
Abstract
Background
Aspergillus nidulans (the asexual form of Emericella nidulans) is a model organism for aspergilli, which are an important group of filamentous fungi that encompasses human and plant pathogens as well as industrial cell factories. Aspergilli have a highly diversified metabolism and, because of their medical, agricultural and biotechnological importance, it would be valuable to have an understanding of how their metabolism is regulated. We therefore conducted a genome-wide transcription analysis of A. nidulans grown on three different carbon sources (glucose, glycerol, and ethanol) with the objective of identifying global regulatory structures. Furthermore, we reconstructed the complete metabolic network of this organism, which resulted in linking 666 genes to metabolic functions, as well as assigning metabolic roles to 472 genes that were previously uncharacterized.
Results
Through combination of the reconstructed metabolic network and the transcription data, we identified subnetwork structures that pointed to coordinated regulation of genes that are involved in many different parts of the metabolism. Thus, for a shift from glucose to ethanol, we identified coordinated regulation of the complete pathway for oxidation of ethanol, as well as upregulation of gluconeogenesis and downregulation of glycolysis and the pentose phosphate pathway. Furthermore, on change in carbon source from glucose to ethanol, the cells shift from using the pentose phosphate pathway as the major source of NADPH (nicotinamide adenine dinucleotide phosphatase, reduced form) for biosynthesis to use of the malic enzyme.
Conclusion
Our analysis indicates that some of the genes are regulated by common transcription factors, making it possible to establish new putative links between known transcription factors and genes through clustering.
Similar content being viewed by others

Background
Aspergillus represents a large and important genus of filamentous fungi comprising human pathogens such as A. fumigatus, plant pathogens such as A. flavus, and important cell factories such as A. niger, A. oryzae, and A. terreus. Furthermore, A. nidulans has been extensively used as a model organism for eukaryotic cells. Despite their importance as human and plant pathogens and their extensive use in food, chemical, and pharmaceutical production, it was only recently that an initiative was undertaken to sequence the genomes of several Aspergillus spp. Thus, the genomes of three Aspergillus spp. have been published (A. nidulans [1], A. oryzae [2], and A. fumigatus [3]), and complete genomic sequencing of several other species has been finished or is ongoing. This has enabled analysis of the function of these important organisms at the genome level.
Aspergilli are natural scavengers and hence they have a very flexible metabolism that enables consumption of a wide range of carbon and nitrogen sources. Considering the high degree of flexibility in the metabolism of aspergilli, it is interesting to evaluate the function of the metabolic network in these organisms during growth on different carbon sources. We therefore undertook a study of the metabolism of A. nidulans at the genome level during growth on three different carbon sources: glucose, glycerol, and ethanol. These three carbon sources enter the central carbon metabolism at different locations, and they have been reported to result in widely different regulatory responses [4–8].
Our study involved genome-wide transcription analysis using in situ synthesized oligonucleotide arrays containing probes for 9,371 out of the 9,541 putative genes in the genome of A. nidulans [9]. In order to map the effects of carbon source on transcription, we used well controlled bioreactors to grow the cells. In recent years a few large-scale transcription studies have been conducted in A. nidulans, but so far none has covered the complete set of predicted genes in the genome. Sims and coworkers [10] used spotted DNA arrays to interrogate 2,080 open reading frames (ORFs) within the genome of A. nidulans, using as probes polymerase chain reaction (PCR) products from expressed sequence tags (ESTs), as well as gene sequences deposited in GenBank. The arrays were initially used in connection with an ethanol-to-glucose upshift batch experiment with a reference strain [10], and subsequently modified to study the effect of recombinant protein secretion on gene expression in A. nidulans by comparing the transcription profiles of a recombinant and a reference strain grown in chemostat cultures [11]. For other species of Aspergillus, a few studies on transcription profiling using microarray technology have been reported in the literature. These made use of spotted DNA arrays fabricated from EST sequences of selected genes (for example, A. oryzae [12], A. flavus [13–15], and A. parasiticus [15]) and other types of arrays (for example, for A. terreus [16]). Furthermore, studies similar to ours (aiming to map differences in gene expression during batch growth on different carbon sources, in particular glucose and ethanol) have been performed with other organisms, such as the filamentous fungi A. oryzae [12] and Trichoderma reesei [17], and the yeast Saccharomyces cerevisiae (many studies, with the first being that of DeRisi and coworkers [18]), with only the latter covering the complete genome.
In this work transcriptome data were analyzed using a recently developed consensus clustering algorithm [19]. Clustering of transcription data is valuable with respect to assigning function to genes, and this is particularly pertinent to A. nidulans because less than 10% of the 9,541 putative genes have been assigned a function (more than 90% of the 9,541 putative genes are called hypothetical or predicted proteins), based on automated gene prediction tools [9]. Using consensus clustering, we identified genes specifically relevant to the metabolism of the different carbon sources and, of particular, interest we identified nearly 200 genes that were significantly upregulated only during growth on glycerol versus growth on glucose and ethanol.
In order to study further the transcriptional response to growth on different carbon sources at the level of the metabolism, we used the transcription data to evaluate the operation of the metabolic network. For this purpose, we reconstructed the metabolic network of A. nidulans at the genome level, based on detailed metabolic reconstructions previously developed for A. niger [20], S. cerevisiae [21], and Mus musculus [22], as well as information on the genetics, biochemistry, and physiology of A. nidulans. The metabolic network reconstructed for A. nidulans contains 1,213 reactions and links 666 genes to metabolic functions. In the process of reconstruction, we assigned metabolic functions to 472 ORFs that had not previously been annotated, by employing tools of comparative genomics based on sequence similarity and using public databases of genes and proteins of established function. The metabolic reconstruction provided a framework for the analysis of transcriptome data. In particular, the metabolic network was used in combination with a recently developed algorithm [23] to identify global regulatory responses of the metabolism to variations in carbon source.
Results
Reconstruction of the metabolic network and ORF annotation
The metabolic network of A. nidulans was reconstructed using a pathway-driven approach, which resulted in the assignment of metabolic roles to 472 ORFs that had not previously been annotated (Table 1). The reconstructed metabolic network linked a total of 666 genes to metabolic functions, including 194 previously annotated ORFs in the Aspergillus nidulans Database [9]. The resulting network comprises 1,213 metabolic reactions, of which 1095 are biochemical transformations and 118 are transport processes (Table 1), as well as 732 metabolites. Out of the 1,213 reactions there are 794 that are unique (681 unique biochemical conversions and 113 unique transport processes), indicating that 419 of the reactions in the metabolic network are redundant. All the reactions in the metabolic network are listed in Additional data file 7 (Table S1), as are the abbreviations assigned to the metabolite names (Table S2). The reconstructed metabolic network is to our knowledge the largest microbial network reported to date [24].
Transcriptional responses to changes in the carbon source
In order to be able to identify primarily the effect of carbon source on transcription, we grew the cells in well controlled bioreactors, which enabled us to perform very reproducible fermentations. Figure 1 shows the biomass and substrate profiles for growth on glucose, glycerol, and ethanol. For the fermentations with glucose and glycerol as the carbon sources, the carbon recoveries were above 90% (>98% for glycerol), whereas it was only about 64% for growth on ethanol because of evaporation of the substrate. The batch fermentations were carried out in three replicates on each of the carbon sources investigated (for standard deviations, see Figure 1). For all of the cultivations, the samples for transcriptome analysis were taken in the early exponential phase of growth, with the biomass concentration being in the range of 1 to 1.5 g dry weight/kg. At this stage, dispersed filamentous growth was observed in all cultivations.

Biomass and substrate profiles for the different batch cultivations carried out with A. nidulans. (a) Cultivation with glucose as carbon source. (b) Cultivation with glycerol as carbon source. (c) Cultivation with ethanol as carbon source. For all cultivations, the time of sampling, the biomass concentration at the time of sampling, and the maximum specific growth rate for the culture are given.
Identification of differentially expressed genes in pair-wise comparisons
The expression data for the three biological replicates on the three carbon sources were normalized (Additional data file 8 [Tables S3 to S5]) and compared in a pair-wise manner, in order to detect genome-wide transcriptional changes in response to a change in carbon source. Differentially expressed genes for each of the comparisons were identified by applying a significance statistical test (see Materials and methods, below) and considering a significance level (or cutoff in P value) of 0.01. Table 2 shows the total number of significantly regulated genes within the genome of A. nidulans for the three possible pair-wise comparisons between carbon sources, as well as the number of upregulated and downregulated genes. Because the change in carbon source is expected to result in changes in carbon metabolism, the number of differentially expressed genes that were comprised in the metabolic reconstruction for A. nidulans is also presented for each case. It is observed that there is an over-representation of metabolic genes that exhibit significant changes in expression (metabolic genes only comprise about 7% of the total number of genes). The complete list of genes whose expression was significantly changed in the pair-wise comparisons can be found in Additional data file 9 (Tables S6 to S8; they are also partly illustrated in Figures S1 to S3 in Additional data files 1, 2 and 3, respectively). The differentially expressed genes were functionally classified based on Gene Ontology (GO) assignments provided by CADRE [25] (Additional data file 10 [Tables S9 and S10]).
Gene clustering
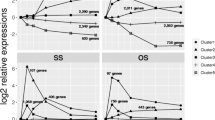
The genes were arranged in clusters, according to their expression profiles. In order to reduce the noise in the expression data before clustering analysis, an analysis of variance (ANOVA) test was performed that considered normalized transcriptome data from all of the replicated experiments on the different carbon sources (Additional data file 11 [Table S11]). The complete list of statistically significant genes for different significance levels is presented in Additional data file 11 (Table S12). For a significance level (or cutoff in P value) of 0.05, it was observed that the expression levels of 1,534 genes were significantly changed, of which 251 represented metabolic genes. Clustering analysis was applied to these 1,534 genes, and a total of eight clusters were identified (along with an additional cluster that included discarded genes). These clusters are represented in Figure 2, and the genes belonging to each group are listed in Additional data file 12 (Table S13). The GO annotation available in CADRE [25] was also used for functional classification of the genes included in the different clusters (Table 3). The transcriptional patterns of these 1,534 differentially expressed genes were also used for hierarchical cluster analysis (data not shown), and it was observed that the replicated experiments clustered together, as expected.

Representation of the eight clusters of genes identified. The numbers of genes in each cluster are as follows: 280 in cluster 1, 146 in cluster 2, 184 in cluster 3, 206 in cluster 4, 92 in cluster 5, 125 in cluster 6, 254 in cluster 7, and 212 in cluster 8. The x-axis represents the different carbon sources investigated: 1, glucose; 2, ethanol; and 3, glycerol. The y-axis represents normalized intensities, according to Grotkjær and coworkers [19]. Cluster 9 contains discarded genes, with low assignment to any of the other clusters.
Identification of metabolic subnetworks
In order to map overall metabolic responses to alterations of the carbon source, we applied the algorithm proposed by Patil and Nielsen [23] to identify the so-called reporter metabolites and to search for highly correlated metabolic subnetworks for each of the three pair-wise comparisons. This analysis relied on the reconstructed genome-scale metabolic network of A. nidulans, and hence we demonstrated how this metabolic network could be used to map global regulatory structures in A. nidulans. The top 15 high-scoring reporter metabolites for each of the cases are listed in Table 4 (also see Additional data files 4, 5 and 6 [Figures S4 to S6, respectively]).
To identify metabolic subnetworks with co-regulated expression patterns we began by finding high-scoring subnetworks, using the whole reaction set in the reconstructed metabolic network for A. nidulans, and subsequently we repeated the algorithm to identify smaller subnetwork structures. The repetition of the algorithm resulted in more robust solutions and in the identification of smaller networks, as demonstrated earlier for yeast data [23]. Table 5 shows the list of enzymes and transporters comprising the 'small' subnetworks for each of the pair-wise comparisons between the three carbon sources investigated (also see Additional data files 4, 5 and 6 [Figures S4 to S6, respectively]). Figure 3 shows key enzymes and transporters comprising the 'small' subnetwork for the glucose versus ethanol comparison. The 'large' subnetworks are given in Additional data file 13 (Tables S14 to S16). The genes in each of the 'small' subnetworks were classified according to the GO-terms assigned, and the results are presented in Additional data file 14 (Table S17).

Small subnetwork identified for the shift from glucose to ethanol as carbon source. Genes marked red are upregulated and genes marked green are downregulated upon the shift. The metabolic map is simplified (many transport reactions are not included and the two steps of the glycoxylate pathway [encoded by the genes acuD and acuE] are placed in the mitochondria even though they are really located in the glyoxysomes). Conversions that involve several steps are indicated by dashed arrows. The metabolites are as follows: ACCOA, acetyl-CoA; ACE, acetate; ACHO, acetaldehyde; CIT, citrate; F16BP, fructose 1,6-bisphosphate; F6P, fructose 6-phosphate; G6P, glucose 6-phosphate; GLY, glyoxylate; ICIT, isocitrate; MAL, malate; OAA, oxaloacetate; PEP, phosphoenolpyruvate; PYR, pyruvate; SUC, succinate.
Discussion
Enzyme complexes
In the process of reconstructing the metabolic network we identified several multi-enzyme complexes (for example, the F0F1 ATP synthase complex or the pyruvate dehydrogenase complex, which consist of several different proteins), and we used the transcriptome data to assess whether there was coordinated control of the expression of genes encoding the proteins of these complexes. Thus, for each enzyme complex included in the metabolic reconstruction of A. nidulans, we investigated whether the corresponding subunits had similar expression profiles. This was checked by verifying whether the genes encoding proteins within each enzyme complex were assigned to the same clusters. Furthermore, we calculated the Pearson correlations for all possible combinations within each enzyme complex (data not shown), in order to evaluate how well the corresponding expression levels correlated to each other. Calculation of Pearson correlations also enabled analysis of genes whose expression did not change significantly in the conditions studied. Based on the clustering and Pearson correlation analyses, we observed that, for about 30% (8/27) of the enzyme complexes considered, the expression profiles of the genes encoding all of the subunits of each enzyme complex were similar. Furthermore, in 11% (3/27) of the cases, the transcription of at least 50% (and <100%) of the subunits within an enzyme complex were highly correlated.
We performed the same analyses for S. cerevisiae using transcription data for similar conditions [26]. Here we observed that for about 21% (4/19) of the enzyme complexes included in the metabolic model for yeast [21], all of the corresponding subunits had similar expression patterns. Moreover, for 11% (2/19) of the enzyme complexes there was high correlation for at least 50% (and <100%) of the genes encoding for the complexes. Despite co-regulation of enzyme complexes in both A. nidulans and yeast, there does not appear to be any conservation in terms of transcriptional regulation of enzyme complexes, because only 7% (2/27) of enzyme complexes in A. nidulans with co-regulation on different carbon sources (either all components or 50% of the components) were also found to be co-regulated in yeast.
Ethanol utilization
The catabolism of ethanol, as well as regulation of the genes involved in this process, is presumably one of the best studied systems in A. nidulans (see Felenbok and coworkers [27] for a recent review). Two genes are responsible for the breakdown of ethanol into acetate via acetaldehyde, namely the genes encoding alcohol dehydrogenase I (alcA; AN8979.2) and aldehyde dehydrogenase (aldA; AN0554.2). The activation of this catabolic pathway is dependent on the transcriptional activator alcR (AN8978.2) [28]. Interestingly, a whole gene cluster composed of seven genes that are responsive to ethanol (or, more specifically, the gratuitous inducer methyl ethyl ketone) has previously been reported [29]. This cluster includes alcA and alcR, as well as five other transcripts (alcP [AN8977.2], alcO, alcM [AN8980.2], alcS [AN8981.2], and alcU [AN8982.2]), whose molecular functions have not yet been identified. In particular, one of these genes (alcO) has not been annotated in the genome sequence of A. nidulans, and similarity searches or gene prediction programs using the DNA sequence of the putative location of this gene were unsuccessful. Because our array design was based on annotated ORFs in the genome, this putative gene was not included in our analysis. However, all of the other genes of this cluster were found to be significantly upregulated on ethanol (alcP, alcR, alcA, alcM, and alcS were found in cluster 7, and alcU was found in cluster 6). Further positional analysis showed that there were no other gene clusters that were significantly regulated under any of the conditions studied (data not shown).
The subnetwork analysis clearly pointed to a coordinated expression of genes involved in ethanol metabolism upon shift from glucose to ethanol (Figure 3), and the response was to a large extent the same in the shift from glycerol to ethanol (Table 5). Ethanol is converted to acetate and is further catabolyzed to acetyl-coenzyme A (CoA), which then enters the mitochondria where it is oxidized (Figure 3). The subnetwork identified (Table 5) includes methylcitrate synthase (encoded by mcsA; AN6650.2), which was upregulated during growth on ethanol. This may point to a role of this enzyme in the catabolism of acetyl-CoA, in addition to the mitochondrial citrate synthase (encoded by citA; AN8275.2), which is expressed during growth both on glucose and ethanol. This is consistent with earlier reports in which it was found that this enzyme also possesses some citrate synthase activity [30].
The list of reporter metabolites (Table 4) is consistent with the identified subnetwork, because several components of the subnetwork are identified as reporter metabolites (CoA, acetyl-CoA, glyoxylate, oxaloacetate, carnitine, and O-acetyl-carnitine).
Besides alcA or ADH I (AN8979.2), A. nidulans has two additional alcohol dehydrogenases, namely alcB or ADH II (AN3741.2) and ADH III (AN2286.2). The former was assigned to cluster 6, whereas the latter did not appear to be significantly regulated in our analysis. It is interesting to observe that several genes in the identified subnetwork are also part of the metabolism of acetate, which is positively regulated by FacB (AN0689.2). Furthermore, facB was found to be significantly upregulated during growth on ethanol and assigned to cluster 7. FacB has been shown to induce directly the transcription of genes that are involved in the catabolism of acetate (acetyl-CoA synthetase, facA [AN5626.2]; carnitine acetyl transferase, facC [AN1059.2]; isocitrate lyase, acuD [AN5634.2]; malate synthase, acuE [AN6653.2]; and acetamidase, amdS [AN8777.2]) [5, 6]. All of these genes were found to be significantly upregulated during growth on ethanol (assigned to cluster 7), and several of them are part of the subnetwork identified from the pair-wise comparison between glucose and ethanol (Table 5).
The subnetwork also included ATP:citrate oxaloacetate-lyase, which catalyzes the formation of acetyl-CoA and oxaloacetate from the reaction of citrate and CoA, with concomitant hydrolysis of ATP to AMP and phosphate. This enzyme represents a major source of cytosolic acetyl-CoA during growth on glucose, which is a precursor for lipid biosynthesis. In A. nidulans, ATP:citrate oxaloacetate-lyase appears to be regulated by the carbon source present in the medium, with high activity in glucose-grown cells and low activity in acetate-grown cells [31]. This may be due to the fact that, during growth on C2 carbon sources, acetyl-CoA is formed directly in the cytosol in connection with the catabolism of the carbon source. The genes encoding the enzyme complex for ATP:citrate oxaloacetate-lyase (AN2435.2 and AN2436.2) were among the most significantly downregulated genes upon shift from glucose to ethanol (decreases of 22.6-fold and 22.2-fold, respectively; Additional data file 9 [Table S6]). Moreover, the genes encoding ATP:citrate oxaloacetate-lyase fell into cluster 2, together with another group of genes that were downregulated upon a shift from glucose to ethanol, namely the major part of the enzymes in the pentose phosphate (PP) pathway (Additional data file 12 [Table S13]). The subnetwork also captured changes in the expression of genes participating in gluconeogenesis, glycolysis, and the PP pathway. It was observed that genes involved in gluconeogenesis (PEP carboxykinase and fructose 1,6-bisphosphatase) were upregulated during growth on ethanol (assigned to clusters 7 and 6, respectively), whereas many of the genes of the PP pathway were downregulated (assigned to cluster 2). This suggests that an energetically more favorable route for supply of NADPH (nicotinamide adenine dinucleotide phosphatase, reduced form) is used during growth on ethanol, namely through the malic enzyme (encoded by maeA [AN6168.2]), which was found to be upregulated during growth on ethanol and was identified in the subnetwork for the glycerol versus ethanol comparison. This is consistent with earlier findings that the activity of malic enzyme is low on glucose and high on ethanol [32], and that maeA may be weakly regulated by carbon catabolite repression [33].
From the above, it is clear that there is coordinated regulation of genes in very different parts of the metabolism, which is important for the cell to maintain homeostasis during growth on different carbon sources. The strength of our analysis based on the metabolic network is that these coordinated expression patterns are clearly captured using a nonsupervised algorithm.
For the ethanol versus glucose comparison, it was interesting to note that the gene with the greatest fold change (151 times) was that of alcS. This is relevant considering that no molecular function has been suggested for this gene so far. In silico analysis suggests that AlcS might be a membrane bound transporter protein (six transmembrane-helix domains; conserved domain [PFAM01184]), indicating that AlcS could be an acetate transporter.
Regulation of transcription factors
As mentioned above, we observed that the gene facB was upregulated during growth on ethanol. However, we also found that several other transcription factors were regulated during growth on ethanol. Thus, we observed that creA (AN6195.2), which is the major mediator of carbon catabolite repression in A. nidulans, was located in cluster 6 and hence was upregulated during growth on ethanol. This might seem surprising, considering that CreA is assumed to be a transcriptional repressor and most active on glucose, but our findings corroborate findings reported by Strauss [34] and Sims [11] and their coworkers, who showed that creA is regulated at the transcriptional level when the mycelium is shifted to or from ethanol. The low expression of creA on glucose could be due to autoregulation, which is presumably elevated on the de-repressing carbon source ethanol, and on the intermediate repressing carbon source glycerol. However, our findings clearly showed that this regulation of creA not only occurs after changing the carbon source but is also reflected in the mRNA abundance of creA, during balanced growth conditions (it is not a transient phenomenon).
Besides the two transcriptional regulators AlcR and FacB, another known positive regulator was found in cluster 7, namely AreA (AN8667.2). AreA was probably the first regulatory gene described in A. nidulans [35], and it is a wide-domain regulator necessary for the activation of genes for the utilization of nitrogen sources. To our knowledge, it has not been reported that AreA is upregulated during growth on ethanol as compared with glucose or glycerol (cluster 7). Our results could indicate crosstalk between carbon repression and nitrogen repression pathways in A. nidulans. Supporting our findings on AreA regulation, we identified the gene uapC (AN6730.2) in cluster 7. This gene encodes a purine permease and has been shown to be regulated by AreA [36]. Another transcription factor assigned to cluster 7, namely metR, encodes a transcriptional activator for sulfur metabolism in A. nidulans [37], and it thereby links yet another branch of central metabolism to the regulatory network that is controlled by the nature of the carbon source.
Glycerol utilization and polyol metabolism
Regulation of the biosynthesis and breakdown of glycerol are less studied in comparison with the metabolism of ethanol, but from our analysis we identified more than 200 genes that were significantly upregulated and another 200 genes that were significantly downregulated only during growth on glycerol as compared with growth on glucose and ethanol (clusters 4 and 8). It was previously described that there are two metabolic pathways that lead to glycerol, from the glycolytic intermediate dihydroxyacetone 3-phosphate. One of these pathways proceeds via dihydroxyacetone kinase to dihydroxyacetone, which is then converted into glycerol, by the action of a glycerol dehydrogenase (NADH [nicotinamide adenine dinucleotide] or NADPH dependent). The alternative route, which has been suggested to be responsible for the catabolism of glycerol [8], includes the formation of glycerol 3-phosphate (catalyzed by glycerol 3-phosphate dehydrogenase), and subsequently its conversion into glycerol, by the action of glycerol 3-phosphate phosphatase.
Several of the genes encoding these enzymes have previously been characterized, and we identified alternative candidates, as well as the missing ones, in our reconstruction of the metabolic network. The data obtained from the transcriptome analysis confirmed that the catabolic pathway via glycerol 3-phosphate is a major route for glycerol catabolism, because a gene putatively encoding the glycerol kinase (AN5589.2), as well as the gene putatively encoding a FADH-dependent glycerol 3-phosphate dehydrogenase (AN1396.2), were both significantly upregulated on glycerol as compared with ethanol and glucose. Moreover, both genes were assigned to cluster 4, which represents genes that are specifically upregulated during growth on glycerol, and were identified in the subnetworks of glycerol comparisons with the two other carbon sources. However, the transcriptome data also showed that the alternative pathway might be involved in the catabolism of glycerol. In fact, a gene that was identified in the metabolic reconstruction process as putatively encoding a NADPH-dependent glycerol dehydrogenase (AN7193.2) was upregulated on glycerol (cluster 3), as well as a gene that was identified as a putative dihydroxyacetone kinase (AN0034.2; cluster 4). Therefore, it seems likely that both pathways are actually involved in the utilization of glycerol. Interestingly, a previously characterized gene encoding a NADPH-dependent glycerol dehydrogenase (gldB; AN5563.2) [38] was also found to be significantly regulated, but exhibited a very different expression pattern from the putative gene encoding NADPH-dependent glycerol dehydrogenase (AN7193.2). Thus, because gldB was downregulated on glycerol, it was assigned to cluster 8.
The biosynthesis of mannitol occurs through routes that are similar to the two metabolic pathways that lead to glycerol. It has been reported that mannitol is implicated in the stress response to heat [39] and that it is the most abundant polyol in conidia of A. nidulans [40]. One of the pathways that lead to mannitol proceeds via mannitol 1-phosphate, from the glycolytic intermediate fructose 6-phosphate, and another one, which has fructose as an intermediate. The metabolic reactions interconverting these four metabolites open the possibility for a cyclic reaction pathway within the cell that allows the conversion of NADH into NADPH at the expense of one molecule of ATP [41]. None of the genes encoding enzymes involved in the mannitol cycle have previously been characterized. However, by applying the comparative genomics approach for the reconstruction of the metabolism, we identified putative candidate ORFs for all the reactions of the mannitol cycle, with the exception of the mannitol 1-phosphate phosphatase. Interestingly, most of these ORFs identified (6/8) showed lower expression levels on ethanol, at least when compared with glycerol (assigned to clusters 2, 3, and 4), and this could point to a role for the mannitol cycle in the formation of NADPH during growth on glycerol. Moreover, the gene that encodes the glucose 6-phosphate dehydrogenase (AN2981.2), which has been shown to be positively correlated with the formation of mannitol, was also assigned to cluster 2 and significantly downregulated on ethanol. This enzyme was identified in the subnetwork for the glucose versus glycerol comparison, and transcription of the corresponding gene was lower during growth on glycerol than during growth on glucose. This could indicate a partial shift from the PP pathway, as the main route for NADPH supply for biosynthesis, to the mannitol cycle.
Glycerol has also been shown to be involved in the response to different osmotic conditions in A. nidulans [42], and it has also been reported that all of the components of the high-osmolarity glycerol (HOG) response pathway that are known in yeast have orthologs in A. nidulans [43, 44]. The analysis of the transcriptional responses of these components to the different growth conditions considered in the present study revealed that only the gene that encodes the sensor protein SlnA (slnA; AN1800.2) was significantly regulated and this was assigned to cluster 4 (slnA seemed to be induced when glycerol was the sole carbon source, as compared with glucose or ethanol).
Metabolism of reserve compounds and cell wall polysaccharides
Another metabolite that has been reported to be related to glycerol metabolism is trehalose. In fact, it has been shown that trehalose, which is stored in the conidiospores, is converted into glycerol upon germination [45].
The biosynthesis of trehalose occurs, via trehalose 6-phosphate, from glucose 6-phosphate and UDP-glucose, whereas it is degraded directly to glucose. Our reconstruction of the metabolic network includes six genes that might be involved in these metabolic pathways, of which four have been confirmed experimentally [45–48]. The cluster analysis showed that the transcription of three of these six genes was significantly changed, with higher levels on glucose, compared with ethanol and glycerol (genes assigned to clusters 1 and 2). Because these three genes encode each of the different steps in the biosynthesis as well as degradation of trehalose, these observations suggest that there may be a higher turnover of trehalose during growth on glucose.
Glycogen is another reserve carbohydrate, similar to trehalose, and interestingly the two genes putatively assigned to its biosynthesis and degradation exhibited their highest expression levels on glycerol (clusters 3 and 4, respectively), which might suggest an effect of this carbon source on glycogen turnover. In this regard, it was also interesting to verify that the GO term analysis for the pair-wise comparisons showed that genes associated with cell wall metabolism were significantly over-represented in the upregulated gene set as well as in the downregulated gene set.
More detailed analysis of the genes that were upregulated on glycerol compared with glucose, and that resulted in the over-representation of the GO terms, revealed that all of them putatively encode enzymes with β-1,3-glucosidase activity, which suggests that specially the β-1,3-glucan fraction of the fungal cell wall undergoes major rearrangements depending on the carbon source. On the other hand, the genes that were downregulated on glycerol and associated with GO terms for the cell wall biosynthesis encoded α-glucosidases (AN8953.2, AN0941.2, and AN4843.2) and were assigned to cluster 5. These enzymes are responsible for the breakdown of α-linked glucans into glucose, and it is therefore surprising that three genes encoding α-glucosidases (one putatively [AN0941.2] and two experimentally confirmed [agdA (AN2017.2) and agdB (AN8953.2)] [49]) exhibited their highest expression levels on glucose, which means that they are not repressed by glucose. It could be speculated that these genes are also involved in the remodeling of the α-glucan fraction of the cell wall, depending on the available carbon source.
One of the α-glucosidases (AN2017.2) is part of a gene cluster that encodes proteins responsible for the breakdown of α-glucans (such as starch). This cluster contains a putative glycosyl transferase (AN2015.2) that was significantly downregulated on ethanol compared with glucose and glycerol (assigned to cluster 3); the previously mentioned agdA, which encodes an α-glucosidase; the regulatory protein amyR (AN2016.2), which appears to be regulated in the same way as agdA (also found in cluster 2 and significantly downregulated on ethanol compared with glucose); and, finally, amyA (AN2018.2), which encodes an α-amylase but which does not appear to be significantly regulated under the conditions investigated in the present study.
AmyR directly controls the expression of agdA by binding to its promoter [50] and the direct correlation between the two mRNA levels suggests that solely the quantity of AmyR within the cell might be responsible for the regulation of agdA, without any further requirement for post-translational activation of AmyR. It is also interesting to note that it has previously been shown that amyR is controlled by CreA [51], which is in agreement with our findings (compare the expression pattern of cluster 6, containing creA, with that of cluster 2, containing amyR).
Ribosome biogenesis
It has been reported that the specific growth rate influences the expression of genes encoding ribosomal proteins in S. cerevisiae, and that the transcription of these genes increases with increasing specific growth rates [52]. Similarly, we observed that the expression profiles of genes encoding ribosomal proteins followed the same trend as the maximal specific growth rate. In fact, in the batch cultivations carried out with A. nidulans on the different carbon sources the cells grew at unlimited conditions, and hence at their maximal specific growth rate possible on the given carbon source. The specific growth rates were highest for growth on glucose and about the same on ethanol and glycerol (Figure 1). According to the GO term analysis, cluster 1 is mainly characterized by genes whose functions are related to ribosome biogenesis, as can be observed in Table 3. Indeed, 59 out of the 280 genes assigned to cluster 1 are associated with the GO term 'ribosome biogenesis'. It is interesting that this cluster includes genes that have higher expression levels during growth on glucose, and this indicates - as observed for yeast - that these genes might indeed be involved in the ribosome biogenesis and that they are possibly regulated in a growth-rate dependent manner.
Materials and methods
Strain
The strain used in this study was the strain Aspergillus nidulans A187 (pabaA1 yA2; obtained from the Fungal Genetics Stock Center, Kansas City, KA, USA).
Growth medium
The medium used in all batch cultivations was a chemically defined medium as described by Agger and coworkers [53], with the following modifications: NH4Cl was used as the nitrogen source, at a concentration of 12.2 g/l, and three different carbon sources were tested, namely glucose, glycerol, and ethanol (10 g/l). Yeast extract was added to the fermenter at a concentration of 3 mg/l in order to encourage the germination of spores. Furthermore, the nutritional supplement p-aminobenzoic acid (PABA) was added to the medium at a concentration of 1 mg/l, as well as the antifoam agent 204 (Sigma, Brøndby, Denmark)) at a concentration of 0.05 ml/l.
Propagation of spores
The fermenters were inoculated with spores of A. nidulans A187 previously propagated on solid minimal medium [54] containing PABA (10 mg/l). The same stock of spores of A. nidulans was used to inoculate all the plates. The spores were cultivated at 37°C, during 3 to 4 days, and harvested by adding distilled water. For the fermentations performed in replicates, the fermenters were inoculated with the same solution of spores, to a final concentration of 5 × 109 spores/l. High concentrations of spores in the inoculum were employed because they favor dispersed filamentous growth. The spore solutions were vortex mixed before introduction into the fermenters, in order to prevent agglomeration of spores and thus pellet formation.
Batch cultivations
All aerobic batch cultivations were performed in 3 L-Braun fermenters with a working volume of 2 l. The bioreactors were equipped with two Rushton four-blade disc turbines and no baffles (thereby reducing the surface available for wall growth). Air was used to sparge the bioreactor. The concentrations of oxygen and carbon dioxide in the exhaust gas were monitored using an acoustic gas analyzer (Brüel & Kjær, Nærum, Denmark). Temperature, pH, agitation, and aeration rate were controlled throughout the cultivations. The temperature was maintained at 30°C. The pH was controlled by automatic addition of 2 N NaOH and 2 N HCl. For the cultivations on glucose and glycerol, the pH was initially set to 3.0 to prevent spore aggregation; only when the spores started to germinate was the pH gradually increased to 6.0. Because pellet formation is unlikely to occur during growth on ethanol, the pH was set to 6.0 from the start, in the cultivations performed with this carbon source. Similarly, the stirrer speed was initially set to 100 rpm and the aeration rate to 0.01 vvm, and only after the spores started to germinate were these parameters progressively increased to 700 rpm and 1 vvm, respectively.
Sampling
For quantification of cell mass and extracellular metabolites, the fermentation broth was withdrawn from the fermenter vessel and filtered through nitrocellulose filters (pore size 0.45 μm; Pall Corporation, East Hills, NY, USA). The filter cakes were immediately processed for determination of cell mass, whereas the filtrates were stored at -20°C until they were analyzed for determination of extracellular metabolites (substrates and metabolic byproducts).
For gene expression analysis, the mycelia were harvested and processed within half a minute. The mycelia were filtered using Miracloth (Calbiochem, San Diego, CA, USA) and washed with phosphate-buffered saline (PBS) solution (137 mmol/l NaCl, 2.7 mmol/l KCl, 10 mmol/l Na2HPO4, and 0.24 mmol/l KH2PO4; pH 7.4). After removal of excess PBS solution, the mycelia were frozen in liquid nitrogen and stored at -80°C until further analysis.
Cell mass determination
Cell dry weight was determined using nitrocellulose filters (pore size 0.45 μm; Pall Corporation). The filters were predried in a microwave oven at 150 W for 10 minutes and subsequently weighed. A known volume of cell culture was filtered and the filter cake was washed with 0.9% NaCl and dried on the filter for 15 minutes in a microwave oven at 150 W. The filter was weighed again to determine the cell mass concentration.
Analysis of extracellular metabolites
The concentrations of sugars, organic acids, and polyols in the filtrates (thawed on ice) were determined using high-pressure liquid-chromatography on an Aminex HPX-87Hm column (Bio-Rad, Hercules, CA, USA). The column was kept at 65°C and eluted at 0.6 ml/minute with 5 mmol/l H2SO4. The compounds were detected refractometrically (410 Differential Refractometer, Waters Corporation, Milford, MA, USA).
Extraction of total RNA
Total RNA was isolated using the Qiagen RNeasy Mini Kit (QIAGEN Nordic, Ballerup, Denmark), according to the protocol for isolation of total RNA from animal tissues. For this purpose, approximately 20 to 30 mg frozen mycelium was placed in a 2 ml Eppendorf tube, precooled in liquid nitrogen, containing three RNase-treated steel balls (two balls with a diameter of 2 mm and one ball with a diameter of 5 mm). The tubes were then shaken in a Retsch Mixer Mill, at 3°C, for 6 to 8 minutes, until the mycelia were ground to powder and thus ready for extraction of total RNA. The quality and quantity of the total RNA extracted were determined by spectrophotometric analysis and by gel electrophoresis. The total RNA was stored at -80°C until further processing.
Microarray manufacturing and design
The microarrays used for the analysis of the transcriptome of A. nidulans were custom-made NimbleExpress™ arrays (NimbleGen Systems Inc., Madison, WI, USA), which were acquired through Affymetrix (Santa Clara, CA, USA). NimbleExpress™ arrays are manufactured using a Maskless Array Synthesizer system, which makes use of a Digital Micromirror Device. This device consists of a system of miniature aluminum mirrors, which are individually controlled, enabling the synthesis of oligomers in a similar way to the photolithographic process used in the manufacture of Affymetrix® GeneChip arrays. The NimbleExpress™ arrays are packaged in an Affymetrix® GeneChip cartridge (49 format), and can be used with GeneChip reagents and processed on the GeneChip® Instrument System (for more information, see [55]).
The selection of the probes for interrogating the ORFs within the genome of A. nidulans was performed by the Affymetrix Chip Design Group, who used the same algorithms employed in the design of standard Affymetrix® GeneChip arrays. The arrays contain only perfect match (PM) probes. Of the 9,541 putative genes identified in the genome of A. nidulans (Aspergillus nidulans Database [9], release 3.1; Broad Institute), 9,444 were represented in the microarray (CBS01CMBA530008N). Each ORF was interrogated with one (8,628), two (815), or three probe sets (1), and each of these probe sets were composed of 11 probes (whenever possible) of 25 oligomers. Different types of probe sets were represented on the array, namely type 1 (if all probes in the set hybridized exclusively with the target sequence), type 2 (if all probes in the set hybridized with the target sequence and cross-hybridized with other sequences), and type 3 (mixed probe set). In the data analysis, only probe sets of type 1 (or, rather, probes that did not cross-hybridize with genes other than the target) were considered, which brought the number of putative genes investigated down to 9,371.
Preparation of biotin-labeled cRNA and microarray processing
Biotin-labeled cRNA was prepared from approximately 10 μg of total RNA, according to the protocol described in the Affymetrix GeneChip® Expression Analysis Technical Manual (2004) [56]. An additional cleanup step was performed before fragmentation, using the Qiagen RNeasy Mini Kit (protocol for RNA Cleanup), in order to guarantee good-quality cRNA samples for subsequent processing. The cRNA was quantified in a spectrophotometer (Amersham Pharmacia Biotech, GE Healthcare Bio-Sciences AB, Uppsala, Sweden) and its quality was assessed by gel electrophoresis and using a bioanalyzer (Agilent Technologies Inc., Santa Clara, CA, USA). The biotin-labeled cRNA was then fragmented and approximately 13 μg of fragmented cRNA was hybridized to the custom-made NimbleExpress™ array (CBS01CMBA530008N), as described in the Affymetrix GeneChip® Expression Analysis Technical Manual (2004) [56]. The array was further processed on a GeneChip® Fluidics Station FS-400 (fluidics protocol EukGE-WS2v4) and on an Agilent GeneArray® Scanner. The scanned probe array images (.DAT files) were converted into .CEL files using the GeneChip® Operating Software (Affymetrix).
Reconstruction of the metabolic network and gene annotation
The metabolic network of A. nidulans was reconstructed by employing a pathway-driven approach described elsewhere [57] and comparative genomics tools based on sequence similarity. The metabolism of A. nidulans was reconstructed using as templates detailed metabolic reconstructions previously developed for A. niger [20], Saccharomyces cerevisiae [21], and Mus musculus [22]. Pathway prediction in A. nidulans was supported by available information on its genetics, biochemistry, and physiology. Thereby, it was possible to identify metabolic functions without a gene associated in the genome of A. nidulans, and thus candidate ORFs for encoding those functions, by employing similarity-based tools of comparative genomic analysis (BLAST [Basic Local Alignment Search Tool]) and using public (nonredundant) databases of genes and proteins of established function [58].
Analysis of transcriptome data
Raw probe intensities were preprocessed by applying the RMA (robust multiarray average) method for background correction (based on PM probe information) [59], and were subsequently normalized using the qspline method [60]. Gene expression index values were calculated using the method developed by Li and Wong [61], using the PM-only model. Normalized gene expression data was deposited at the GEO database [62], with accession numbers GPL3604 (platform), GSM102371-GSM102379 (samples), and GSE4578 (series).
Statistical analysis was applied to determine differentially expressed genes between the categories of replicated experiments. For all of the possible pair-wise comparisons within the three different categories, the logit-t method [63] was employed, whereas ANOVA was used to compare gene expression levels among all categories. The genes whose expression was found to be significantly changed using the ANOVA test were further arranged into clusters, by applying ClusterLustre, a Bayesian consensus clustering method recently developed and available in the Matlab application [19].
Furthermore, reporter metabolites (metabolites around which the most significant changes in transcription occur) and highly correlated metabolic subnetworks (sets of connected genes with significant and coordinated transcriptional response to a perturbation) were identified for each of the pair-wise comparisons possible within the three categories, as described by Patil and Nielsen [23]. For this purpose, information on the topology of the reconstructed metabolic network of A. nidulans was used in combination with the P values from the logit-t test performed for each of the pair-wise comparisons of expression data.
Data deposition
Normalized gene expression data were deposited at the GEO database [62], with accession numbers GPL3604 (platform), GSM102371-GSM102379 (samples), and GSE4578 (series).
Additional data files
The following additional data are available with the online version of this paper. Additional data file 1 illustrates genes whose expression was significantly changed in the pair-wise comparison: glucose versus ethanol. Additional data file 2 illustrates genes whose expression was significantly changed in the pair-wise comparison: glycerol versus ethanol. Additional data file 3 illustrates genes whose expression was significantly changed in the pair-wise comparisons glucose versus glycerol. Additional data file 4 illustrates reporter metabolites and enzymes comprising the 'small' subnetwork identified by comparing expression data on glucose and ethanol. Additional data file 5 illustrates reporter metabolites and enzymes comprising the 'small' subnetwork identified by comparing expression data on glycerol and ethanol. Additional data file 6 illustrates reporter metabolites and enzymes comprising the 'small' subnetwork identified by comparing expression data on glucose and glycerol. Additional data file 7 lists all of the reactions in the metabolic network reconstructed for A. nidulans and gives the abbreviations assigned to the metabolite names. Additional data file 8 lists the normalized intensities for the three biologic replicates on the three carbon sources studied. Additional data file 9 includes complete lists of genes whose expression was significantly changed in the pair-wise comparisons. Additional data file 10 presents the functional classification of the differentially expressed genes based on GO assignments provided by CADRE. Additional data file 11 provides results from the ANOVA test performed, considering normalized transcriptome data from all of the replicated experiments on the different carbon sources. Additional data file 12 lists the genes assigned to each gene cluster. Additional data file 13 presents the 'large' subnetworks for each of the pair-wise comparisons between the three carbon sources investigated. Additional data file 14 presents the functional classification of the genes in each of the 'small' subnetworks, based on GO assignments provided by CADRE.
References
Galagan JE, Calvo SE, Cuomo C, Ma LJ, Wortman JR, Batzoglou S, Lee SI, Basturkmen M, Spevak CC, Clutterbuck J, et al: Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature. 2006, 438: 1105-1115. 10.1038/nature04341.
Machida M, Asai K, Sano M, Tanaka T, Kumagai T, Terai G, Kusumoto K, Arima T, Akita O, Kashiwagi Y, et al: Genome sequencing and analysis of Aspergillus oryzae. Nature. 2005, 438: 1157-1161. 10.1038/nature04300.
Nierman WC, Pain A, Anderson MJ, Wortman JR, Kim HS, Arroyo J, Berriman M, Abe K, Archer DB, Bermejo C, et al: Genomic sequence of the pathogenic and allergenic filamentous fungus Aspergillus fumigatus. Nature. 2005, 438: 1151-1156. 10.1038/nature04332.
Flipphi M, Mathieu M, Cirpus I, Panozzo C, Felenbok B: Regulation of the aldehyde dehydrogenase gene (aldA) and its role in the control of the coinducer level necessary for induction of the ethanol utilization pathway in Aspergillus nidulans. J Biol Chem. 2001, 276: 6950-6958. 10.1074/jbc.M005769200.
Todd RB, Andrianopoulos A, Davis MA, Hynes MJ: FacB, the Aspergillus nidulans activator of acetate utilization genes, binds dissimilar DNA sequences. EMBO J. 1998, 17: 2042-2054. 10.1093/emboj/17.7.2042.
Stemple CJ, Davis MA, Hynes MJ: The facC gene of Aspergillus nidulans encodes an acetate-inducible carnitine acetyltransferase. J Bacteriol. 1998, 180: 6242-6251.
Ruijter GJ, Visser J: Carbon repression in aspergilli. FEMS Microbiol Lett. 1997, 151: 103-114. 10.1111/j.1574-6968.1997.tb12557.x.
Hondmann DH, Busink R, Witteveen CF, Visser J: Glycerol catabolism in Aspergillus nidulans. J Gen Microbiol. 1991, 137: 629-636.
Aspergillus nidulans Database. [http://www.broad.mit.edu/annotation/fungi/aspergillus/index.html]
Sims AH, Robson GD, Hoyle DC, Oliver SG, Turner G, Prade RA, Russell HH, Dunn-Coleman NS, Gent ME: Use of expressed sequence tag analysis and cDNA microarrays of the filamentous fungus Aspergillus nidulans. Fungal Genet Biol. 2004, 41: 199-212. 10.1016/j.fgb.2003.11.005.
Sims AH, Gent ME, Lanthaler K, Dunn-Coleman NS, Oliver SG, Robson GD: Transcriptome analysis of recombinant protein secretion by Aspergillus nidulans and the unfolded-protein response in vivo. Appl Environ Microbiol. 2005, 71: 2737-2747. 10.1128/AEM.71.5.2737-2747.2005.
Maeda H, Sano M, Maruyama Y, Tanno T, Akao T, Totsuka Y, Endo M, Sakurada R, Yamagata Y, Machida M, et al: Transcriptional analysis of genes for energy catabolism and hydrolytic enzymes in the filamentous fungus Aspergillus oryzae using cDNA microarrays and expressed sequence tags. Appl Microbiol Biotechnol. 2004, 65: 74-83. 10.1007/s00253-004-1608-4.
Guo BZ, Yu J, Holbrook CC, Lee RD, Lynch RE: Application of differential display RT-PCR and EST-microarray technologies to the analysis of gene expression in response to drought stress and elimination of aflatoxin contamination in corn and peanut. J Toxicol. 2003, 22: 287-312. 10.1080/10915810305121.
Scheidegger KA, Payne GA: Unlocking the secrets behind secondary metabolism: a review of Aspergillus flavus from pathogenicity to functional genomics. J Toxicol. 2003, 22: 423-459.
OBrian GR, Fakhoury AM, Payne GA: Identification of genes differentially expressed during aflatoxin biosynthesis in Aspergillus flavus and Aspergillus parasiticus. Fungal Genet Biol. 2003, 39: 118-127. 10.1016/S1087-1845(03)00014-8.
Askenazi M, Driggers EM, Holtzman DA, Norman TC, Iverson S, Zimmer DP, Boers ME, Blomquist PR, Martinez EJ, Monreal AW, et al: Integrating transcriptional and metabolite profiles to direct the engineering of lovastatin-producing fungal strains. Nat Biotechnol. 2003, 21: 150-156. 10.1038/nbt781.
Chambergo FS, Bonaccorsi ED, Ferreira AJ, Ramos AS, Ferreira Junior JR, Abrahao-Neto J, Farah JP, El-Dorry H: Elucidation of the metabolic fate of glucose in the filamentous fungus Trichoderma reesei using expressed sequence tag (EST) analysis and cDNA microarrays. J Biol Chem. 2002, 277: 13983-13988. 10.1074/jbc.M107651200.
DeRisi JL, Iyer VR, Brown PO: Exploring the metabolic and genetic control of gene expression on a genomic scale. Science. 1997, 278: 680-686. 10.1126/science.278.5338.680.
Grotkjaer T, Winther O, Regenberg B, Nielsen J, Hansen LK: Robust multi-scale clustering of large DNA microarray datasets with the consensus algorithm. Bioinformatics. 2006, 22: 58-67. 10.1093/bioinformatics/bti746.
David H, Akesson M, Nielsen J: Reconstruction of the central carbon metabolism of Aspergillus niger. Eur J Biochem. 2003, 270: 4243-4253. 10.1046/j.1432-1033.2003.03798.x.
Forster J, Famili I, Fu P, Palsson BO, Nielsen J: Genome-scale reconstruction of the Saccharomyces cerevisiae metabolic network. Genome Res. 2003, 13: 244-253. 10.1101/gr.234503.
Sheik K, Forster J, Nielsen L: Modeling hybridoma cell metabolism using a generic genome-scale metabolic model of Mus musculus. Biotechnol Prog. 2005, 21: 112-121. 10.1021/bp0498138.
Patil KR, Nielsen J: Uncovering transcriptional regulation of metabolism by using metabolic network topology. Proc Natl Acad Sci USA. 2005, 102: 2685-2689. 10.1073/pnas.0406811102.
Borodina I, Nielsen J: From genomes to in silico cells via metabolic networks. Curr Opin Biotechnol. 2005, 16: 350-355. 10.1016/j.copbio.2005.04.008.
CADRE - Central Aspergillus Data Repository. [http://www.cadre.man.ac.uk]
Westergaard SL, Oliveira AP, Bro C, Olsson L, Nielsen J: A systems biology approach to study glucose repression in the yeast Saccharomyces cerevisiae. Biotechnol Bioeng. 2007, 96: 134-145. 10.1002/bit.21135.
Felenbok B, Flipphi M, Nikolaev I: Ethanol catabolism in Aspergillus nidulans: a model system for studying gene regulation. Prog Nucleic Acid Res Mol Biol. 2001, 69: 149-204.
Kulmburg P, Mathieu M, Dowzer C, Kelly J, Felenbok B: Specific binding sites in the alcR and alcA promoters of the ethanol regulon for the CREA repressor mediating carbon catabolite repression in Aspergillus nidulans. Mol Microbiol. 1993, 7: 847-857. 10.1111/j.1365-2958.1993.tb01175.x.
Fillinger S, Felenbok B: A newly identified gene cluster in Aspergillus nidulans comprises five novel genes localized in the alc region that are controlled both by the specific transactivator AlcR and the general carbon-catabolite repressor CreA. Mol Microbiol. 1996, 20: 475-488. 10.1046/j.1365-2958.1996.5301061.x.
Brock M, Fischer R, Linder D, Buckel W: Methylcitrate synthase from Aspergillus nidulans: implications for propionate as an antifugal agent. Mol Microbiol. 2000, 35: 961-973. 10.1046/j.1365-2958.2000.01737.x.
Adams IP, Dack S, Dickinson FM, Ratledge C: The distinctiveness of ATP:citrate lyase from Aspergillus nidulans. Biochim Biophys Acta. 2002, 1597: 36-41.
Wynn JP, Kendrick A, Hamid AA, Ratledge C: Malic enzyme: a lipogenic enzyme in fungi. Biochem Soc Trans. 1997, 25: S669-
Kelly JM, Hynes MJ: The regulation of phosphoenolpyruvate carboxykinase and the NADP-linked malic enzyme in Aspergillus nidulans. J Gen Microbiol. 1981, 123: 371-375.
Strauss J, Horvath HK, Abdallah BM, Kindermann J, Mach RL, Kubicek CP: The function of CreA, the carbon catabolite repressor of Aspergillus nidulans, is regulated at the transcriptional and post-transcriptional level. Mol Microbiol. 1999, 32: 169-178. 10.1046/j.1365-2958.1999.01341.x.
Arst HN, Cove DJ: Nitrogen metabolite repression in Aspergillus nidulans. Mol Gen Genet. 1973, 126: 111-141. 10.1007/BF00330988.
Diallinas G, Gorfinkiel L, Arst HN, Cecchetto G, Scazzocchio C: Genetic and molecular characterization of a gene encoding a wide specificity purine permease of Aspergillus nidulans reveals a novel family of transporters conserved in prokaryotes and eukaryotes. J Biol Chem. 1995, 270: 8610-8622. 10.1074/jbc.270.15.8610.
Natorff R, Sienko M, Brzywczy J, Paszewski A: The Aspergillus nidulans metR gene encodes a bZIP protein which activates transcription of sulphur metabolism genes. Mol Microbiol. 2003, 49: 1081-1094. 10.1046/j.1365-2958.2003.03617.x.
de Vries RP, Flitter SJ, van de Vondervoort PJ, Chaveroche MK, Fontaine T, Fillinger S, Ruijter GJ, d'Enfert C, Visser J: Glycerol dehydrogenase, encoded by gldB is essential for osmotolerance in Aspergillus nidulans. Mol Microbiol. 2003, 49: 131-141. 10.1046/j.1365-2958.2003.03554.x.
Noventa-Jordao MA, Couto RM, Goldman MH, Aguirre J, Iyer S, Caplan A, Terenzi HF, Goldman GH: Catalase activity is necessary for heat-shock recovery in Aspergillus nidulans germlings. Microbiology. 1999, 145: 3229-3234.
Hallsworth JE, Prior BA, Nomura Y, Iwahara M, Timmis KN: Compatible solutes protect against chaotrope (ethanol)-induced, nonosmotic water stress. Appl Environ Microbiol. 2003, 69: 7032-7034. 10.1128/AEM.69.12.7032-7034.2003.
Singh M, Scrutton NS, Scrutton MC: NADPH generation in Aspergillus nidulans: is the mannitol cycle involved?. J Gen Microbiol. 1988, 134: 643-654.
Beever RE, Laracy EP: Osmotic adjustment in the filamentous fungus Aspergillus nidulans. J Bacteriol. 1986, 168: 1358-1365.
Han KH, Prade RA: Osmotic stress-coupled maintenance of polar growth in Aspergillus nidulans. Mol Microbiol. 2002, 43: 1065-1078. 10.1046/j.1365-2958.2002.02774.x.
Furukawa K, Hoshi Y, Maeda T, Nakajima T, Abe K: Aspergillus nidulans HOG pathway is activated only by two-component signalling pathway in response to osmotic stress. Mol Microbiol. 2005, 56: 1246-1261. 10.1111/j.1365-2958.2005.04605.x.
d'Enfert C, Fontaine T: Molecular characterization of the Aspergillus nidulans treA gene encoding an acid trehalase required for growth on trehalose. Mol Microbiol. 1997, 24: 203-216. 10.1046/j.1365-2958.1997.3131693.x.
Fillinger S, Chaveroche MK, van Dijck P, de Vries R, Ruijter G, Thevelein J, d'Enfert C: Trehalose is required for the acquisition of tolerance to a variety of stresses in the filamentous fungus Aspergillus nidulans. Microbiology. 2001, 147: 1851-1862.
Borgia PT, Miao Y, Dodge CL: The orlA gene from Aspergillus nidulans encodes a trehalose-6-phosphate phosphatase necessary for normal growth and chitin synthesis at elevated temperatures. Mol Microbiol. 1996, 20: 1287-1296. 10.1111/j.1365-2958.1996.tb02647.x.
d'Enfert C, Bonini BM, Zapella PD, Fontaine T, da Silva AM, Terenzi HF: Neutral trehalases catalyse intracellular trehalose breakdown in the filamentous fungi Aspergillus nidulans and Neurospora crassa. Mol Microbiol. 1999, 32: 471-483. 10.1046/j.1365-2958.1999.01327.x.
Kato N, Murakoshi Y, Kato M, Kobayashi T, Tsukagoshi N: Isomaltose formed by alpha-glucosidases triggers amylase induction in Aspergillus nidulans. Curr Genet. 2002, 42: 43-50. 10.1007/s00294-002-0325-8.
Tani S, Itoh T, Kato M, Kobayashi T, Tsukagoshi N: In vivo and in vitro analyses of the AmyR binding site of the Aspergillus nidulans agdA promoter; requirement of the CGG direct repeat for induction and high affinity binding of AmyR. Biosci Biotechnol Biochem. 2001, 65: 1568-1574. 10.1271/bbb.65.1568.
Tani S, Katsuyama Y, Hayashi T, Suzuki H, Kato M, Gomi K, Kobayashi T, Tsukagoshi N: Characterization of the amyR gene encoding a transcriptional activator for the amylase genes in Aspergillus nidulans. Curr Genet. 2001, 39: 10-15. 10.1007/s002940000175.
Regenberg B, Grotkjaer T, Winther O, Fausboll A, Akesson M, Bro C, Hansen LK, Brunak S, Nielsen J: Growth-rate regulated genes have profound impact on interpretation of transcriptome profiling in Saccharomyces cerevisiae. Genome Biol. 2006, 14;7 (11): R107-10.1186/gb-2006-7-11-r107.
Agger T, Petersen JB, O'Connor SM, Murphy RL, Kelly JM, Nielsen J: Physiological characterisation of recombinant Aspergillus nidulans strains with different creA genotypes expressing A. oryzae alpha-amylase. J Biotechnol. 2002, 92: 279-285. 10.1016/S0168-1656(01)00366-2.
Fundamentals of growth, storage, genetics and microscopy of Aspergillus nidulans. [http://www.fgsc.net/fgn48/Kaminskyj.htm]
NimbleGen Systems, Inc. [http://www.nimblegen.com]
Affymetrix GeneChip® Expression Analysis Technical Manual (2004). [http://www.affymetrix.com/support/technical/manual/expression_manual.affx]
Osterman A, Overbeek R: Missing genes in metabolic pathways: a comparative genomics approach. Curr Opin Chem Biol. 2003, 7: 238-251. 10.1016/S1367-5931(03)00027-9.
NCBI - National Center for Biotechnology Inofrmation. [http://www.ncbi.nlm.nih.gov]
Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP: Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003, 4: 249-264. 10.1093/biostatistics/4.2.249.
Workman C, Jensen LJ, Jarmer H, Berka R, Gautier L, Nielser HB, Saxild HH, Nielsen C, Brunak S, Knudsen S: A new non-linear normalization method for reducing variability in DNA microarray experiments. Genome Biol. 2002, 3: research0048-10.1186/gb-2002-3-9-research0048.
Li C, Wong WH: Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci USA. 2001, 98: 31-36. 10.1073/pnas.011404098.
GEO - Gene Expression Omnibus. [http://www.ncbi.nlm.nih.gov/geo/]
Lemon WJ, Liyanarachchi S, You M: A high performance test of differential gene expression for oligonucleotide arrays. Genome Biol. 2003, 4: R67-10.1186/gb-2003-4-10-r67.
Acknowledgements
The authors thank Michael Lynge Nielsen for his contribution to the the design of the array. Jesper Mogensen and Steen Lund Westergaard are thanked for their guidance in the sample preparation and microarray processing, and Lene Christiansen, Kristine Bøje Dahlin and Thomas Jensen for technical assistance. Kiran Patil and Thomas Grotkjær are acknowledged for helping with the data analysis. HD was funded through a research fellowship (SFRH/BD/3110/2000) of the III Community Support Framework financed by the European Social Fund and by a Portuguese National Fund from the Ministry of Science and Technology. Much of this work was funded by the Danish Technical Research Council through basic funding for Center for Microbial Biotechnology.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
13059_2006_1376_MOESM1_ESM.pdf
Additional data file 1: Differentially expressed genes in the central metabolism of A. nidulans between the replicated experiments on glucose and ethanol, revealed by the logit-t method (see Additional data file 9 [Table S6] for a full list of genes. Upregulated and downregulated genes are represented in red and green, respectively. Fold changes greater than 10 are highlighted in bold. (PDF 34 KB)
13059_2006_1376_MOESM2_ESM.pdf
Additional data file 2: Differentially expressed genes in the central metabolism of A. nidulans between the replicated experiments on glycerol and ethanol, revealed by the logit-t method (see Additional data file 9 [Table S7] for a full list of genes. Upregulated and downregulated genes are represented in red and green, respectively. Fold changes greater than 10 are highlighted in bold. (PDF 33 KB)
13059_2006_1376_MOESM3_ESM.pdf
Additional data file 3: Differentially expressed genes in the central metabolism of A. nidulans between the replicated experiments on glucose and glycerol, revealed by the logit-t method (see Additional data file 9 [Table S8] for a full list of genes. Upregulated and downregulated genes are represented in red and green, respectively. Fold changes greater than 10 are highlighted in bold. (PDF 31 KB)
13059_2006_1376_MOESM4_ESM.pdf
Additional data file 4: Reporter metabolites and enzymes comprising the 'small' subnetwork identified by comparing expression data on glucose and ethanol (represented in blue). Also shown are the top 15 high-scoring reporter metabolites. (PDF 33 KB)
13059_2006_1376_MOESM5_ESM.pdf
Additional data file 5: Reporter metabolites and enzymes comprising the 'small' subnetwork identified by comparing expression data on glycerol and ethanol (represented in blue). Also shown are the top 15 high-scoring reporter metabolites. (PDF 35 KB)
13059_2006_1376_MOESM6_ESM.pdf
Additional data file 6: Reporter metabolites and enzymes comprising the 'small' subnetwork identified by comparing expression data on glucose and glycerol (represented in blue). Also shown are the top 15 high-scoring reporter metabolites. (PDF 42 KB)
13059_2006_1376_MOESM7_ESM.xls
Additional data file 7: Table S1 lists the reactions comprising the metabolic reconstruction developed for A. nidulans. Each reaction is associated to an ORF within the genome of A. nidulans (when available), and to the EC number of the corresponding enzyme (when available). Table S2 lists metabolites, abbreviations used, and full names. (In the abbreviations, the suffixes m, g, and e stand for metabolites localized in the mitochondria, glyoxysomes and extracellular medium, respectively. No suffix was added to cytosolic metabolites.) (XLS 226 KB)
13059_2006_1376_MOESM8_ESM.xls
Additional data file 8: Tables S3, S4, and S5 give normalized intensities considering the replicated experiments on glucose and ethanol, on glycerol and ethanol, and on glucose and glycerol, respectively. (XLS 4 MB)
13059_2006_1376_MOESM9_ESM.xls
Additional data file 9: Tables S6, S7 and S8 list differentially expressed genes between the replicated experiments on glucose and ethanol, revealed by the logit-t method. The genes are sorted according to ascending P value from the statistical analysis. The average fold changes of expression between ethanol and glucose (Table S6), between ethanol and glycerol (Table S7), and between glycerol and glucose (Table S8) are also presented (a fold change equal to 2 [-2] indicates a twofold upregulation [downregulation] in ethanol relative to glucose [Table S6], in ethanol relative to glycerol [Table S7], or in glycerol relative to glucose [Table S8]). (XLS 3 MB)
13059_2006_1376_MOESM10_ESM.pdf
Additional data file 10: Tables S9 and S10 present the classifications of the upregulated and downregulated genes, respectively, given in Table 2 into GO categories (provided by CADRE), according to the three most important biological processes and molecular functions. (PDF 9 KB)
13059_2006_1376_MOESM11_ESM.xls
Additional data file 11: Table S11 gives the normalized intensities considering all of the categories of replicated experiments (glucose, glycerol, and ethanol). Table S12 provides P values from the ANOVA test between all categories of replicated experiments (glucose, glycerol, and ethanol). The genes are sorted according to ascending P value. (XLS 3 MB)
13059_2006_1376_MOESM12_ESM.xls
Additional data file 12: Table S13 summarizes gene clusters identified by applying ClusterLustre to the genes that were found to be significantly changed in the ANOVA test. (XLS 225 KB)
13059_2006_1376_MOESM13_ESM.xls
Additional data file 13: Tables S14, S15, and S16 provide lists of enzymes and transporters comprising the 'large' subnetwork, obtained by comparing expression data on glucose and ethanol, on glycerol and ethanol, and on glucose and glycerol, respectively. (XLS 78 KB)
13059_2006_1376_MOESM14_ESM.pdf
Additional data file 14: Table S17 summarizes the classification of the genes included in the 'small' highly correlated subnetworks into GO categories (provided by CADRE), according to the three most important biological processes and molecular functions. (PDF 5 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
David, H., Hofmann, G., Oliveira, A.P. et al. Metabolic network driven analysis of genome-wide transcription data from Aspergillus nidulans. Genome Biol 7, R108 (2006). https://doi.org/10.1186/gb-2006-7-11-r108
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/gb-2006-7-11-r108