficacy of coenzyme Q10 in migraine prophylaxis: A randomized controlled trial. Neurol 64(4):713. https://doi.org/10.1212/01.WNL.0000151975.03598.ED
Zeng Z, Li Y, Lu S, Huang W, Di W (2019) Efficacy of CoQ10 as supplementation for migraine: A meta-analysis. Acta Neurol Scand 139(3):284–293. https://doi.org/10.1111/ane.13051
Nattagh-Eshtivani E, Dahri M, Hashemilar M, Tarighat-Esfanjani A (2018) The effect of Coenzyme Q10 supplementation on serum levels of lactate, pyruvate, matrix metalloproteinase 9 and nitric oxide in women with migraine. A double blind, placebo, controlled randomized clinical trial. Eur J Integr Med 21:70–6. https://doi.org/10.1016/j.eujim.2018.06.009
Dahri M, Tarighat-Esfanjani A, Asghari-Jafarabadi M, Hashemilar M (2019) Oral coenzyme Q10 supplementation in patients with migraine: Effects on clinical features and inflammatory markers. Nutr Neurosci 22(9):607–615. https://doi.org/10.1080/1028415x.2017.1421039
Boehnke C, Reuter U, Flach U, Schuh-Hofer S, Einhaupl KM, Arnold G (2004) High-dose riboflavin treatment is efficacious in migraine prophylaxis: an open study in a tertiary care centre. Eur J Neurol 11(7):475–477. https://doi.org/10.1111/j.1468-1331.2004.00813.x
Schoenen J, Jacquy J, Lenaerts M (1998) Effectiveness of high-dose riboflavin in migraine prophylaxis A randomized controlled trial. Neurol 50(2):466–470. https://doi.org/10.1212/wnl.50.2.466
Rainero I, Limone P, Ferrero M, Valfrè W, Pelissetto C, Rubino E et al (2005) Insulin Sensitivity is Impaired in Patients with Migraine. Cephalalgia 25(8):593–597. https://doi.org/10.1111/j.1468-2982.2005.00928.x
Cavestro C, Rosatello A, Micca G, Ravotto M, Pia Marino M, Asteggiano G et al (2007) Insulin metabolism is altered in migraineurs: a new pathogenic mechanism for migraine? Headache: J Head Face Pain 47(10):1436–42. https://doi.org/10.1111/j.1526-4610.2007.00719.x
Fava A, Pirritano D, Consoli D, Plastino M, Casalinuovo F, Cristofaro S et al (2014) Chronic migraine in women is associated with insulin resistance: a cross-sectional study. Eur J Neurol 21(2):267–272. https://doi.org/10.1111/ene.12289
Bhoi SK, Kalita J, Misra UK (2012) Metabolic syndrome and insulin resistance in migraine. J Headache Pain 13(4):321–326. https://doi.org/10.1007/s10194-012-0416-y
McCarthy LC, Hosford DA, Riley JH, Bird MI, White NJ, Hewett DR et al (2001) Single-nucleotide polymorphism alleles in the insulin receptor gene are associated with typical migraine. Genomics 78(3):135–149. https://doi.org/10.1006/geno.2001.6647
Rosta J, Tóth M, Friedrich N, Sántha P, Jancsó G, Dux M (2022) Insulin sensitizes neural and vascular TRPV1 receptors in the trigeminovascular system. J Headache Pain 23(1):7. https://doi.org/10.1186/s10194-021-01380-x
Mimaki Y, Kawasaki H, Okazaki M, Nakatsuma A, Araki H, Gomita Y (1998) Involvement of calcitonin gene-related peptide (CGRP) receptors in insulin-induced vasodilatation in mesenteric resistance blood vessels of rats. Br J Pharmacol 123(8):1684–1690
Gray ALH, Antevska A, Link BA, Bogin B, Burke SJ, Dupuy SD et al (2021) α-CGRP disrupts amylin fibrillization and regulates insulin secretion: implications on diabetes and migraine. Chem Sci 12(16):5853–5864. https://doi.org/10.1039/d1sc01167g
Zhong B, Ma S, Wang DH (2019) TRPV1 mediates glucose-induced insulin secretion through releasing neuropeptides. In Vivo 33(5):1431. https://doi.org/10.21873/invivo.11621
Moore MC, Lin DW, Colburn CA, Goldstein RE, Neal DW, Cherrington AD (1999) Insulin- and glucagon-independent effects of calcitonin gene—related peptide in the conscious dog. Metab - Clin Exp 48(5):603–610. https://doi.org/10.1016/S0026-0495(99)90058-6
Choi SB, Frontoni S, Rossetti L (1991) Mechanism by which calcitonin gene-related peptide antagonizes insulin action in vivo. Ame J Physiol-Endocrinol Metab 260(2):E321–E325. https://doi.org/10.1152/ajpendo.1991.260.2.E321
Leighton B, Cooper GJS (1988) Pancreatic amylin and calcitonin gene-related peptide cause resistance to insulin in skeletal muscle in vitro. Nat 335(6191):632–635. https://doi.org/10.1038/335632a0
Halloran J, Lalande A, Zang M, Chodavarapu H, Riera CE (2020) Monoclonal therapy against calcitonin gene-related peptide lowers hyperglycemia and adiposity in type 2 diabetes mouse models. Metab Open 8:100060. https://doi.org/10.1016/j.metop.2020.100060
Köhli P, Appelt J, Otto E, Jahn D, Baranowsky A, Bahn A et al (2021) Effects of CGRP receptor antagonism on glucose and bone metabolism in mice with diet-induced obesity. Bone 143:115646. https://doi.org/10.1016/j.bone.2020.115646
Walker CS, Li X, Whiting L, Glyn-Jones S, Zhang S, Hickey AJ et al (2010) mice lacking the neuropeptide α-calcitonin gene-related peptide are protected against diet-induced obesity. Endocrinol 151(9):4257–4269. https://doi.org/10.1210/en.2010-0284
Tanaka H, Kashiwagi R, Koizumi T (2013) Inhibition of calcitonin gene-related peptide (CGRP) has the potential to extend first-phase insulin secretion. Exp Clin Endocrinol Diabetes 121(5):280–285. https://doi.org/10.1055/s-0033-1341441
Kahn SE, D’Alessio DA, Schwartz MW, Fujimoto WY, Ensinck JW, Taborsky GJ Jr et al (1990) Evidence of cosecretion of islet amyloid polypeptide and insulin by beta-cells. Diabetes 39(5):634–638. https://doi.org/10.2337/diab.39.5.634
Westermark P, Wernstedt C, Wilander E, Sletten K (1986) A novel peptide in the calcitonin gene related peptide family as an amyloid fibril protein in the endocrine pancreas. Biochem Biophys Res Commun 140(3):827–831. https://doi.org/10.1016/0006-291x(86)90708-4
Walker CS, Eftekhari S, Bower RL, Wilderman A, Insel PA, Edvinsson L et al (2015) A second trigeminal CGRP receptor: function and expression of the AMY1 receptor. Ann Clin Transl Neurol 2(6):595–608. https://doi.org/10.1002/acn3.197
Irimia P, Martínez-Valbuena I, Mínguez-Olaondo A, Domínguez-Vivero C, Sánchez-Arias JA, Martínez-Vila E et al (2021) Interictal amylin levels in chronic migraine patients: A case-control study. Cephalalgia 41(5):604–612. https://doi.org/10.1177/0333102420977106
Ghanizada H, Al-Karagholi MA, Walker CS, Arngrim N, Rees T, Petersen J et al (2021) Amylin analog pramlintide induces migraine-like attacks in patients. Ann Neurol 89(6):1157–1171. https://doi.org/10.1002/ana.26072
Rea BJ, Sowers LP, Davison AL, Fairbanks AM, Wattiez A-S, Poolman P, et al. Female mice exhibit a more sensitive automated squint response to pain induced by CGRP and amylin. bioRxiv. 2021:2021.05.26.445893. https://journals.lww.com/pain/Abstract/2022/08000/Automated_detection_of_squint_as_a_sensitive_assay.12.aspx.
Ryan GJ, Jobe LJ, Martin R (2005) Pramlintide in the treatment of type 1 and type 2 diabetes mellitus. Clin Ther 27(10):1500–1512. https://doi.org/10.1016/j.clinthera.2005.10.009
Sangiorgi S, Mochi M, Riva R, Cortelli P, Monari L, Pierangeli G et al (1994) Abnormal platelet mitochondrial function in patients affected by migraine with and without aura. Cephalalgia 14(1):21–23
Koponen H, Kautiainen H, Leppänen E, Mäntyselkä P, Vanhala M (2015) Association between suicidal behaviour and impaired glucose metabolism in depressive disorders. BMC Psychiatry 15(1):163. https://doi.org/10.1186/s12888-015-0567-x
Acknowledgements
Not applicable.
Funding
O.G is funded by a Brain Research UK PhD studentship. G.T reports funding from Dutch Brain Foundation, Dutch Research Council. GGL is supported by a Wellcome Trust Senior Fellowship (104612/Z/14/Z), A.J.S was funded by a National Institute for Health Research (NIHR) clinician scientist fellowship (NIHR-CS-011–028) and the Medical Research Council, UK (MR/K015184/1) for the duration of the study. A.J.S is funded by a Sir Jules Thorn Award for Biomedical Science.
Author information
Authors and Affiliations
Contributions
O.G wrote the main manuscript and prepared the figure. M.S also contributed towards the writing of the main manuscript. G.T, G.G.L, S.P and A.J all contributed towards reviewing and editing the manuscript. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
O.G reports scientific consultancy fees from Invex therapeutics. G.T reports consultancy fees Novartis, Teva, Allergan, Lilly, Lundbeck, royalties from the Dutch Neurology Handbook, other from CGRP Education and Research Forum and presenter for webinars with Springer Media, Mednet, Ashfield MedComms, Remedica, Cygnea. S.P reports other Invex Therapeutics, other Heidelberg engineering; other from Chugai-Roche Ltd, other from Janssen, other from Allergan, other from Santen, other from Roche, other from Neurodiem, outside the submitted work. A.J.S reports consulting fees and stockholding with Invex therapeutics, during the conduct of the study. Other from Allergan, Amgen, Novartis and Cheisi All other authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Grech, O., Sassani, M., Terwindt, G. et al. Alterations in metabolic flux in migraine and the translational relevance. J Headache Pain 23, 127 (2022). https://doi.org/10.1186/s10194-022-01494-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s10194-022-01494-w