
Abstract
Ever since their identification, interest in the role of transient receptor potential (TRP) channels in health and disease has steadily increased. Robust evidence has underlined the role of TRP channels expressed in a subset of primary sensory neurons of the trigeminal ganglion to promote, by neuronal excitation, nociceptive responses, allodynia and hyperalgesia. In particular, the TRP vanilloid 1 (TRPV1) and the TRP ankyrin 1 (TRPA1) are expressed in nociceptive neurons, which also express the sensory neuropeptides, tachykinins, and calcitonin gene-related peptide (CGRP), which mediate neurogenic inflammatory responses. Of interest, CGRP released from the trigeminovascular network of neurons is currently recognized as a main contributing mechanism of migraine attack. The ability of TRPA1 to sense and to be activated by an unprecedented series of exogenous and endogenous reactive molecules has now been extensively documented. Several of the TRPA1 activators are also known as triggers of migraine attack. Thus, TRP channels, and particularly TRPA1, may be proposed as novel pathways in migraine pathophysiology and as possible new targets for its treatment.
Similar content being viewed by others

Review
TRP channels
The observation that the stimulation of a specific receptor, and the consequent, associated transient inward current [1, 2] are necessary to vision in Drosophila melanogaster has been the primal evidence of the transient receptor potential (TRP) family of channels, which currently encompasses more than 50 different channels [3]. TRP channels represent a heterogeneous system oriented towards environment perception, and participating in sensing visual, gustatory, olfactive, auditive, mechanical, thermal, and osmotic stimuli. TRP channels consist of six transmembrane domains (S1-S6) with both the NH2 and COOH termini localized into the cytosol. The COOH region is highly conserved among TRPs, whereas the NH2 region usually contains different numbers of ankyrin repeats, the 33-residue motifs with a conserved backbone and variable residues that mediate protein-protein interactions [4]. The entry of cations through homo- or heterotetramers occurs via the pore formed by loops between the S5 and S6 domains. TRPs are commonly described as nonselective Ca2+-permeable channels, however, their Ca2+/Na+ permeability ratio may vary widely between different members of the TRP family [5].
TRP channel gating is operated by both the direct action on the channel of a plethora of exogenous and endogenous physicochemical stimuli, and changes in the intracellular machinery, including activation of G-protein coupled receptor (GPCR) or tyrosine kinase receptor [6]. In mammals, the TRP family consists of 28 proteins grouped into 6 subfamilies according to sequence identity, namely TRP canonical (TRPC), TRP vanilloid (TRPV), TRP melastatin (TRPM), TRP polycystin (TRPP), TRP mucolipin (TRPML), and TRP ankyrin (TRPA) [7, 8]. The mammalian TRPC subfamily enlists 7 members (TRPC1-7), activated by the stimulation of GPCR and receptor tyrosine kinases [9], although TRPC1 seems to be directly activated by membrane stretch [10]. The TRPM has 8 members (TRPM1-8), and TRPM8 is activated by menthol and low temperatures (< 25°C). Of the three TRPML members, TRPML1 is widely expressed and has been described as an H+-sensor of endosomes/lysosomes, where it probably prevents overacidification [11]. The TRPP family can be subdivided into PKD1-like (TRPP1-like) and PKD2-like (TRPP2-like) proteins, which couple to act as a signaling complex at the plasma membrane plasma membrane [12]. Various diseases have been attributed to mutations of TRP channels. However, only a few TRP channelopathies, commonly known as “TRPpathies”, have been conclusively identified so far. TRPpathies include neurological disorders, renal diseases, and complex skeletal dysplasias [13].
TRPA1 and other thermo-TRPs
Primary sensory neurons express different TRP channels, including four of the six members of the TRPV subfamily. TRPV1, TRPV2, TRPV3, and TRPV4 channels sense warm-hot temperatures and are chemosensors for a large series of naturally occurring and synthetic ligands. TRPV1, the receptor of the vanilloid compound capsaicin (which promoted the labeling of the group with the V), is responsive to high proton concentrations (pH 5–6) [14, 15], anandamide [16] and various lipid derivatives [17]. Camphor and hypotonic solutions are non-selective activators of the TRPV3 and TRPV4, respectively [18, 19]. The synthetic compound 4α-phorbol 12,13-didecanoate (4α-PDD), low pH, citrate, endocannabinoids, and arachidonic acid metabolites may also gate TRPV4 [20, 21]. Activators of TRPV2 are not well identified, although the uricosuric agent probenecid can activate the channel [22]. Additional TRPs expressed in nociceptors are TRPA1 and TRPM8. Finally, there is also evidence that TRPM3, rather uniquely activated by pregnenolone sulfate, seems to be also expressed in primary sensory neurons [23].
A large NH2-terminal domain with 17 predicted ankyrin repeat domains characterizes TRPA1, the sole member of the TRPA subfamily. TRPA1, first cloned from human fetal lung fibroblasts, is widely expressed in mammals, where it has been found in hair cells, pancreas, heart, brain, keratinocytes [24], urinary bladder [25], prostate [26], arteries [27], enterochromaffin cells [28], odontoblasts and dental pulp [29, 30], synovial fibroblasts [31], and epithelial and smooth muscle cells of the airways and lung [32]. A large amount of evidence shows that TRPA1 plays a key role in the detection of pungent or irritant compounds, including principles contained in different spicy foods, such as allyl isothiocyanate (mustard oil) in horseradish [33], allicin and diallyldisulfide in garlic [34], and cinnamaldehyde in cinnamon [35]. Gingerol (in ginger), eugenol (in cloves), methyl salicylate (in wintergreen), carvacrol (in oregano), thymol (in thyme and oregano) [36], are also able to gate TRPA1. In addition, environmental irritants and industry pollutants, such as acetaldehyde, formalin, hydrogen peroxide, hypochlorite, isocyanates, ozone, carbon dioxide, ultraviolet light, and acrolein (a highly reactive α,β-unsatured aldehyde present in tear gas, cigarette smoke, smoke from burning vegetation, and vehicle exhaust), have been recognized as TRPA1 activators [37–45]. The dispute regarding the role of TRPA1 as a sensor of mechanical stimuli and noxious cold (< 17°C) remains unresolved [36]. Electrophilic molecules have been found to activate TRPA1 via a unique mechanism, mediated by a Michael addition with specific cysteine and lysine residues identified in both the rat and human channel [46, 47]. Finally, TRPA1-expressing neurons also express other TRP channels, in particular TRPV1, and, even more importantly, the sensory neuropeptides substance P (SP), neurokinin A (NKA), and calcitonin gene-related peptide (CGRP). SP/NKA and CGRP release from peripheral endings of nociceptors promoted by TRPV1 or TRPA1 activation produces a series of responses collectively described as neurogenic inflammation [48].
TRPA1, pain and neurogenic inflammation
It is generally recognized that C and Aδ-fibre sensory neurons convey pain signals, while neurons with larger-size fibres mediate touch sensation. Thermo-TRPs, which under normal conditions are expressed in C and Aδ-fibre neurons, have been proposed to contribute to transmission and modulation of nociceptive signals. This conclusion originates from empirical observation that TRPV1 or TRPA1 agonists, derived from foods and spices, are able to cause, in a dose-dependent fashion, a range from appreciated hot feelings to unpleasant pain sensation, as in the case of capsaicin, the selective TRPV1 agonist contained in hot peppers, and piperine, another TRPV1 agonist present in black pepper, or allyl isothiocyanate, the TRPA1 agonist contained in mustard or wasabi [5, 49]. Another finding that enlists thermo-TRPs as major pain controlling mechanisms is the clinical use of topical (cutaneous) capsaicin application that, by defunctionalizing sensory nerve terminals, alleviates several pain conditions, including post-herpetic neuralgias or pain associated with diabetic neuropathy [50].
Generation of deleted mice and, more importantly, identification and preclinical development of selective antagonists for thermo-TRPs, have greatly increased our knowledge of the role of these channels in the regulation of acute nociceptive responses and the development of allodynia and hyperalgesia. While TRPV1-deleted mice exhibit decreased hyperalgesia to elevated temperatures [51], TRPA1-deletion abrogated the two classical nociceptive phases to formalin [38]. However, gene deletion may not completely recapitulate the effects of channel blockade, as compensatory mechanism may counterbalance the function of the absent gene-protein. Thus, findings produced by using selective thermo-TRP antagonists may better unveil the role of these channels in models of pain diseases.
For detailed information on other thermo-TRPs, the reader is referred to previous review articles [5–8] whereas we will focus on the increasing evidence that supports the role of TRPA1 in models of both inflammatory and neuropathic pain. Mechanical hyperalgesia [52] and ongoing neuronal discharge [53, 54] evoked by complete Freund’s adjuvant (CFA) and tumour necrosis factor-α (TNFα), and mechanical hyperalgesia induced by low doses of monosodium iodoacetate (MIA) [55] are inhibited by TRPA1 receptor antagonists in rodents. Convergent findings indicate that TRPA1 contributes to carrageenan-evoked inflammatory hyperalgesia [56, 57]. Carrageenan administration, among a number of lipid derivatives, notably increases metabolites of 12-lipoxygenases, particularly hepoxilins A3 (HXA3) and HXB3, whose hyperalgesic/allodynic effect is abrogated by TRPA1 antagonism [58]. It has also been found that TRPA1 mediates ongoing nociception in chronic pancreatitis [59], and that both TRPV1 and TRPA1 initiate key pathways to transform acute into chronic inflammation and hyperalgesia in pancreatitis [60]. The clinical observation that an antioxidant afforded protection in patients with pancreatitis [61] indirectly supports the role of the oxidative stress sensor, TRPA1, in this condition.
The underlying mechanisms that from neural tissue injury produce the chronic allodynia and hyperalgesia typical of neuropathic pain are largely unknown. However, recent reports have pointed to the role of TRPA1 in different models of neuropathic pain. Several metabolic pathways in glycolysis or lipid peroxidation produce methylglyoxal (MG), which appears in the plasma in diabetic patients as hyperglycemia strongly enhances MG accumulation. MG has been recently described to react reversibly with cysteine residues, and probably due to this property stimulates TRPA1, thus representing a likely candidate metabolite to promote neuropathic pain in metabolic disorders [62]. Chemotherapeutic induced peripheral neuropathy (CIPN) is a scarcely understood and poorly treated condition, which, characterized by spontaneous pain, and mechanical and cold allodynia and hyperalgesia, causes significant discomfort, and often therapy discontinuation. CIPN may outlast the time period of chemotherapeutic drug administration for weeks or months [63].
Alteration of several ion channels has been advocated to explain this painful condition, but a univocal consensus has not emerged. Recently, in mouse models of CIPN produced by a single administration of oxaliplatin, paclitaxel or bortezomib, TRPA1 has been shown to play a major role in the development and maintenance of cold and mechanical [64–66]. In a therapeutic perspective, it is of relevance that treatment with a TRPA1 antagonist just before and shortly after (about 6 hours) the administration of bortezomib or oxaliplatin totally prevented the development and maintenance (for 10–15 days) of mechanical and cold hypersensitivity [66]. This finding suggests that TRPA1 is key in initiating CIPN and promoting the transition from an acute to a chronic condition. In addition, TRPA1 antagonists could represent novel therapeutic strategies of CIPN. In this context, it could be better understood that the unexpected attenuation by etodolac, a nonsteroidal anti-inflammatory drug of mechanical allodynia in a mouse model of neuropathic pain, might be due to its ability to inhibit TRPA1 [67]. In addition, to contribute to mechanical hyperalgesia, TRPA1 seems to be involved in the development of cold allodynia [68]. Cold allodynia was reduced in models of neuropathic (peripheral nerve injury) and inflammatory (CFA) pain [69, 70] or in models of CIPN, which typically exhibit this type of hypersensitivity [64–66]. Interestingly, a familial episodic pain syndrome has been attributed to a gain of function mutation of TRPA1 TRPA1 [71].
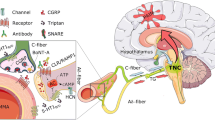
Coincidence between TRPA1 and neuropeptide expression has been suggested [72, 73], although evidence for coexistence of isolectin B4-positive and non-peptidergic neurons with TRPA1 has also been reported [74, 75]. Notwithstanding, exposure of tissues containing either peripheral or central sensory nerve terminals to TRPA1 agonists invariably results in a calcium-dependent release of SP/NKA and CGRP. Thus, TRPA1 stimulation has been proven to increase sensory neuropeptide release from oesophagus and urinary bladder, meninges, or dorsal spinal cord [76–78]. The outcome of such a release in peripheral tissues encompasses the series of responses commonly referred to as ‘neurogenic inflammation’ [48] (Figure 1). Although implication in transmission of nociceptive signals has been proposed, the pathophysiological outcome of the central release of sensory neuropeptides within the dorsal spinal cord or brain stem is less clear (Figure 1).

Schematic representation of the probable mechanisms of the action of antimigraine remedies, either currently used, or proven effective in clinical trials (gray boxes) or of novel medicines (empty box), regarding their ability to modulate the release of calcitonin gene related peptide (CGRP) or the activation of its receptor (CGRP-R). (1) Non steroidal antiinflammatory drugs (NSAIDs) block prostaglandin synthesis and the ensuing nociceptor sensitization and CGRP release evoked by prostaglandin receptor (PG-R) activation. (2) Triptans, by activating neuronal 5HT1D receptors, inhibit CGRP release. (3) CGRP-R antagonists inhibit the action of CGRP on effector cells. (4) Antagonists of transient receptor potential channels (TRPs), including TRP ankyrin 1 (TRPA1), block the ability of a series of stimulants (for TRPA1, cigarette smoke, acrolein, nitric oxide, umbellulone, and others) to release CGRP. All medicines may act at both peripheral and central endings of trigeminal nociceptors. Receptor/channel activation may trigger/facilitate (+) or inhibit (−) CGRP release.
TRPA1, TRPV1, and migraine
One of the first findings that, although indirectly, suggested the role of TRP channels in cluster headache and migraine was represented by the protective effect of the topical, desensitizing application of capsaicin to the patient nasal mucosa [79, 80]. Capsaicin treatment couples the unique ability of the drug to first activate, and, subsequently, upon repeated administration, desensitize both the afferent pathway that from channel activation conveys nociceptive signals, and the ‘efferent’ function that results in sensory neuropeptide release [48]. Within the fifth cranial nerve, capsaicin desensitization causes the defunctionalization of peripheral and possibly central nerve endings, thus preventing SP/NKA-dependent plasma protein extravasation within the dura mater, CGRP-dependent dilatation of meningeal arterioles, and inhibition of afferent nociceptive impulses. Further support to the contribution of TRPV1 to migraine mechanism derived from the observation that ethanol, a known trigger of migraine attacks, activates CGRP release from sensory neurons, and promotes CGRP-dependent menin-geal vasodilatation by reducing the threshold temperature for channel activation, a phenomenon that eventually results in TRPV1 activation [81, 82].
TRPV4 is stimulated by hypoosmotic stimuli that cause plasma membrane stretch and mechanical distension. Although this effect could, in principle, be implicated in the throbbing pain often described by migraine patients during their headache attacks, no evidence has yet been reported in support of TRPV4 in any model of head pain. In contrast, a series of observational findings has recently been obtained regarding a possible association between TRPA1 and migraine. In this respect, it is of interest that a number of compounds, recently identified as TRPA1 agonists, including cigarette smoke, ammonium chloride, formaldehyde, chlorine, garlic and others [34, 38, 41, 78, 83] are known triggers of migraine attacks in susceptible individuals [84–89].
Nitric oxide (NO) donor drugs, including nitroglycerine, are known inducers of migraine attacks and have been extensively used in migraine provocation studies in humans [90]. Although their pro-migraine action has been attributed to their intrinsic vasodilatatory action [91], the temporal mismatch between vasodilatation and the onset of migraine-like attacks argues against a close association between the two phenomena. In fact, at the time of the maximum vasodilatation, a mild to moderate headache develops both in migraineurs (more) and in healthy subjects (less), while delayed migraine-like attacks, present only in migraine patients, are observed only 5–6 hours after nitroglycerine administration [92, 93]. Although not confirmed in vitro [94]. NO may release CGRP from trigemino-vascular neurons [95]. More recently, NO has been revealed to target TRPA1 by nitrosylation of channel cysteine residues [96], that seem to differ from those targeted by other reactive molecules [97], and this novel molecular mechanism could contribute to the nociceptive response evoked by NO [98]. It is possible that the S-nitrosylation process [99], produced by NO, contributes to channel sensitization to eventually (hours after the exposure to NO) amplify CGRP release by other agents, thus leading to exaggerated neurogenic inflammation and potentiation of pain responses.
The environmental pollution agent, acrolein, is produced by the combustion of organic material which causes its accidental inhalation, which, however, occurs also with cigarette smoking [100]. Several components of cigarette smoke, such as acrolein, crotonaldehyde [78], acetaldehyde [37] and nicotine [101], are TRPA1 agonists. Recently, acrolein application to the rat nasal mucosa has been shown to produce ipsilateral meningeal vasodilatation by a TRPA1- and CGRP-dependent mechanism [102], thus offering a mechanistic explanation for the association between the exposure to cigarette smoke and migraine attack appearance or worsening [103, 104].
Herbalism has been instrumental for the development of pharmacology and also to a better understanding of pathophysiological mechanisms. These principles also apply to the migraine field. Umbellularia californica (California bay laurel) is also known as the ‘headache tree’ because of the ability of its scent to trigger headache attacks in susceptible individuals [105]. A case of cluster headache-like attacks preceded by cold sensations perceived in the ipsilateral nostril following inhalation of Umbellularia californica scent has recently been described in a cluster headache patient whose attacks had ceased 10 years before [106]. The irritant monoterpene ketone, umbellulone, one of the most abundant reactive molecules of Umbellularia californica, was found to activate the human recombinant and the constitutive rat/mouse TRPA1 in trigeminal ganglia (TG) neurons, and via this mechanism to produce nociceptive behaviour and the release of CGRP from TG or meningeal tissue in rats [77]. In addition, similar to acrolein, umbellulone application to the rat nasal mucosa evoked ipsilateral TRPA1- and CGRP-dependent meningeal vasodilatation [77].
Ligustilide, an electrophilic volatile dihydrophthalide of dietary and medicinal relevance, has been found to produce a moderate activation of TRPA1, but also to inhibit allyl isothiocyanate-evoked stimulation of TRPA1 [107]. This newly identified target of ligustilide offers a novel mechanistic explanation for the use of the compound in traditional medicine to treat pain diseases, including headaches. Finally, although several hypotheses have been advanced, the mechanism of the analgesic action of acetaminophen (paracetamol) in different pain conditions is far from clear. The reactive metabolite of acetaminophen, N-acetyl-p-benzo-quinoneimine (NAPQI), is able to activate the TRPA1 channel and thereby evoke a moderate and reversible neurogenic inflammatory response, which, in susceptible individuals, may contribute to emphasizing inflammation in peripheral tissues [76]. However, NAPQI may also be produced by cytochrome activity within the spinal cord, and NAPQI action at the spinal level results in channel desensitization [108]. Inhibition of central TRPA1 has been advocated as the mechanism, which may be responsible the hitherto unexplained analgesic and possibly antimigraine action of its parent molecule [108]. Importantly, this novel spinal mechanism could be of general relevance also for other TRPA1 agonists which share with NAPQI the ability of desensitizing the channel.
A host of endogenous inflammatory mediators possibly released during migraine attacks can activate and sensitize peripheral and central sensory neurons, including trigeminal neurons. Sensitization of first-order neurons is involved in the perception of headache throbbing pain [109], while sensitization of second‒order neurons contributes to cephalic allodynia and muscle tenderness [110, 111]. Recently, it has been shown that innocuous brush and heat stimuli induce larger activation in the thalamus of patients who exhibit allodynia during mi-graine, as compared to pain‒free state, and that topical application of inflammatory molecules on the rat meninges sensitizes thalamic trigeminovascular neurons [112]. Each component of the nociceptive pathways could contribute differently to sensitization of the neural tissues. TRP channels could be sensitized by different compounds and via different mechanisms [5, 36, 66, 81, 113]. Although there is no specific information on the role of these channels to peripheral nociceptor sensitization or central sensitization in migraine, it is possible that TRP channels, and in particular TRPV1 and TRPA1, contribute to this key mechanism.
Conclusions
The still largely unfinished puzzle of the migraine mechanism is being completed by novel unexpected pieces of information, which compose a clearer picture. The first, corner of the picture, known for decades, is that cyclooxygenase (namely cyclooxygenase 2) inhibition has a beneficial effect on migraine attack. The second corner, predicted by scientists and appreciated by clinicians and patients, is that targeting 5-HT1 receptors is also highly effective. The third corner is that a series of clinical trials show that CGRP antagonists afford a protection similar to that of triptans. To complete at least the frame, a fourth corner, which should reconcile the other three in an intelligible sequence of events, is needed. While prostaglandins sensitize nociceptors and eventually promote CGRP release, triptans inhibit such release, thus producing an indirect anti-migraine effect, and CGRP antagonists abrogate the final common pathway of migraine mechanism. The pathway, which, sensitized by prostaglandins and inhibited by serotonin receptor stimulation, results in trigeminal neuron activation and the pro-migraine release of CGRP could represent the fourth corner of the picture. Emerging information on TRP channels, and particularly TRPA1, which, targeted by migraine triggers, contribute, by activating the trigeminal CGRP-dependent pathway, to the genesis of pain and the accompanying symptoms of the attack, seems to be of paramount importance to solve what still remains the enigma of the migraine mechanism.
References
Minke B: Drosophila mutant with a transducer defect. Biophys Struct Mech 1977, 3: 59–64. 10.1007/BF00536455
Montell C, Jones K, Hafen E, Rubin G: Rescue of the Drosophila phototransduction mutation trp by germline transformation. Science 1985, 230: 1040–1043. 10.1126/science.3933112
Vriens J, Owsianik G, Voets T, Droogmans G, Nilius B: Invertebrate TRP proteins as functional models for mammalian channels. Pflugers Arch 2004, 449: 213–226.
Sedgwick SG, Smerdon SJ: The ankyrin repeat: a diversity of interactions on a common structural framework. Trends Biochem Sci 1999, 24: 311–316. 10.1016/S0968-0004(99)01426-7
Nilius B, Owsianik G, Voets T, Peters JA: Transient receptor potential cation channels in disease. Physiol Rev 2007, 87: 165–217. 10.1152/physrev.00021.2006
Ramsey IS, Delling M, Clapham DE: An introduction to TRP channels. Annu Rev Physiol 2006, 68: 619–647. 10.1146/annurev.physiol.68.040204.100431
Clapham DE: TRP channels as cellular sensors. Nature 2003, 426: 517–524. 10.1038/nature02196
Montell C, Birnbaumer L, Flockerzi V: The TRP channels, a remarkably functional family. Cell 2002, 108: 595–598. 10.1016/S0092-8674(02)00670-0
Montell C: Visual transduction in Drosophila. Annu Rev Cell Dev Biol 1999, 15: 231–268. 10.1146/annurev.cellbio.15.1.231
Maroto R, Raso A, Wood TG, Kurosky A, Martinac B, Hamill OP: TRPC1 forms the stretch-activated cation channel in vertebrate cells. Nat Cell Biol 2005, 7: 179–185. 10.1038/ncb1218
Soyombo AA, Tjon-Kon-Sang S, Rbaibi Y, Bashllari E, Bisceglia J, Muallem S, Kiselyov K: TRP-ML1 regulates lysosomal pH and acidic lysosomal lipid hydrolytic activity. J Biol Chem 2006, 281: 7294–7301. 10.1074/jbc.M508211200
Hanaoka K, Qian F, Boletta A, Bhunia AK, Piontek K, Tsiokas L, Sukhatme VP, Guggino WB, Germino GG: Co-assembly of polycystin-1 and −2 produces unique cation-permeable currents. Nature 2000, 408: 990–994. 10.1038/35050128
Nilius B, Owsianik G: Transient receptor potential channelopathies. Pflugers Arch 2010, 460: 437–450. 10.1007/s00424-010-0788-2
Bevan S, Geppetti P: Protons: small stimulants of capsaicin-sensitive sensory nerves. Trends Neurosci 1994, 17: 509–512. 10.1016/0166-2236(94)90149-X
Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, Raumann BE, Basbaum AI, Julius D: The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron 1998, 21: 531–543. 10.1016/S0896-6273(00)80564-4
Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sorgard M, Di Marzo V, Julius D, Hogestatt ED: Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 1999, 400: 452–457. 10.1038/22761
Shin J, Cho H, Hwang SW, Jung J, Shin CY, Lee SY, Kim SH, Lee MG, Choi YH, Kim J, Haber NA, Reichling DB, Khasar S, Levine JD, Oh U: Bradykinin-12-lipoxygenase-VR1 signaling pathway for inflammatory hyperalgesia. Proc Natl Acad Sci USA 2002, 99: 10150–10155. 10.1073/pnas.152002699
Moqrich A, Hwang SW, Earley TJ, Petrus MJ, Murray AN, Spencer KS, Andahazy M, Story GM, Patapoutian A: Impaired thermosensation in mice lacking TRPV3, a heat and camphor sensor in the skin. Science 2005, 307: 1468–1472. 10.1126/science.1108609
Everaerts W, Nilius B, Owsianik G: The vanilloid transient receptor potential channel TRPV4: from structure to disease. Prog Biophys Mol Biol 2010, 103: 2–17. 10.1016/j.pbiomolbio.2009.10.002
Watanabe H, Vriens J, Prenen J, Droogmans G, Voets T, Nilius B: Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature 2003, 424: 434–438. 10.1038/nature01807
Vriens J, Owsianik G, Fisslthaler B, Suzuki M, Janssens A, Voets T, Morisseau C, Hammock BD, Fleming I, Busse R, Nilius B: Modulation of the Ca2 permeable cation channel TRPV4 by cytochrome P450 epoxygenases in vascular endothelium. Circ Res 2005, 97: 908–915. 10.1161/01.RES.0000187474.47805.30
Bang S, Kim KY, Yoo S, Lee SH, Hwang SW: Transient receptor potential V2 expressed in sensory neurons is activated by probenecid. Neurosci Lett 2007, 425: 120–125. 10.1016/j.neulet.2007.08.035
Wagner TF, Loch S, Lambert S, Straub I, Mannebach S, Mathar I, Dufer M, Lis A, Flockerzi V, Philipp SE, Oberwinkler J: Transient receptor potential M3 channels are ionotropic steroid receptors in pancreatic beta cells. Nat Cell Biol 2008, 10: 1421–1430. 10.1038/ncb1801
Atoyan R, Shander D, Botchkareva NV: Non-neuronal expression of transient receptor potential type A1 (TRPA1) in human skin. J Invest Dermatol 2009, 129: 2312–2315. 10.1038/jid.2009.58
Streng T, Axelsson HE, Hedlund P, Andersson DA, Jordt SE, Bevan S, Andersson KE, Hogestatt ED, Zygmunt PM: Distribution and function of the hydrogen sulfide-sensitive TRPA1 ion channel in rat urinary bladder. Eur Urol 2008, 53: 391–399. 10.1016/j.eururo.2007.10.024
Gratzke C, Weinhold P, Reich O, Seitz M, Schlenker B, Stief CG, Andersson KE, Hedlund P: Transient receptor potential A1 and cannabinoid receptor activity in human normal and hyperplastic prostate: relation to nerves and interstitial cells. Eur Urol 2010, 57: 902–910. 10.1016/j.eururo.2009.08.019
Earley S, Gonzales AL, Crnich R: Endothelium-dependent cerebral artery dilation mediated by TRPA1 and Ca2+−Activated K+ channels. Circ Res 2009, 104: 987–994. 10.1161/CIRCRESAHA.108.189530
Nozawa K, Kawabata-Shoda E, Doihara H, Kojima R, Okada H, Mochizuki S, Sano Y, Inamura K, Matsushime H, Koizumi T, Yokoyama T, Ito H: TRPA1 regulates gastrointestinal motility through serotonin release from enterochromaffin cells. Proc Natl Acad Sci USA 2009, 106: 3408–3413. 10.1073/pnas.0805323106
El Karim IA, Linden GJ, Curtis TM, About I, McGahon MK, Irwin CR, Lundy FT: Human odontoblasts express functional thermo-sensitive TRP channels: implications for dentin sensitivity. Pain 2010, 152: 2211–2223.
El Karim IA, Linden GJ, Curtis TM, About I, McGahon MK, Irwin CR, Killough SA, Lundy FT: Human dental pulp fibroblasts express the “cold-sensing” transient receptor potential channels TRPA1 and TRPM8. J Endod 2011, 37: 473–478. 10.1016/j.joen.2010.12.017
Kochukov MY, McNearney TA, Fu Y, Westlund KN: Thermosensitive TRP ion channels mediate cytosolic calcium response in human synoviocytes. Am J Physiol Cell Physiol 2006, 291: C424-C432. 10.1152/ajpcell.00553.2005
Nassini R, Pedretti P, Moretto N, Fusi C, Carnini C, Facchinetti F, Viscomi AR, Pisano AR, Stokesberry S, Brunmark C, Svitacheva N, McGarvey L, Patacchini R, Damholt AB, Geppetti P, Materazzi S: Transient receptor potential ankyrin 1 channel localized to non-neuronal airway cells promotes non-neurogenic inflammation. PLoS One 2012, 7: e42454. 10.1371/journal.pone.0042454
Jordt SE, Bautista DM, Chuang HH, McKemy DD, Zygmunt PM, Hogestatt ED, Meng ID, Julius D: Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature 2004, 427: 260–265. 10.1038/nature02282
Bautista DM, Movahed P, Hinman A, Axelsson HE, Sterner O, Hogestatt ED, Julius D, Jordt SE, Zygmunt PM: Pungent products from garlic activate the sensory ion channel TRPA1. Proc Natl Acad Sci USA 2005, 102: 12248–12252. 10.1073/pnas.0505356102
Bandell M, Story GM, Hwang SW, Viswanath V, Eid SR, Petrus MJ, Earley TJ, Patapoutian A: Noxious cold ion channel TRPA1 is activated by pungent compounds and bradykinin. Neuron 2004, 41: 849–857. 10.1016/S0896-6273(04)00150-3
Nilius B, Appendino G, Owsianik G: The transient receptor potential channel TRPA1: from gene to pathophysiology. Pflugers Arch 2012, 464: 425–458. 10.1007/s00424-012-1158-z
Bang S, Kim KY, Yoo S, Kim YG, Hwang SW: Transient receptor potential A1 mediates acetaldehyde-evoked pain sensation. Eur J Neurosci 2007, 26: 2516–2523. 10.1111/j.1460-9568.2007.05882.x
McNamara CR, Mandel-Brehm J, Bautista DM, Siemens J, Deranian KL, Zhao M, Hayward NJ, Chong JA, Julius D, Moran MM, Fanger CM: TRPA1 mediates formalin-induced pain. Proc Natl Acad Sci USA 2007, 104: 13525–13530. 10.1073/pnas.0705924104
Andersson DA, Gentry C, Moss S, Bevan S: Transient receptor potential A1 is a sensory receptor for multiple products of oxidative stress. J Neurosci 2008, 28: 2485–2494. 10.1523/JNEUROSCI.5369-07.2008
Sawada Y, Hosokawa H, Matsumura K, Kobayashi S: Activation of transient receptor potential ankyrin 1 by hydrogen peroxide. Eur J Neurosci 2008, 27: 1131–1142. 10.1111/j.1460-9568.2008.06093.x
Bessac BF, Sivula M, von Hehn CA, Escalera J, Cohn L, Jordt SE: TRPA1 is a major oxidant sensor in murine airway sensory neurons. J Clin Invest 2008, 118: 1899–1910. 10.1172/JCI34192
Bessac BF, Sivula M, von Hehn CA, Caceres AI, Escalera J, Jordt SE: Transient receptor potential ankyrin 1 antagonists block the noxious effects of toxic industrial isocyanates and tear gases. Faseb J 2009, 23: 1102–1114. 10.1096/fj.08-117812
Taylor-Clark TE, Undem BJ: Ozone activates airway nerves via the selective stimulation of TRPA1 ion channels. J Physiol 2010, 588: 423–433. 10.1113/jphysiol.2009.183301
Wang YY, Chang RB, Liman ER: TRPA1 is a component of the nociceptive response to CO2. J Neurosci 2010, 30: 12958–12963. 10.1523/JNEUROSCI.2715-10.2010
Hill K, Schaefer M: Ultraviolet light and photosensitising agents activate TRPA1 via generation of oxidative stress. Cell Calcium 2009, 45: 155–164. 10.1016/j.ceca.2008.08.001
Macpherson LJ, Dubin AE, Evans MJ, Marr F, Schultz PG, Cravatt BF, Patapoutian A: Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature 2007, 445: 541–545. 10.1038/nature05544
Hinman A, Chuang HH, Bautista DM, Julius D: TRP channel activation by reversible covalent modification. Proc Natl Acad Sci USA 2006, 103: 19564–19568. 10.1073/pnas.0609598103
Geppetti P, Holzer P: Neurogenic inflammation. Boca Raton: CRC Press; 1996.
Preti D, Szallasi A, Patacchini R: TRP channels as therapeutic targets in airway disorders: a patent review. Expert Opin Ther Pat 2012, 22: 663–695. 10.1517/13543776.2012.696099
Derry S, Sven-Rice A, Cole P, Tan T, Moore RA: Topical capsaicin (high concentration) for chronic neuropathic pain in adults. Cochrane Database Syst Rev 2013., 2: CD007393
Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D: The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 1997, 389: 816–824. 10.1038/39807
Fernandes ES, Russell FA, Spina D, McDougall JJ, Graepel R, Gentry C, Staniland AA, Mountford DM, Keeble JE, Malcangio M, Bevan S, Brain SD: A distinct role for transient receptor potential ankyrin 1, in addition to transient receptor potential vanilloid 1, in tumor necrosis factor alpha-induced inflammatory hyperalgesia and Freund’s complete adjuvant-induced monarthritis. Arthritis Rheum 2011, 63: 819–829. 10.1002/art.30150
McGaraughty S, Chu KL, Perner RJ, Didomenico S, Kort ME, Kym PR: TRPA1 modulation of spontaneous and mechanically evoked firing of spinal neurons in uninjured, osteoarthritic, and inflamed rats. Mol Pain 2010, 6: 14. 10.1186/1744-8069-6-14
da Costa DS, Meotti FC, Andrade EL, Leal PC, Motta EM, Calixto JB: The involvement of the transient receptor potential A1 (TRPA1) in the maintenance of mechanical and cold hyperalgesia in persistent inflammation. Pain 2010, 148: 431–437. 10.1016/j.pain.2009.12.002
Okun A, Liu P, Davis P, Ren J, Remeniuk B, Brion T, Ossipov MH, Xie J, Dussor GO, King T, Porreca F: Afferent drive elicits ongoing pain in a model of advanced osteoarthritis. Pain 2012, 153: 924–933. 10.1016/j.pain.2012.01.022
Bonet IJ, Fischer L, Parada CA, Tambeli CH: The role of transient receptor potential A 1 (TRPA1) in the development and maintenance of carrageenan-induced hyperalgesia. Neuropharmacology 2013, 65: 206–212.
Moilanen LJ, Laavola M, Kukkonen M, Korhonen R, Leppanen T, Hogestatt ED, Zygmunt PM, Nieminen RM, Moilanen E: TRPA1 contributes to the acute inflammatory response and mediates carrageenan-induced paw edema in the mouse. Sci Rep 2012, 2: 380.
Gregus AM, Doolen S, Dumlao DS, Buczynski MW, Takasusuki T, Fitzsimmons BL, Hua XY, Taylor BK, Dennis EA, Yaksh TL: Spinal 12-lipoxygenase-derived hepoxilin A3 contributes to inflammatory hyperalgesia via activation of TRPV1 and TRPA1 receptors. Proc Natl Acad Sci USA 2013, 109: 6721–6726.
Cattaruzza F, Johnson C, Leggit A, Grady EF, Schenk AK, Cevikbas F, Cedron WJ, Bondada S, Kirkwood R, Malone BJ, Steinhoff M, Bunnett NW, Kirkwood KS: Transient Receptor Potential Ankyrin 1 (TRPA1) Mediates Chronic Pancreatitis Pain In Mice. Am J Physiol Gastrointest Liver Physiol 2013, 304: G1002-G1012. 10.1152/ajpgi.00005.2013
Schwartz ES, La JH, Scheff NN, Davis BM, Albers KM, Gebhart GF: TRPV1 and TRPA1 antagonists prevent the transition of acute to chronic inflammation and pain in chronic pancreatitis. J Neurosci 2013, 33: 5603–5611. 10.1523/JNEUROSCI.1806-12.2013
Bhardwaj P, Garg PK, Maulik SK, Saraya A, Tandon RK, Acharya SK: A randomized controlled trial of antioxidant supplementation for pain relief in patients with chronic pancreatitis. Gastroenterology 2009, 136: 149–159. 10.1053/j.gastro.2008.09.028
Eberhardt MJ, Filipovic MR, Leffler A, de la Roche J, Kistner K, Fischer MJ, Fleming T, Zimmermann K, Ivanovic-Burmazovic I, Nawroth PP, Bierhaus A, Reeh PW, Sauer SK: Methylglyoxal activates nociceptors through transient receptor potential channel A1 (TRPA1): a possible mechanism of metabolic neuropathies. J Biol Chem 2012, 287: 28291–28306. 10.1074/jbc.M111.328674
Cavaletti G, Marmiroli P: Chemotherapy-induced peripheral neurotoxicity. Nat Rev Neurol 2010, 6: 657–666. 10.1038/nrneurol.2010.160
Nassini R, Gees M, Harrison S, De Siena G, Materazzi S, Moretto N, Failli P, Preti D, Marchetti N, Cavazzini A, Mancini F, Pedretti P, Nilius B, Patacchini R, Geppetti P: Oxaliplatin elicits mechanical and cold allodynia in rodents via TRPA1 receptor stimulation. Pain 2011, 152: 1621–1631. 10.1016/j.pain.2011.02.051
Materazzi S, Fusi C, Benemei S, Pedretti P, Patacchini R, Nilius B, Prenen J, Creminon C, Geppetti P, Nassini R: TRPA1 and TRPV4 mediate paclitaxel-induced peripheral neuropathy in mice via a glutathione-sensitive mechanism. Pflugers Arch 2012, 463: 561–569. 10.1007/s00424-011-1071-x
Trevisan G, Materazzi S, Fusi C, Altomare A, Aldini G, Lodovici M, Patacchini R, Geppetti P, Nassini R: Novel Therapeutic Strategy to Prevent Chemotherapy-Induced Persistent Sensory Neuropathy By TRPA1 Blockade. Cancer Res 2013, 73: 3120–3131. 10.1158/0008-5472.CAN-12-4370
Inoue N, Ito S, Nogawa M, Tajima K, Kyoi T: Etodolac blocks the allyl isothiocyanate-induced response in mouse sensory neurons by selective TRPA1 activation. Pharmacology 2012, 90: 47–54. 10.1159/000338756
Karashima Y, Talavera K, Everaerts W, Janssens A, Kwan KY, Vennekens R, Nilius B, Voets T: TRPA1 acts as a cold sensor in vitro and in vivo. Proc Natl Acad Sci USA 2009, 106: 1273–1278. 10.1073/pnas.0808487106
del Camino D, Murphy S, Heiry M, Barrett LB, Earley TJ, Cook CA, Petrus MJ, Zhao M, D’Amours M, Deering N, Brenner GJ, Costigan M, Hayward NJ, Chong JA, Fanger CM, Woolf CJ, Patapoutian A, Moran MM: TRPA1 contributes to cold hypersensitivity. J Neurosci 2010, 30: 15165–15174. 10.1523/JNEUROSCI.2580-10.2010
Chen J, Joshi SK, DiDomenico S, Perner RJ, Mikusa JP, Gauvin DM, Segreti JA, Han P, Zhang XF, Niforatos W, Bianchi BR, Baker SJ, Zhong C, Simler GH, McDonald HA, Schmidt RG, McGaraughty SP, Chu KL, Faltynek CR, Kort ME, Reilly RM, Kym PR: Selective blockade of TRPA1 channel attenuates pathological pain without altering noxious cold sensation or body temperature regulation. Pain 2011, 152: 1165–1172. 10.1016/j.pain.2011.01.049
Kremeyer B, Lopera F, Cox JJ, Momin A, Rugiero F, Marsh S, Woods CG, Jones NG, Paterson KJ, Fricker FR, Villegas A, Acosta N, Pineda-Trujillo NG, Ramirez JD, Zea J, Burley MW, Bedoya G, Bennett DL, Wood JN, Ruiz-Linares A: A gain-of-function mutation in TRPA1 causes familial episodic pain syndrome. Neuron 2010, 66: 671–680. 10.1016/j.neuron.2010.04.030
Bhattacharya MR, Bautista DM, Wu K, Haeberle H, Lumpkin EA, Julius D: Radial stretch reveals distinct populations of mechanosensitive mammalian somatosensory neurons. Proc Natl Acad Sci USA 2008, 105: 20015–20020. 10.1073/pnas.0810801105
Story GM, Peier AM, Reeve AJ, Eid SR, Mosbacher J, Hricik TR, Earley TJ, Hergarden AC, Andersson DA, Hwang SW, McIntyre P, Jegla T, Bevan S, Patapoutian A: ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell 2003, 112: 819–829. 10.1016/S0092-8674(03)00158-2
Barabas ME, Kossyreva EA, Stucky CL: TRPA1 is functionally expressed primarily by IB4-binding, non-peptidergic mouse and rat sensory neurons. PLoS One 2012, 7: e47988. 10.1371/journal.pone.0047988
Kim YS, Son JY, Kim TH, Paik SK, Dai Y, Noguchi K, Ahn DK, Bae YC: Expression of transient receptor potential ankyrin 1 (TRPA1) in the rat trigeminal sensory afferents and spinal dorsal horn. J Comp Neurol 2010, 518: 687–698. 10.1002/cne.22238
Nassini R, Materazzi S, Andre E, Sartiani L, Aldini G, Trevisani M, Carnini C, Massi D, Pedretti P, Carini M, Cerbai E, Preti D, Villetti G, Civelli M, Trevisan G, Azzari C, Stokesberry S, Sadofsky L, McGarvey L, Patacchini R, Geppetti P: Acetaminophen, via its reactive metabolite N-acetyl-p-benzo-quinoneimine and transient receptor potential ankyrin-1 stimulation, causes neurogenic inflammation in the airways and other tissues in rodents. Faseb J 2010, 24: 4904–4916. 10.1096/fj.10-162438
Nassini R, Materazzi S, Vriens J, Prenen J, Benemei S, De Siena G, la Marca G, Andre E, Preti D, Avonto C, Sadofsky L, Di Marzo V, De Petrocellis L, Dussor G, Porreca F, Taglialatela-Scafati O, Appendino G, Nilius B, Geppetti P: The ‘headache tree’ via umbellulone and TRPA1 activates the trigeminovascular system. Brain 2012, 135: 376–390. 10.1093/brain/awr272
Andre E, Campi B, Materazzi S, Trevisani M, Amadesi S, Massi D, Creminon C, Vaksman N, Nassini R, Civelli M, Baraldi PG, Poole DP, Bunnett NW, Geppetti P, Patacchini R: Cigarette smoke-induced neurogenic inflammation is mediated by alpha, beta-unsaturated aldehydes and the TRPA1 receptor in rodents. J Clin Invest 2008, 118: 2574–2582.
Fusco BM, Barzoi G, Agro F: Repeated intranasal capsaicin applications to treat chronic migraine. Br J Anaesth 2003, 90: 812. 10.1093/bja/aeg572
Fusco BM, Marabini S, Maggi CA, Fiore G, Geppetti P: Preventative effect of repeated nasal applications of capsaicin in cluster headache. Pain 1994, 59: 321–325. 10.1016/0304-3959(94)90017-5
Trevisani M, Geppetti P, Davis JB, Bianchi A, Harrison S, Randall AD, Smith GD, Owen D, Brough SJ, Jerman JC, Gray J, Amadesi S, Campi B, Barbieri M, Tognetto M, Gunthorpe MJ, Smart D: Ethanol elicits and potentiates nociceptor responses via the vanilloid receptor-1. Nat Neurosci 2002, 5: 546–551. 10.1038/nn0602-852
Nicoletti P, Trevisani M, Manconi M, Gatti R, De Siena G, Zagli G, Benemei S, Capone JA, Geppetti P, Pini LA: Ethanol causes neurogenic vasodilation by TRPV1 activation and CGRP release in the trigeminovascular system of the guinea pig. Cephalalgia 2008, 28: 9–17.
Fujita F, Uchida K, Moriyama T, Shima A, Shibasaki K, Inada H, Sokabe T, Tominaga M: Intracellular alkalization causes pain sensation through activation of TRPA1 in mice. J Clin Invest 2008, 118: 4049–4057. 10.1172/JCI35957
Courteau JP, Cushman R, Bouchard F, Quevillon M, Chartrand A, Bherer L: Survey of construction workers repeatedly exposed to chlorine over a three to six month period in a pulpmill: I. Exposure and symptomatology. Occup Environ Med 1994, 51: 219–224. 10.1136/oem.51.4.219
Peatfield RC: Relationships between food, wine, and beer-precipitated migrainous headaches. Headache 1995, 35: 355–357. 10.1111/j.1526-4610.1995.hed3506355.x
Wantke F, Focke M, Hemmer W, Bracun R, Wolf-Abdolvahab S, Gotz M, Jarisch R, Gotz M, Tschabitscher M, Gann M, Tappler P: Exposure to formaldehyde and phenol during an anatomy dissecting course: sensitizing potency of formaldehyde in medical students. Allergy 2000, 55: 84–87. 10.1034/j.1398-9995.2000.00307.x
Irlbacher K, Meyer BU: Nasally triggered headache. Neurology 2002, 58: 294. 10.1212/WNL.58.2.294
Kelman L: The triggers or precipitants of the acute migraine attack. Cephalalgia 2007, 27: 394–402. 10.1111/j.1468-2982.2007.01303.x
Roussos AP, Hirsch AR: Alliaceous Migraines. Headache 2013, 2013: 12091.
Iversen HK, Olesen J: Headache induced by a nitric oxide donor (nitroglycerin) responds to sumatriptan. A human model for development of migraine drugs. Cephalalgia 1996, 16: 412–418. 10.1046/j.1468-2982.1996.1606412.x
Shevel E: The extracranial vascular theory of migraine–a great story confirmed by the facts. Headache 2011, 51: 409–417. 10.1111/j.1526-4610.2011.01844.x
Iversen HK, Olesen J: Nitroglycerin-induced headache is not dependent on histamine release: support for a direct nociceptive action of nitric oxide. Cephalalgia 1994, 14: 437–442. 10.1046/j.1468-2982.1994.1406437.x
Thomsen LL, Olesen J: Nitric oxide in primary headaches. Curr Opin Neurol 2001, 14: 315–321. 10.1097/00019052-200106000-00009
Eltorp CT, Jansen-Olesen I, Hansen AJ: Release of calcitonin gene-related peptide (CGRP) from guinea pig dura mater in vitro is inhibited by sumatriptan but unaffected by nitric oxide. Cephalalgia 2000, 20: 838–844. 10.1046/j.1468-2982.2000.00131.x
Wei EP, Moskowitz MA, Boccalini P, Kontos HA: Calcitonin gene-related peptide mediates nitroglycerin and sodium nitroprusside-induced vasodilation in feline cerebral arterioles. Circ Res 1992, 70: 1313–1319. 10.1161/01.RES.70.6.1313
Yoshida T, Inoue R, Morii T, Takahashi N, Yamamoto S, Hara Y, Tominaga M, Shimizu S, Sato Y, Mori Y: Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat Chem Biol 2006, 2: 596–607. 10.1038/nchembio821
Takahashi N, Mizuno Y, Kozai D, Yamamoto S, Kiyonaka S, Shibata T, Uchida K, Mori Y: Molecular characterization of TRPA1 channel activation by cysteine-reactive inflammatory mediators. Channels (Austin) 2008, 2: 287–298. 10.4161/chan.2.4.6745
Miyamoto T, Dubin AE, Petrus MJ, Patapoutian A: TRPV1 and TRPA1 mediate peripheral nitric oxide-induced nociception in mice. PLoS One 2009, 4: e7596. 10.1371/journal.pone.0007596
Takahashi N, Mori Y: TRP Channels as Sensors and Signal Integrators of Redox Status Changes. Front Pharmacol 2011, 2: 58.
Facchinetti F, Amadei F, Geppetti P, Tarantini F, Di Serio C, Dragotto A, Gigli PM, Catinella S, Civelli M, Patacchini R: Alpha, beta-unsaturated aldehydes in cigarette smoke release inflammatory mediators from human macrophages. Am J Respir Cell Mol Biol 2007, 37: 617–623. 10.1165/rcmb.2007-0130OC
Talavera K, Gees M, Karashima Y, Meseguer VM, Vanoirbeek JA, Damann N, Everaerts W, Benoit M, Janssens A, Vennekens R, Viana F, Nemery B, Nilius B, Voets T: Nicotine activates the chemosensory cation channel TRPA1. Nat Neurosci 2009, 12: 1293–1299. 10.1038/nn.2379
Kunkler PE, Ballard CJ, Oxford GS, Hurley JH: TRPA1 receptors mediate environmental irritant-induced meningeal vasodilatation. Pain 2011, 152: 38–44. 10.1016/j.pain.2010.08.021
Lima AM, Sapienza GB, Giraud Vde O, Fragoso YD: Odors as triggering and worsening factors for migraine in men. Arq Neuropsiquiatr 2011, 69: 324–327. 10.1590/S0004-282X2011000300011
Friedman DI, De ver Dye T: Migraine and the environment. Headache 2009, 49: 941–952. 10.1111/j.1526-4610.2009.01443.x
California Laurel (2006) USDA Natural Resources Conservation Service. . Accessed 16 Apr 2009 http://plants.usda.gov/plantguide/pdf/cs_umca.pdf
Benemei S, Appendino G, Geppetti P: Pleasant natural scent with unpleasant effects: cluster headache-like attacks triggered by Umbellularia californica. Cephalalgia 2009, 30: 744–746.
Zhong J, Minassi A, Prenen J, Taglialatela-Scafati O, Appendino G, Nilius B: Umbellulone modulates TRP channels. Pflugers Arch 2011, 462: 861–870. 10.1007/s00424-011-1043-1
Andersson DA, Gentry C, Alenmyr L, Killander D, Lewis SE, Andersson A, Bucher B, Galzi JL, Sterner O, Bevan S, Hogestatt ED, Zygmunt PM: TRPA1 mediates spinal antinociception induced by acetaminophen and the cannabinoid Delta(9)-tetrahydrocannabiorcol. Nat Commun 2011, 2: 551.
Strassman AM, Raymond SA, Burstein R: Sensitization of meningeal sensory neurons and the origin of headaches. Nature 1996, 384: 560–564. 10.1038/384560a0
Burstein R, Cutrer MF, Yarnitsky D: The development of cutaneous allodynia during a migraine attack clinical evidence for the sequential recruitment of spinal and supraspinal nociceptive neurons in migraine. Brain 2000,123(Pt 8):1703–1709.
Burstein R, Jakubowski M, Garcia-Nicas E, Kainz V, Bajwa Z, Hargreaves R, Becerra L, Borsook D: Thalamic sensitization transforms localized pain into widespread allodynia. Ann Neurol 2010, 68: 81–91. 10.1002/ana.21994
Noseda R, Burstein R: Migraine pathophysiology: anatomy of the trigeminovascular pathway and associated neurological symptoms, CSD, sensitization and modulation of pain. Pain 2013. Epub ahead of print
Selescu T, Ciobanu AC, Dobre C, Reid G, Babes A: Camphor activates and sensitizes transient receptor potential melastatin 8 (TRPM8) to cooling and icilin. Chem Senses 2013. Epub ahead of print
Acknowledgements
We acknowledge Regione Toscana (Regional Health Research Program 2009, P.G.).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contribution
FDC, CF, ER, and CL searched for the literature. SB and PG wrote and revised the text. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Benemei, S., De Cesaris, F., Fusi, C. et al. TRPA1 and other TRP channels in migraine. J Headache Pain 14, 71 (2013). https://doi.org/10.1186/1129-2377-14-71
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1129-2377-14-71