
Abstract
The Sesame Genome Working Group (SGWG) has been formed to sequence and assemble the sesame (Sesamum indicum L.) genome. The status of this project and our planned analyses are described.
Similar content being viewed by others
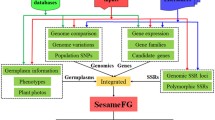


The importance of the sesame genome
Sesame (Sesamum indicum L., 2n = 26), which belongs to the Sesamum genus of the Pedaliaceae family, is one of the oldest oilseed crops and is cultivated in tropical and subtropical regions of Asia, Africa and South America [1, 2]. Its cultivation history can be traced back to between 5,000 and 5,500 years ago in the Harappa Valley of the Indian subcontinent [3]. The total area of sesame harvested in the world is currently 7.8 million hectares, and annual production is 3.84 million tons (2010, UN Food and Agriculture Organization data). Being one of the four main sesame-producing countries, China has contributed 15.2 to 32.5% of the total world sesame production over the past 10 years (2001 to 2010, UN Food and Agriculture Organization data). Sesame has one of the highest oil contents: decorticated seeds contain 45 to 63% oil [2]. The seed is also rich in protein, vitamins, including niacin, minerals and lignans, such as sesamolin and sesamin [4–7], and it is a popular food and medicine [8–13]. Sequencing and analysis of the sesame genome is essential if we are to elucidate the evolutionary origins and characteristics of the sesame species.
Sesamum is the main genus in the family Pedaliaceae, which contains 17 genera and 80 species of annual and perennial herbs that are distributed in the Old World tropics and subtropics [14]. The taxonomy and cytogenetics of the Sesamum genus has been reviewed and debated for a long time [1, 14–17], and many heterogeneous landraces present in various growing areas still need to be distinguished [1, 18]. S. indicum is the sole cultivar in the Sesamum genus and evolved from wild populations [14, 19]. However, the origin and evolution of cultivated sesame is still unclear and requires more detailed investigation [1, 15]. Evidence suggests that sesame may have originated in either India or Africa [3, 20–26]. Bedigian reported that sesame was derived from the Indian subcontinent (the western Indian peninsula and parts of Pakistan) thousands of years ago, and believed that the progenitor of sesame is a taxon named S. orientale var. malabaricum Nar. [22, 23], although most species of Sesamum and genera of the Pedaliaceae are native to Africa [27–29]. We hope to clarify the origin and phylogeny of S. indicum by applying comparative genomics and morphological and cytological analyses.
Sesame seed is commonly known as the 'Queen of the oil seeds', perhaps for its resistance to oxidation and rancidity [3]. As it contains lignans, sesame oil also exerts anti-cancer properties both in vitro and in animal bioassays [30–34]. Compared with peanut (Arachis hypogaea), soybean (Glycine max), oilseed rape (Brassica napus), sunflower (Helianthus annuus L.) and other oilseed crops, sesame seed oil has an ideal nearly equal content of oleic acid (18:1) (39.6%) and linoleic acid (18:2) (46.0%), and has desirable physiological effects, including antioxidant activity, and blood pressure- and serum lipid-lowering potential [2, 35, 36]. Studies of the genome and functional genome of sesame are essential for elucidating the regulatory mechanisms underlying fatty acid and storage protein composition and content, and the secondary metabolism of antioxidant lignans [37–40].
Sesame grows well and gives good yields in both tropical and temperate climates. Its tolerance of drought and high temperatures make sesame well suited to land where few other crops can survive. However, compared with other oilseed crops, sesame seed production is not consistent, as it is susceptible to pathogens, waterlogging and low temperature conditions [41]. Sesame breeding objectives, like those for other seed-producing crops, especially oil crops, are to create new varieties with high quality and yield potential, and resistance to pathogens (including Fusarium wilt and Charcoal rot diseases), insect pests, waterlogging, drought and low temperature stress [37, 42–45]. However, identification of genes or gene families and marker loci associated with yield, quality, and resistance to disease and abiotic stresses has been hampered due to a lack of information on the sesame genome. Only a few functional genes, mainly involved in the formation and regulation of fatty acids, seed storage proteins and secondary metabolites, and salt stress response, have been investigated [46–54]. With the exception of a sole amplified fragment length polymorphism (AFLP) marker associated with the indehiscent-capsule trait reported in 2003 [55], no quantitative trait loci have been found in the linkage map of sesame, let alone used for molecular-assisted selection (MAS) in sesame breeding programs. Integrating desirable qualities from the few available excellent germplasm resources, including wild species, will not be achievable rapidly unless considerably more genomic and functional genomic information is available. In addition, sequencing of the sesame genome will facilitate studies of other genera of the Pedaliaceae family by providing a closely related reference genome.
We therefore plan to implement a Sesame Genome Project and sequence the S. indicum genome using the Chinese domestic cultivar, Yuzhi 11, which represents S. indicum cultivars with a simple stem, three flowers per axilla, oblong-quadrangular capsules, and white flower and seed-coat color. Yuzhi 11 is one of the most important Chinese cultivars due to its high oil content (56.66%), resistance to fungal pathogens such as Fusarium wilt, charcoal rot and Alternaria leaf spot, and waterlogging stress. It is cultivated in the main production regions of China [56, 57].
Phylogenetic position of sesame
S. indicum is located in the asterids clade of the core eudicotyledons of Angiosperm Phylogeny Group 2 (APG 2) [58]. Its phylogenetic position determined using sesame chloroplast genomic data indicates that Sesamum (Pedaliaceae family) is a sister genus to the Olea and Jasminum (Oleaceae family) clade and represents the core lineage of the Lamiales families [59]. Compared with the 19 families shown in Figure 1 (adapted from the NCBI taxonomy database [60]), Sesamum, which has 36 available genomes, is closely related to the Solanaceae and Phrymaceae families, but distantly related to other oil crops such as soybean (Glycine max), castor (Ricinus communis) and rape (Brassica rapa). At present, genomic information on the Pedaliaceae family is quite limited, as genomes from this family have not previously been sequenced.

Phylogenetic positions of sesame and the 36 land plants with available genome sequences. aRefers to sesame (S. indicum L.), a member of the Pedaliaceae family, only 34 genera of which have been entered in the NCBI taxonomy database.
Overview of the Sesame Genome Project
The Sesame Genome Working Group (SGWG) comprises six major sesame research teams in China involved in investigating genetic diversity of germplasm resources, functional genomics, and biotic and abiotic resistance, in addition to sesame genome sequencing. All members of the SGWG work under the Toronto Statement for prepublication data release [61]. The main goal of the Sesame Genome Project is to provide a fine map of S. indicum and facilitate global genomic and functional genomic studies. We have already released a preliminary draft assembly [62] of the sesame genome that can be used according to the conditions outlined in this letter. A detailed plan for the Sesame Genome Project has been made available on our website [62].
Properties of the S. indicumgenome and available genomic resources
Natural sesame species can be divided into three types based on chromosome numbers, that is, 2n = 26 (for example, S. indicum, S. alatum), 2n = 32 (for example, S. protratum, S. angolense) and 2n = 64 (for example, S. radiatum, S. schinzianum) [14, 37]. The basic chromosome number in the Sesamum genus is X = 8 and 13, with X = 13 probably resulting from ancient polyploidy [37]. The size of a haploid genome of S. indicum (2n = 26) was reported to be about 0.95 Gb, with a mass of 0.97 pg [63], which is out of proportion with the 0.51 Gb and 0.97 Gb of Cerathoteca sesamoides (2n = 32) and S. radiatum (2n = 64), respectively [64]. Before beginning this genome project, we examined the characteristics of sesame chromosomes using cv. Yuzhi 11. Results showed that its karyotype formula is 2n = 2x = 26 = 6m + 16sm + 4st, and chromosome length ranges from 1.21 to 2.48 μm (H Zhang, unpublished data). We distinguished and numbered the chromosomes with 45S rRNA, simple sequence repeats (SSR) and bacterial artificial chromosome (BAC) sequence probes using fluorescent in situ hybridization (FISH) and BAC-FISH techniques to facilitate super-scaffold assembly in the sesame genome (H Zhang, unpublished data). Comparing genome size with that of Arabidopsis thaliana [65], soybean (cv. William 82) [66] and rice (cv. Nipponbare) [67], the genome size of S. indicum cv. Yuzhi 11 is estimated by flow cytometry to be about 369 Mb (H Zhang, unpublished data). From our preliminary sequencing data, we estimate the genome size to be approximately 354 Mb, close to this result (see below).
The sesame chloroplast genome was published recently [59]. Sequencing of the chloroplast genome of S. indicum cv. Yuzhi 11 has also been performed (H Zhang, unpublished data), and will be used for raw read filtering and genome assembly in our Sesame Genome Project. A total of 86,222 unigenes with an average length of 629 bp are available and 46,584 (54.03%) unigenes have a significant similarity with proteins in the NCBI nonredundant protein database and Swiss-Prot database (E-value <10-5) [39]. Before the beginning of this project, we sequenced sesame transcriptomes from 24 groups of S. indicum materials and treatments using Illumina paired-end sequencing technology to greatly enrich available information on the functional genome [40, 68], obtaining a 40G dataset containing 42,566 unitranscript sequences. We also constructed a BIBAC (pCLD 04541) library of 80,000 clones with an insert size of 120 kb and a BAC (CopyControl™ pCC1BAC™) library of 57,600 clones with an insert size of 85 kb. The genome coverage of both BAC libraries was 27- and 13-fold, respectively (H Zhang, unpublished data). There are 45,093 S. indicum expressed sequence tags (ESTs) available in the NCBI EST database. Prior to our work, only two other S. indicum seed-specific cDNA libraries, including one full-length cDNA library, had been constructed, some clones of which were chosen at random and sequenced [38, 69]. In order to explore more genes involved in sesame growth and development, we constructed a full-length cDNA library of S. indicum cv. Yuzhi 11 containing 300,000 clones, 1,200 clones of which were selected randomly and sequenced (H Zhang, unpublished data). The genomic and transcriptomic data from these studies should facilitate genome assembly and analysis. The first sesame linkage map, which contains 284 microsatellite polymorphic loci, was set up in 2009 and has been used as a landmark frame for assembly of the whole genome [70]. We recently updated this high-density linkage map with 653 SSR, SNP, AFLP and random selective amplification of microsatellite polymorphic loci (RSAMPL) markers falling into 14 linkage groups to facilitate sesame genome assembly and anchoring of trait loci (H Zhang, unpublished data).
Sequencing strategy for the S. indicumgenome
The Sesame Genome Project is divided into three phases. The first phase, which has already been completed, involves high coverage Illumina sequencing and draft genome assembly. We constructed five types of Illumina libraries, including two paired-end libraries with insert sizes of 300 and 500 bp, and three mate-pair libraries with insert sizes of 2, 3 and 5 kb. In order to avoid bias in library construction, at least two libraries for each insert length were constructed. Illumina technology was used to generate 98 Gb of reads, giving a 276× coverage of the estimated genome (Table 1). Subsequently, the draft genome was assembled using ABySS (v 1.3.3) [71]. Paired-end Illumina reads were first assembled into contigs. Mate-pair reads with insert sizes of 2, 3 and 5 kb were then aligned into the contigs, and the relationship between mate-pair reads was used to join contigs and construct scaffolds. As a result, a preliminary assembly of 293.7 Mb was generated (Table 2).
The second phase will involve Roche 454 pyrosequencing and BAC sequencing and fine map construction. We have constructed Roche 454 paired-end libraries with an insert size of 20 kb and will generate 3.5 Gb of data giving a 250× coverage of the estimated genome. We also plan to end-sequence 40,000 sesame BAC clones using conventional Sanger sequencing, giving a 12× coverage of the estimated genome. To ensure hybrid de novo assembly of the best possible quality, we will use a modified Celera Assembler pipeline [72]. Roche 454 paired-end reads and BAC-end reads are better for spanning longer repetitive elements and joining scaffolds into superscaffolds. We will use BAC-end information to retrieve and select 1,000 specific BAC clones, one end of which aligns well to the scaffold while the other end is located in a gap region, for full-length sequencing using the Illumina BAC polling method. The full-length BAC sequences will fill in the gaps within superscaffolds and greatly improve genome integrity. At this stage, we expect to obtain a fine map of Yuzhi 11 with 800 to 1,000 superscaffolds of a putative N50 length of 1 Mb and N90 length of 250 kb.
In the final phase, the superscaffolds will be anchored to chromosomes. We will first anchor the BACs containing mapped SSR markers on the updated linkage map [70] (H Zhang, unpublished data). Physical distances between landmarks will then be determined. Furthermore, we will construct a physical chromosome map based on at least 1,000 BAC clones using information obtained from BAC-FISH and BAC-end. At least one BAC will be anchored on the chromosomes per superscaffold to ensure all superscaffolds are anchored onto the 13 chromosomes. In order to validate the accuracy and integrity of the sesame genome assembly, several quality control parameters, such as read depth of coverage, average quality values per contig, discordant read pairs and gene footprint coverage, will be examined. To check the accuracy of the assembly of scaffolds, we will also complete full-length sequencing of 15 BAC clones using conventional Sanger sequencing and align them to the scaffolds.
Timeline and goals of the Sesame Genome Project
The blueprint for the Sesame Genome Project was conceived and designed by the SGWG in 2009. We completed the goals of the first phase in March 2012. In the second phase, Roche 454 paired-ends reads will be sequenced by December 2012, and the double-ended sequencing of the 40,000 BAC clones and full-length sequencing of 1,000 BAC clones will be completed by June 2013. The final phase of scaffold anchoring will proceed in parallel with bioinformatics analysis. We expect to complete all the goals of Sesame Genome Project and submit a paper by December 2013. To make our data broadly available prior to publication, the completion of each goal of these phases will be publicly communicated via our website [62]. Updated versions of assembly data will be made available to any independent research groups performing non-genome-scale analyses. Sequence data and the preliminary assembly produced in the first phase are already available on the website.
Status of current preliminary genome assemblies
The current draft assembly of Yuzhi 11 is 293.7 Mb in length, with a GC content of 34.65%. The N50 and N90 sizes of the scaffolds are 22.6 kb and 4.3 kb, respectively (Table 2). Genome size was estimated to be 354 Mb using the well-established 17-mer method [73], in line with flow cytometry data that suggest it is 369 Mb (H Zhang, unpublished data). The 17-mer distribution frequency in 16.77 Gb of trimmed Illumina PE reads was calculated using Jellyfish (v1.1.4) [74]. We identified a total of 13,931,658,332 unique k-mers, and 87,207,553 k-mers that had a frequency <10. The frequency of peak k-mers was 39 (Figure 2).

K-mer (17mer) frequency analysis of the S. indicum genomic sequence. Data produced from 500 bp insert libraries. The peak k-mer frequency is 39 and its minimum point is 10. Genome size was estimated with the formula: Estimated genome size (bp) = total number of k-mers with a frequency >10/peak k-mer frequency.
In order to determine the frequency and complexity of repetitive elements in the draft assembly, we compared the assembly information with the Arabidopsis repetitive elements database from the RepeatMasker library (version 20120418) and the sesame de novo database constructed for the Yuzhi 11 draft assembly (RepeatModeler, version 1.0.5) using RepeatMasker (version open-3.2.9) [75, 76]. Thirty-eight percent of the draft assembly was identified as repetitive elements (Table 3), only approximately 5.7% of which shared homology with the Arabidopsis database.
Quality control the raw data and intermediate datasets
In order to control the quality of raw data, the SolexaQA package was used to verify the sequence data generated from each of the 17 Illumina-Solexa libraries [77]. The raw reads were trimmed by DynamicTrim (quality threshold Q ≈ 20) and then filtered by LengthSort (the length cutoff set as 25). Unpaired reads would be screened and discarded in this system. Meanwhile, Roche 454 reads data, which are kept in Standard Flowgram Format (SFF), were converted into FastQ format and evaluated using the traditional quality metrics. As Sanger reads may contain vector sequences, the Lucy package was used to search and trim for cutting off the vector sequence contamination [78]. Low-quality bases and chimeric reads would be tracked with trim modules of the Celera Assembler.
We validated the coding region coverage of the draft assembly using two different gene footprint coverage methods. Using the Core Eukaryotic Genes Mapping Approach (CEGMA) [79], 444 (96.9%) of the 458 core eukaryotic genes (CEGs) mapped against the draft assembly were identified. An RNA sequence based method employing Velvet [80] and OASES [81] allowed us to assemble 3.5 Gb of RNA-Seq reads (NCBI accession SRX061117) [39] into 99,589 putative transcripts. Putative transcripts were then translated into 82,549 peptides using ESTScan (version 2.1) [82]. These peptides were aligned against the SWISS-PROT [83] database using BLAST (E-value 10-5) to obtain high-confidence peptides. Redundant peptides (such as alternative-splicing transcripts) were filtered according to BLAST scores and the names of the hits. More than 99.5% of the 3,584 peptides obtained could be aligned to the draft assembly using GMAP [84]. The above results indicate that the draft assembly has a high coverage of the coding region.
Gene prediction for the draft assembly was performed using InchWorm [85]: 3.5 Gb of RNA-Seq reads [GenBank: SRX061117] were assembled into 472,257 contigs and mapped to the draft genome using GMAP. The GMAP mapping results were used as a training set for ab initio prediction using AUGUSTUS [86]. As a result, 23,713 gene models were obtained with a total length of 28 Mb (Table 4). Average coding sequence length was 1.2 kb and average GC content was 45%. We obtained functional annotations of all genes using InterProScan [87], which also determines motifs and domains. Gene Ontology (GO) annotations were given to 10,656 genes using corresponding InterPro entries and the Pfam database [88]. Visualization of the functional categories of these 10,656 genes was performed using WEGO [89] (Figure 3).

Functional catalogues of sesame genes in the preliminary assembly. Results are summarized in three main categories: biological processes, cellular components and molecular functions. A total of 10,656 genes have been assigned with Gene Ontology terms.
Biological questions to be addressed
We plan to address several key biological questions specific to sesame using this new genome and transcriptome data. We will compare the sesame genome with the genomes of monocotyledonous and other dicotyledonous plants to elucidate the phylogeny of the Sesamum genus and the origin of S. indicum. We will also perform more detailed investigations on the formation and regulation of fatty acids, storage proteins and secondary metabolites (including sesamin) in sesame. We will apply the bio-information obtained in this genome project in sesame breeding programs, paying particular attention to the induction and regulation of resistance to the main sesame diseases, including Fusarium wilt and charcoal rot diseases, and the environmental stress of waterlogging. Other possible uses of the genomics dataset, such as determining the regulatory mechanisms of biological characteristics in Sesamum, including simple stem or branch, leaf shape, indeterminate growth habit, flower number per axilla, capsule carpel number, flower color and other species-specific traits, will not form part of our analysis. We believe that the main achievement of this project will be to markedly accelerate sesame genetic research and breeding. Members of the SGWG also hope to address additional questions about the relationship between sesame growth and environmental conditions, such as identifying which genes regulate low temperature responses and drought sensitivity.
Joining the SGWG and using our early release data
This project is being conducted by the SGWG. We invite other research groups to access and use the draft assembly and raw read data, which have already been released. Any group performing non-genome-scale analyses, or investigating the above biological questions, is welcome to use our data without restriction. As a matter of courtesy and to avoid duplication of effort, we request that competing genome-scale projects or studies that overlap with the above stated research areas disclose their status to the SGWG consortium. Formal inquiries and requests to join the working group should be made to HZ. Updated versions of the genome assembly, further project descriptions and a complete list of current SGWG members dedicated to this project can be accessed on our website [62].
Abbreviations
- AFLP:
-
amplified fragment length polymorphism
- BAC:
-
bacterial artificial chromosome
- EST:
-
expressed sequence tags
- FISH:
-
fluorescent in situ hybridization
- RSAMPL:
-
random selective amplification of microsatellite polymorphic loci
- SGWG:
-
Sesame Genome Working Project
- SNP:
-
single nucleotide polymorphism
- SSR:
-
simple sequence repeats.
References
Ashri A: Sesame breeding. Plant Breeding Reviews. Edited by: Janick J. 1998, Oxford: Oxford UK, 79-228.
Anilakumar KR, Pal A, Khanum F, Bawa AS: Nutritional, medicinal and industrial uses of sesame (Sesamum indicum L.) seeds an overview. Agric Conspec Sci. 2010, 75: 159-168.
Bedigian D, Harlan JR: Evidence for cultivation of sesame in the ancient world. Econ Bot. 1986, 40: 137-154. 10.1007/BF02859136.
Budowski P, Markley KS: The chemical and physiological properties of sesame oil. Chem Rev. 1951, 48: 125-151. 10.1021/cr60149a005.
Moazzami AA, Kamal-Eldin A: Sesame seed is a rich source of dietary lignans. J Am Oil Chem Soc. 2006, 8: 719-723.
Nakimi M: The chemistry and physiological functions of sesame. Food Rev Int. 1995, 11: 281-329. 10.1080/87559129509541043.
Yamashita K, Nohara Y, Katayama K, Namiki M: Sesame seed lignans and γ-tocopherol act synergistically to produce vitamin E activity in rats. J Nutr. 1992, 122: 2440-2246.
Mochizuki M, Tsuchie Y, Yamada N, Miyake Y, Osawa T: Effect of sesame lignans on TNF-alpha-induced expression of adhesion molecules in endothelial cells. Biosci Biotechnol Biochem. 2010, 74: 1539-1544. 10.1271/bbb.100095.
Liao CD, Hung WL, Lu WC, Jan KC, Shih DY, Yeh AI, Ho CT, Hwang LS: Differential tissue distribution of sesaminol triglucoside and its metabolites in rats fed with lignan glycosides from sesame meal with or without nano/submicrosizing. J Agric Food Chem. 2010, 58: 563-569. 10.1021/jf9028046.
Jan KC, Hwang LS, Ho CT: Biotransformation of sesaminol triglucoside to mammalian lignans by intestinal microbiota. J Agric Food Chem. 2009, 57: 6101-6106. 10.1021/jf901215j.
Jan KC, Ku KL, Chu YH, Hwang LS, Ho CT: Tissue distribution and elimination of estrogenic and anti-inflammatory catechol metabolites from sesaminol triglucoside in rats. J Agric Food Chem. 2010, 58: 7693-7700. 10.1021/jf1009632.
Jan KC, Ku KL, Chu YH, Hwang LS, Ho CT: Intestinal distribution and excretion of sesaminol and its tetrahydrofuranoid metabolites in rats. J Agric Food Chem. 2011, 59: 3078-3086. 10.1021/jf105012v.
Coulman KD, Liu Z, Hum WQ, Michaelides J, Thompson LU: Whole sesame seed is as rich a source of mammalian lignan precursors as whole flaxseed. Nutr Cancer. 2009, 52: 156-165.
Nimmakayala P, Perumal R, Mulpuri S, Reddy UK: Sesamum. Wild Crop Relatives: Genomic and Breeding Resources Oilseeds. Edited by: Kole C. 2011, Berlin Heidelberg: Springer-Verlag, 261-273.
Joshi AB: Sesamum. 1961, Hyderabad: Examiner Press
Bruce EA: Notes on African Pedaliaceae. Kew Bull. 1953, 67: 417-429.
Kobayashi T: The wild and cultivated species in the genus Sesamum. Sesame: Status and Improvement. Proceedings of Expert Consultation: 8-12 December 1980; Rome. Edited by: Amram A. 1981, Rome, 157-163.
Weiss EA: Castor, Sesame and Safflower. 1971, New York: Barnes & Noble Press
Zhang H, Miao H, Li C, Wei L, Ma Q: Analysis of sesame karyotype and resemblance-near coefficient. Chinese Plant Bullet. 2012, 47: 602-614.
Bedigian D, Seigler DS, Harlan JR: Sesamin, sesamolin and the origin of sesame. Biochem Systemat Ecol. 1985, 13: 133-139. 10.1016/0305-1978(85)90071-7.
Bedigian D: Evolution of sesame revisited: domestication, diversity and prospects. Genet Resour Crop Evol. 2003, 50: 779-787. 10.1023/A:1025029903549.
Bedigian D: Slimy leaves and oily seeds: distribution and use of wild relatives of sesame in Africa. Economic Botany. 2004, 58 (Suppl): 3-33.
Bedigian D: Characterization of sesame (Sesamum indicum L.) germplasm: a critique. Genet Resour Crop Evol. 2010, 57: 641-647. 10.1007/s10722-010-9552-x.
Nanthakumar G, Singh KN, Vaidyanathan P: Relationships between cultivated sesame (Sesamum sp.) and the wild relatives based on morphological characters, isozymes and RAPD markers. J Genet Breeding. 2000, 54: 5-12.
Fuller DQ: Further evidence on the prehistory of sesame. Asian Agri-History. 2003, 7: 127-137.
Kumar AKMS, Hiremath SC: Cytological analysis of interspecific hybrid between Sesamum indicum L × S. Orientale L. Var. malabaricum. Karnataka J Agric Sci. 2008, 21: 498-502.
Ashri A: Sesame research overview: Current status, perspective and priorities. Proceeding of the 1st Australian Sesame Workshop: 21-23 March 1995; Darwin-Katherine. Edited by: Bennett MR. 1995, Wood IM: Darwin, 1-17.
Grubben GJH, Denton OA: Plant Resources of Tropical Africa: Vegetables. 2004, Leiden: Backhuys Publishers
Purseglove JW: Tropical Crops: Dicotyledons. 1968, London: Longmans Press
Nakano D, Itoh C, Takaoka M, Kiso Y, Tanaka T, Matsumura Y: Antihypertensive effect of sesamin inhibition of vascular superoxide production by sesamin. Biol Pharm Bull. 2002, 25: 1247-1249. 10.1248/bpb.25.1247.
Kanu PJ, Bahsoon JZ, Kanu JB, Kandeh JB: Nutraceutical importance of sesame seed and oil A review of the contribution of their lignans. Sierra Leone J Biomed Res. 2010, 2: 4-16.
Salerno JW, Smith DE: The use of sesame oil and other vegetable oils in the inhibition of human colon cancer growth in vitro. Anticancer Res. 1991, 11: 209-215.
Kapadia GJ, Azuine MA, Tokuda H, Takasaki M, Mukainaka T, Konoshima T, Nishino H: Chemopreventive effect of resveratrol, sesamol, sesame oil and sunflower oil in the Epstein-Barr Virus early antigen activation assay and the mouse two state carcinogenesis. Pharmacol Res. 2002, 45: 499-505. 10.1006/phrs.2002.0992.
Fukuda Y, Nagata M, Osawa T, Namiki M: Chemical aspects of the antioxidative activity of roasted sesame seed oil and the effect of using the oil for frying. Agric Biol Chem. 1986, 50: 857-862. 10.1271/bbb1961.50.857.
Yermanos DM, Hemstreet S, Saleeb W, Huszar CK: Oil content and composition of the seed in the world collection of sesame introductions. J Am Oil Chem Soc. 1972, 49: 20-23. 10.1007/BF02545131.
Nweke FN, Ubi BE, Kunert K: Application of microsatellite polymorphisms to study the diversity in seed oil content and fatty acid composition in Nigerian sesame (Sesamum indicum L.) accessions. Afr J Biotech. 2012, 11: 8820-8830.
Ashi A: Sesame (Sesamum indicum L.). Genetic Resources, Chromosome Engineering, and Crop Improvement. Edited by: Signh RJ. 2006, Boca Raton: CRC Press, 231-280.
Suh MC, Kim MJ, Hur C-G, Bae JM, Park YI, Chung C-H, Kang C-W, Ohlrogge JB: Comparative analysis of expressed sequence tags from Sesamum indicum and Arabidopsis thaliana developing seeds. Plant Mol Biol. 2003, 52: 1107-1123.
Wei WL, Qi XQ, Wang LH, Zhang YX, Hua W, Li DH, Lv HX, Zhang XR: Characterization of the sesame (Sesamum indicum L.) global transcriptome using Illumina paired-end sequencing and development of EST-SSR markers. BMC Genomics. 2011, 12: 451-10.1186/1471-2164-12-451.
Wei L, Miao H, Zhang H: De novo transcriptome sequencing and analysis of sesame growth and development. Scientia Agricultura Sinica. 2012, 45: 1246-1256.
Verma ML, Mehta N, Snagwan MS: Fungal and baterial diseases of sesame. Diseases of Oilseed Crops. Edited by: Saharan GS, Mehta N. 2005, New Delhi; Sangwan Indus, 269-301.
Baydar H: Breeding for the improvement of the ideal plant type of sesame. Plant Breed. 2005, 124: 263-267. 10.1111/j.1439-0523.2005.01080.x.
El-Bramawy MAS, Shaban WI: Nature of gene action for yield, yield components and major diseases resistance in sesame (Sesamum indicum L.). Res J Agric Biol Sci. 2007, 3: 821-826.
Mensah JK, Obadoni BO, Eruotor PG, Onome-Irieguna F: Simulated flooding and drought effects on germination, growth, and yield parameters of sesame (Sesamum indicum L.). Afr J Biotechnol. 2006, 5: 1249-1253.
Kuol BG: Breeding for Drought Tolerance in Sesame (Sesamum indicum L.) in Sudan. 2004, Göttingen: Cuvillier Press
Yukawa Y, Takaiwa F, Shoji K, Masuda K, Yamada K: Structure and expression of two seed-specfic cDNA clones encoding stearoyl-acyl carrier protein desaturase from sesame, Sesamum indicum L. Plant Cell Physiol. 1996, 37: 201-205. 10.1093/oxfordjournals.pcp.a028932.
Chen JCF, Lin RH, Huang HC, Tzen JTC: Cloning, expression and isoform classification of a minor oleosin in sesame oil bodies. J Biochem. 1997, 122: 819-824. 10.1093/oxfordjournals.jbchem.a021828.
Jin UH, Lee JW, Chung YS, Lee JH, Yi YB, Kim YK, Hyung NI, Pyee JH, Chung CH: Characterization and temporal expression of a ω-6 fatty acid desaturase cDNA from sesame (Sesamum indicum L.) seeds. Plant Sci. 2001, 161: 935-941. 10.1016/S0168-9452(01)00489-7.
Lee TTT, Chung MC, Kao YW, Wang CS, Chen LJ, Tzen JTC: Specific expression of a sesame storage protein in transgenic rice bran. J Cereal Sci. 2005, 41: 23-29. 10.1016/j.jcs.2004.08.014.
Chyan CL, Lee TTT, Liu CP, Yang YC, Tzen JTC, Chou WM: Cloning and expression of a seed-specific mtallothione-like protein from sesame. Biosci Biotech Biochem. 2005, 69: 2319-2325. 10.1271/bbb.69.2319.
Hsiao ESL, Lin LJ, Li FY, Wang MMC, Liao MY, Tzen JTC: Gene families encoding isoforms of two major sesame seed storage proteins, 11S globulin and 2S albumin. J Agirc Food Chem. 2006, 54: 9544-9550. 10.1021/jf061505x.
Kim MJ, Kim JK, Shin JS, Suh MC: The SebHLH transcription factor mediates trans-activation of the SeFAD2 gene promoter through binding to E-and G-box elements. Plant Mol Biol. 2007, 64: 453-466. 10.1007/s11103-007-9165-8.
Kim MJ, Go YS, Lee SB, Kim YS, Shin JS, Min MK, Hwang I, Suh MC: Seed-expressed casein kinase I acts as a positive regulator of the SeFAD2 promoter via phosphorylation of the SebHLH transcription factor. Plant Mol Biol. 2010, 73: 425-437. 10.1007/s11103-010-9630-7.
Hata N, Hayashi Y, Okazawa A, Ono E, Satake H, Kobayashi A: Comparison of sesamin contents and CYP81Q1 gene expressions in aboveground vegetative organs between two Japanese sesame (Sesamum indicum L.) varieties differing in seed sesamin contents. Plant Sci. 2010, 178: 510-516. 10.1016/j.plantsci.2010.02.020.
Uzun B, Lee D, Donini P, Cagirgan MI: Identification of a molecular marker linked to the closed capsule mutant trait in sesame using AFLP. Plant Breeding. 2003, 122: 95-97. 10.1046/j.1439-0523.2003.00787.x.
Wei W, Wei S, Zhang H, Ding F, Zhang T, Lu F: Breeding of a new sesame variety Yuzhi 11. J Henan Agri Sci. 1999, 7: 3-4.
Zhang T, Zhang H, Wei S, Zheng Y, Zhang Z, Wang Z: Analysis of Integrated characteristics of Yuzhi 11. Chinese Agri Sci Bullet. 2003, 19: 44-46.
Angiosperm Phylogeny Group: An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG II. Bot J Linn Soc. 2003, 141: 399-436.
Yi D-K, Kim K-J: Complete chloroplast genome sequences of important oilseed crop Sesamum indicum L. PloS One. 2011, 7: e35872-
Sayers EW, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, Church DM, DiCuccio M, Edgar R, Federhen S, Feolo M, Geer LY, Helmberg W, Kapustin Y, Landsman D, Lipman DJ, Madden TL, Maglott DR, Miller V, Mizrachi I, Ostell J, Pruitt KD, Schuler GD, Sequeira E, Sherry ST, Shumway M, Sirotkin K, Souvorov A, Starchenko G, Tatusova TA, et al: Database resources of the National Center for Biotechnology Information. Nucleic Acid Res. 2009, 37 (Database issue): 5-15.
Toronto 2009 Data Release Workshop Authors: Benefits and best practices of rapid re-publication data release. Nature. 2009, 461: 168-170.
The Sesame Genome Working Group. [http://www.sesamum.org]
Test system for the qualitative detection of sesame DNA in food products by PCR Real Time. I-007.9a E Rev.007. Code: A-02-1081, [http://www.sacace.com/]
Adéoti K, Rival A, Dansi A, Santoni S, Brown S, Beule T, Nato A, Henry Y, Vodouhe R, Loko LY, Sanni A: Genetic characterization of two traditional leafy vegetables (Sesamum radiatum Thonn. ex Hornem and Ceratotheca sesamoides Endl.) of Benin, using flow cytometry and amplified fragment length polymorphism (AFLP) markers. Afr J Biotechnol. 2011, 10: 14264-14275.
The Arabidopsis Genome Initiative: Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000, 408: 796-815. 10.1038/35048692.
Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, Hyten DL, Song Q, Thelen JJ, Cheng J, Xu D, Hellsten U, May GD, Yu Y, Sakurai T, Umezawa T, Bhattacharyya MK, Sandhu D, Valliyodan B, Lindquist E, Peto M, Grant D, Shu S, Goodstein D, Barry K, Futrell-Griggs M, Abernathy B, Du J, Tian Z, Zhu L, Gill N, et al: Genome sequence of the palaeopolyploid soybean. Nature. 2010, 463: 178-10.1038/nature08670.
Goff SA, Ricke D, Lan T-H, Presting G, Wang R, Dunn M, Glazebrook J, Sessions A, Oeller P, Varma H, Hadley D, Hutchison D, Martin C, Katagiri F, Lange BM, Moughamer T, Xia Y, Budworth P, Zhong J, Miguel T, Paszkowski U, Zhang S, Colbert M, Sun W-L, Chen L, Cooper B, Park S, Wood TC, Mao L, Quail P, et al: A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science. 2002, 296: 92-100. 10.1126/science.1068275.
Zhang H, Wei L, Miao H, Zhang T, Wang C: Development and validation of genic-SSR markers in sesame by RNA-seq. BMC Genomics. 2012, 13: 316-10.1186/1471-2164-13-316.
Ke T, Dong C, Mao H, Zhao Y, Chen H, Liu H, Dong X, Tong C, Liu S: Analysis of expression sequence tags from a full-length-enriched cDNA library of developing sesame seeds (Sesamum indicum). BMC Plant Biol. 2011, 11: 180-10.1186/1471-2229-11-180.
Wei L-B, Zhang H-Y, Zheng Y-Z, Miao H-M, Zhang T-Z, Guo W-Z: A genetic linkage map construction for sesame (Sesamum indicum L.). Genes Genomics. 2009, 31: 199-208. 10.1007/BF03191152.
Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I: ABySS: A parallel assembler for short read sequence data. Genome Res. 2009, 19: 1117-1123. 10.1101/gr.089532.108.
Miller JR, Delcher AL, Koren S, Venter E, Walenz BP, Brownley A, Johnson J, Li K, Mobarry C, Sutton G: Aggressive assembly of pyrosequencing reads with mates. Bioinformatics. 2008, 24: 2818-2824. 10.1093/bioinformatics/btn548.
Xu X, Pan S, Cheng S, Zhang B, Mu D, Ni P, Zhang G, Yang S, Li R, Wang J, Orjeda G, Guzman F, Torres M, Lozano R, Ponce O, Martinez D, De la Cruz G, Chakrabarti SK, Patil VU, Skryabin KG, Kuznetsov BB, Ravin NV, Kolganova TV, Beletsky AV, Mardanov AV, Di Genova A, Bolser DM, Martin DM, Li G, Yang Y, et al: Genome sequence and analysis of the tuber crop potato. Nature. 2011, 475: 189-195. 10.1038/nature10158.
Marçais G, Kingsford CE: A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011, 27: 764-70. 10.1093/bioinformatics/btr011.
Smit AFA, Green P: RepeatMasker Open-3.0. [http://www.repeatmasker.org]
RepeatModeler, version 1.0.5. [http://www.repeatmasker.org/RepeatModeler.html]
Cox MP, Peterson DA, Biggs P: SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics. 2010, 11: 485-10.1186/1471-2105-11-485.
Chou HH, Holmes MH: DNA sequence quality trimming and vector removal. Bioinformatics. 2001, 17: 1093-1104. 10.1093/bioinformatics/17.12.1093.
Parral G, Bradnaml K, Korfl I: CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007, 23: 1061-1067. 10.1093/bioinformatics/btm071.
Zerbino DR, Birney E: Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18: 821-829. 10.1101/gr.074492.107.
Schulz1 MH, Zerbino DR, Vingron M, Birney E: Oases: Robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics. 2012, 28: 1086-1092. 10.1093/bioinformatics/bts094.
Iseli C, Jongeneel CV, Bucher P: ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc Int Conf Intell Syst Mol Biol. 1999, 7: 138-148.
Boeckmann B, Bairoch A, Apweiler R, Blatter M-C, Estreicher A, Gasteiger E, Martin MJ, Michoud K, O'Donovan C, Phan I, Pilbout S, Schneider M: The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003, 31: 365-370. 10.1093/nar/gkg095.
Wu TD, Watanabe CK: GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005, 21: 1859-1875. 10.1093/bioinformatics/bti310.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, Palma F, Birren BW, Nusbaum C, Friedman N, Regev A: Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011, 29: 644-652. 10.1038/nbt.1883.
Stanke M, Keller O, Gunduz I, Hayes A, Waack S, Morgenstern B: AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34: W435-239. 10.1093/nar/gkl200.
Zdobnov EM, Apweiler R: InterProScan an integration platform for the signature- recognition methods in InterPro. Bioinformatics. 2001, 17: 847-848. 10.1093/bioinformatics/17.9.847.
Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths-Jones S, Khanna A, Marshall M, Moxon S, Sonnhammer ELL, Studholme DJ, Yeats C, Eddy SR: The Pfam protein families database. Nucleic Acids Res. 2004, 32 (Suppl 1): D138-141.
Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, Wang J, Li S, Li R, Bolund L, Wan J: WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34 (Suppl 2): W293-297.
Acknowledgements
This work was supported by the earmarked fund for the China Agriculture Research System (CARS-15), China National '973' Project (2011CB109304), and Henan Zhongyuan Scholar Fund (092101211100) to HZ. HM was supported by a grant from the China National Key Technology R & D program (2009BADA8B04-03) and the earmarked fund for China Agriculture Research System (CARS-15). HL, QW and MY were individually supported by the earmarked fund for China Agriculture Research System (CARS-15). Special thanks to Dr Joy Fleming for helpful discussions and suggestions in the manuscript revision process.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Zhang, H., Miao, H., Wang, L. et al. Genome sequencing of the important oilseed crop Sesamum indicumL. Genome Biol 14, 401 (2013). https://doi.org/10.1186/gb-2013-14-1-401
Published:
DOI: https://doi.org/10.1186/gb-2013-14-1-401