
Abstract
Introduction
A recent genome-wide association study (GWAS) comprising a French cohort of systemic sclerosis (SSc) reported several non-HLA single-nucleotide polymorphisms (SNPs) showing a nominal association in the discovery phase. We aimed to identify previously overlooked susceptibility variants by using a follow-up strategy.
Methods
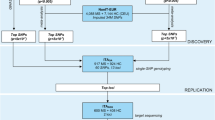
Sixty-six non-HLA SNPs showing a P value <10-4 in the discovery phase of the French SSc GWAS were analyzed in the first step of this study, performing a meta-analysis that combined data from the two published SSc GWASs. A total of 2,921 SSc patients and 6,963 healthy controls were included in this first phase. Two SNPs, PPARG rs310746 and CHRNA9 rs6832151, were selected for genotyping in the replication cohort (1,068 SSc patients and 6,762 healthy controls) based on the results of the first step. Genotyping was performed by using TaqMan SNP genotyping assays.
Results
We observed nominal associations for both PPARG rs310746 (PMH = 1.90 × 10-6, OR, 1.28) and CHRNA9 rs6832151 (PMH = 4.30 × 10-6, OR, 1.17) genetic variants with SSc in the first step of our study. In the replication phase, we observed a trend of association for PPARG rs310746 (P value = 0.066; OR, 1.17). The combined overall Mantel-Haenszel meta-analysis of all the cohorts included in the present study revealed that PPARG rs310746 remained associated with SSc with a nominal non-genome-wide significant P value (PMH = 5.00 × 10-7; OR, 1.25). No evidence of association was observed for CHRNA9 rs6832151 either in the replication phase or in the overall pooled analysis.
Conclusion
Our results suggest a role of PPARG gene in the development of SSc.
Similar content being viewed by others


Introduction
Systemic sclerosis (SSc) is a complex autoimmune disease with heterogeneous clinical manifestations characterized by extensive fibrosis in the skin and multiple internal organs, vascular damage, and immune imbalance with autoantibody production [1]. SSc patients are commonly classified in two major subtypes: limited cutaneous SSc (lcSSc) and diffuse cutaneous SSc (dcSSc), the latter with more progressive fibrosis of the skin, lungs, and other internal organs and, ultimately, with worse prognosis [2].
The etiology of this disorder is still unclear. However, epidemiologic and genetic studies clearly reflect the existence of a complex genetic component together with the influence of environmental factors [1]. During recent years, great advances have been made in our knowledge of the genetic basis of SSc [3, 4], in part, thanks to the two independent genome-wide associations studies (GWASs) conducted in Caucasian populations that have been recently published [5, 6], and several consequent follow-up studies [7–10].
However, despite these advances, the number of currently known loci explaining the genetic component of SSc is limited. To date, 13 loci have been identified as genetic risk factors for SSc at the genome-wide significance level. In other autoimmune diseases with multifactorial inheritance, such as Crohn disease, ulcerative colitis, or systemic lupus erythematosus, individual GWAS scans and follow-up meta-analyses have identified more than 71, 47, and 35 susceptibility loci, respectively [11–13]. Therefore, it is expected that additional risk factors for SSc remain to be discovered, and further meta-analyses and large replication studies are needed to identify part of the missing heritability of this disease.
Follow-up studies focused on the so-called grey zone of the GWASs, where SNPs with tier 2 associations (P values between 5 × 10-8 and 5 × 10-3) are located, constitute one of the most useful GWAS data-mining methods, because possible real association signals could be masked in that area because of a lack of statistical power. On this basis, we aimed to perform a follow-up study of the SNPs located in the grey zone of the GWAS by Allanore et al. [6], taking advantage of our GWAS data sets. We hypothesize that using a larger cohort would increase the statistical power and might lead to the identification of new suitable SSc genetic risk factors.
Methods
Study design
In the first step of this study, we focused on the 90 GWAS-genotyped SNPs that reached a P value < 10-4 in the discovery phase of the GWAS carried out by Allanore et al. [6]. Then, we analyzed the SNPs overlapping with those included in Radstake et al. [5]. After excluding those SNPs located within MHC genes or in previously associated loci, data for 66 SNPs were selected. A meta-analysis including these 66 SNPs was performed on the combined data set from the two SSc GWASs, showing only two SNPs (rs310746 PPARG and rs6832151 CHRNA9 genetic variants) with a P value < 10-5 (see later). These two genetic variants were genotyped in independent replication cohorts. Finally, we performed a meta-analysis for these two selected SNPs combining genotype data from both first and replication steps.
Study population
The first step of the study comprised a total of 2,921 SSc patients and 6,963 healthy controls of Caucasian ancestry from two previously published GWASs (European, USA, and French) [5, 6]. The replication cohort was composed of 1,068 SSc patients and 1,490 healthy controls from two case–control sets of European ancestry (Italy and United Kingdom). We also included 5,272 extra English controls from The Wellcome Trust Case Control Consortium for the replication step comprising a total of 6,762 controls for this stage.
All SSc patients fulfilled the classification criteria by LeRoy et al. [2]. Approval from the local ethical committees (Comité de Bioética del Consejo Superior de Investigaciones Científicas, U.O. Comitato di Etica e Sperimentazione Farmaci Fondazione IRCCS Ca’ Granda, Ospedale Maggiore Policlinico di Milano, Comitato Etico Azienda Ospedaliera Universitaria Integrata di Verona, The Ethics Committee of the Spedali Civili, Brescia, Royal Free Hospital and Medical School Research Ethics Committee, Manchester University Research Ethics Committee, Local Research Ethics Committee at Glasgow Royal Infirmary, Newcastle University Ethics Committee, Ethical Committee of the University Erlangen-Nuremberg, Local Ethics Committee of the Radboud University Nijmegen Medical Centre, Medical Ethics Review Committee of the VU University, Medische Ethische Commissie Leids Universitair Medisch Centrum, Ethics Review Board of the Ruhr University Bochum, Ethics Committee of the University of Cologne, Ethical Committee from the Charité University Hospital, Ethik-Kommission der MHH, Internal Review Board of Texas University), and written informed consents from all participants were obtained in accordance with the tenets of the Declaration of Helsinki.
Genotyping
In the first stage, genotype data for the 66 selected SNPs were obtained from both published SSc GWASs [5, 6]. QC filters and principal component analysis were applied to the GWASs data, as described in Radstake et al. [5] and Allanore et al.[6].
In the replication phase, DNA from patients and controls was obtained by using standard methods. Genotyping was performed by using TaqMan 5′ allele discrimination predesigned assays from Applied Biosystems (rs310746 ID: C___8756618_10; rs6832151 ID: C__29224385_10, Foster City, CA, USA) in both 7900HT Fast Real-Time PCR System (Applied Biosystems, Foster City, C A, USA), and LightCycler 480 Real-Time PCR System (Roche Applied Science, Mannheim, Germany). Genotyping call rate was > 98% for both genotyped SNPs.
Statistical analysis
Association analyses of the genotype data was carried out with StatsDirect V.2.6.6 (StatsDirect, Altrincham, UK) and PLINK V.1.07 [14] software. Statistical significance was calculated by 2 × 2 contingency tables and χ2 or Fisher Exact test, when necessary, to obtain P values, odds ratios (ORs), and 95% confidence intervals (CIs) in the population-specific analyses. Mantel-Haenszel tests under fixed effects or random effects, when appropriate, were performed to meta-analyze the combined data. Breslow–Day method (BD) was used to assess the homogeneity of the associations among the different populations (Breslow–Day P values <0.05 were considered statistically significant). Hardy-Weinberg equilibrium (HWE) was tested for all cohorts (HWE P values lower than 0.01 were considered to show significant deviation from the equilibrium). None of the included cohorts showed significant deviation from HWE for the two genotyped SNPs. Since the analyses were performed by using GWAS data, the statistical threshold for considering a P value as a significant P value in the allelic association analyses was set at 5 × 10-8.
The statistical power of the combined analysis was 70% for the PPARG rs310746 and 100% for the CHRNA9 rs6832151 to detect associations with OR = 1.3 and a statistical significance of 5 × 10-8, according to Power Calculator for Genetic Studies 2006 software [15].
Results
Table 1 shows the results of the 66 GWAS-genotyped SNPs selected for the combined meta-analysis of the two GWAS data sets performed in the first step of this study (see Additional file 1: Table S1 provides the results from both GWASs and the combined meta-analysis for the 66 selected SNPs). Two SNPs showed a P value lower than 10-5 (PPARG rs310746: PMH = 1.90 × 10-6; OR, 1.28; CI, 95%, 1.12 to 1.47; and CHRNA9 rs6832151: PMH = 4.30 × 10-6, OR, 1.17; CI 95%, 1.08 to 1.27), and presented no significant Breslow-Day P values (PBD) showing homogeneity in the ORs among populations. Therefore, these two SNPs were selected to genotype in independent cohorts. Patients and healthy controls were found to be in HWE at 1% significance level for both selected SNPs.
In the replication phase, we observed a trend of association for the PPARG rs310746 genetic variant (P value = 0.066; OR = 1.17; CI 95%, 0.99 to 1.38) in the combined analysis of the two replication cohorts (Table 2, upper rows). However, no evidence of association was observed for CHRNA9 rs6832151 either in the pooled analysis (Table 2, upper rows) or in the analysis of each individual population (see Additional file 2: Table S2).
Finally, we combined the results from both steps of the study and performed a Mantel-Haenszel meta-analysis observing that the PPARG genetic variant showed suggestive evidence of association with SSc (PMH = 5.00 × 10-7; OR = 1.25; CI, 95%, 1.15 to 1.37) (Table 2, lower rows; Figure 1). However, CHRNA9 rs6832151 showed no evidence of association with the disease when the meta-analysis was performed either under a random-effects model (heterogeneity of the ORs was observed for this SNP; P value = 5.10 × 10-2, OR = 1.10; CI 95%, 0.99 to 1.22), or a fixed-effects model (P value = 1.07 × 10-4; OR = 1.12; CI 95%, 1.06 to 1.19) (Table 2, lower rows; Figure 1).

Forest plots of PPARG rs310746 and CHRNA9 rs6832151. Forest plots showing the odds ratios and confidence intervals of both PPARG rs310746 (under a fixed-effects model) and CHRNA9 rs6832151 (under a random-effects model) in all the populations included in the combined analysis.
Discussion
In this study we conducted a meta-analysis combining previously published SSc GWASs data for 66 SNPs and analyzed the possible role of two selected SNPs, PPARG rs310746 and CHRNA9 rs6832151, in SSc risk by using independent replication cohorts.
Meta-analyses are a useful tool to increase the statistical power of genetic studies, thus improving the accuracy of the estimations of statistical significance. Of note, associations identified from a single GWAS often tend to have inflated effect sizes [16]. On this basis, our data suggest that most signals from the grey zone observed in the discovery phase of the GWAS by Allanore et al. [6] presented inflated effect sizes, also called the winner’s curse. In fact, this effect was already observed in the replication study conducted by our group for the novel SSc genetic risk factors identified by Allanore et al. [6], in which we could not replicate the association described for RHOB[17].
Our overall combined meta-analysis showed that the association of the PPARG rs310746 genetic variant with SSc remained with a nominal but non-genome-wide significant P value. This SNP is located upstream of PPARG, which encodes the peroxisome proliferator-activated receptor gamma (PPARG). PPARG was initially identified in adipose tissue, where this nuclear receptor plays important roles in adipogenesis, insulin sensitivity, and homeostasis [18]. Interestingly, during recent years, several studies have identified a novel role of PPARG as an antifibrotic effector. Thus, it has been reported that fibroblasts exposure to pharmacologic PPARG ligands give rise to suppression of collagen synthesis, myofibroblast differentiation, and other TGF-β-induced fibrotic responses in vitro[19–21]. Moreover, functional studies showed that PPARG agonist attenuated dermal fibrosis in mice with bleomycin-induced scleroderma [22, 23].
These findings are remarkable in SSc, in which fibrosis is one of the main hallmarks of the disease. In this regard, Wei et al. [24] demonstrated that PPARG expression and function are impaired in SSc patients. Therefore, defects in PPARG expression may influence the uncontrolled progression of fibrosis in SSc. In addition, PPARG has been associated with other autoimmune diseases, such as inflammatory bowel disease [25, 26] and psoriatic arthritis [27], and it is also a confirmed susceptibility locus in type 2 diabetes mellitus [28].
Although PPARG was the most likely biologic candidate gene for the reported suggestive association signal, we could not rule out TIMP4 as another possible gene for this signal. Further analyses are required to elucidate the functional implication of the reported signal.
Regarding the CHRNA9 genetic variant, despite the suggestive association found in the first step of the present study, the overall combined meta-analysis did not show evidence of association with SSc. Moreover, the effect size of the analyzed genetic variant was heterogeneous between the different populations. Although our data showed heterogeneity and lack of association in this locus, a slight or modest effect of CHRNA9 cannot be ruled out, and further studies will be required to determine whether this region is associated with SSc.
It is worth mentioning that the analyzed CHRNA9 SNP has been previously associated with Graves disease (first, through a GWAS performed in the Chinese Han population [29], and subsequently, in a replication study performed in a Polish Caucasian population [30]), but this is the only reported association between this gene and an autoimmune disease.
Conclusion
In conclusion, we report a suggestive association between PPARG rs310746 and SSc. However, further studies are needed to establish this locus firmly as a new susceptibility SSc genetic risk factor.
Abbreviations
- BD test:
-
Breslow-day test
- CHRNA9:
-
cholinergic receptor nicotinic, Alpha 9
- CI:
-
Confidence interval
- dcSSc:
-
diffuse cutaneous systemic sclerosis
- DNA:
-
deoxyribonucleic acid
- GWAS:
-
genome-wide association study
- HLA:
-
Human leukocyte antigen
- HWE:
-
Hardy-Weinberg equilibrium
- lcSSc:
-
limited cutaneous systemic sclerosis
- MAF:
-
minor allele frequency
- MHC:
-
major histocompatibility complex
- OR:
-
Odds ratio
- PCR:
-
polymerase chain reaction
- PPARG:
-
peroxisome proliferator-activated receptor gamma
- SNP:
-
single nucleotide polymorphism
- SSc:
-
systemic sclerosis
- TGF:
-
transforming growth factor.
References
Gabrielli A, Avvedimento EV, Krieg T: Scleroderma. N Engl J Med. 2009, 360: 1989-2003. 10.1056/NEJMra0806188.
LeRoy EC, Black C, Fleischmajer R, Jablonska S, Krieg T, Medsger TA, Rowell N, Wollheim F: Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol. 1988, 15: 202-205.
Martin JE, Bossini-Castillo L, Martin J: Unraveling the genetic component of systemic sclerosis. Hum Genet. 2012, 131: 1023-1037. 10.1007/s00439-011-1137-z.
Assassi S, Radstake TR, Mayes MD, Martin J: Genetics of scleroderma: implications for personalized medicine?. BMC Med. 2013, 11: 9-10.1186/1741-7015-11-9.
Radstake TR, Gorlova O, Rueda B, Martin JE, Alizadeh BZ, Palomino-Morales R, Coenen MJ, Vonk MC, Voskuyl AE, Schuerwegh AJ, Broen JC, Van Riel PL, Van’t Slot R, Italiaander A, Ophoff RA, Riemekasten G, Hunzelmann N, Simeon CP, Ortego-Centeno N, Gonzalez-Gay MA, Gonzalez-Escribano MF, Airo P, Van Laar J, Herrick A, Worthington J, Hesselstrand R, Smith V, De Keyser F, Houssiau F, Chee MM: Genome-wide association study of systemic sclerosis identifies CD247 as a new susceptibility locus. Nat Genet. 2010, 42: 426-429. 10.1038/ng.565.
Allanore Y, Saad M, Dieude P, Avouac J, Distler JH, Amouyel P, Matucci-Cerinic M, Riemekasten G, Airo P, Melchers I, Hachulla E, Cusi D, Wichmann HE, Wipff J, Lambert JC, Hunzelmann N, Tiev K, Caramaschi P, Diot E, Kowal-Bielecka O, Valentini G, Mouthon L, Czirjak L, Damjanov N, Salvi E, Conti C, Muller M, Muller-Ladner U, Riccieri V, Ruiz B: Genome-wide scan identifies TNIP1, PSORS1C1, and RHOB as novel risk loci for systemic sclerosis. PLoS Genet. 2011, 7: e1002091-10.1371/journal.pgen.1002091.
Gorlova O, Martin JE, Rueda B, Koeleman BP, Ying J, Teruel M, Diaz-Gallo LM, Broen JC, Vonk MC, Simeon CP, Alizadeh BZ, Coenen MJ, Voskuyl AE, Schuerwegh AJ, Van Riel PL, Vanthuyne M, Van’t Slot R, Italiaander A, Ophoff RA, Hunzelmann N, Fonollosa V, Ortego-Centeno N, Gonzalez-Gay MA, Garcia-Hernandez FJ, Gonzalez-Escribano MF, Airo P, Van Laar J, Worthington J, Hesselstrand R, Smith V: Identification of novel genetic markers associated with clinical phenotypes of systemic sclerosis through a genome-wide association strategy. PLoS Genet. 2011, 7: e1002178-10.1371/journal.pgen.1002178.
Bossini-Castillo L, Martin JE, Broen J, Gorlova O, Simeon CP, Beretta L, Vonk MC, Callejas JL, Castellvi I, Carreira P, Garcia-Hernandez FJ, Fernandez Castro M, Coenen MJ, Riemekasten G, Witte T, Hunzelmann N, Kreuter A, Distler JH, Koeleman BP, Voskuyl AE, Schuerwegh AJ, Palm O, Hesselstrand R, Nordin A, Airo P, Lunardi C, Scorza R, Shiels P, van Laar JM, Herrick A: A GWAS follow-up study reveals the association of the IL12RB2 gene with systemic sclerosis in Caucasian populations. Hum Mol Genet. 2012, 21: 926-933. 10.1093/hmg/ddr522.
Martin JE, Broen JC, Carmona FD, Teruel M, Simeon CP, Vonk MC, Van’t Slot R, Rodriguez-Rodriguez L, Vicente E, Fonollosa V, Ortego-Centeno N, Gonzalez-Gay MA, Garcia-Hernandez FJ, De la Pena PG, Carreira P, Voskuyl AE, Schuerwegh AJ, van Riel PL, Kreuter A, Witte T, Riemekasten G, Airo P, Scorza R, Lunardi C, Hunzelmann N, Distler JH, Beretta L, van Laar J, Chee MM, Worthington J: Identification of CSK as a systemic sclerosis genetic risk factor through Genome Wide Association Study follow-up. Hum Mol Genet. 2012, 21: 2825-2835. 10.1093/hmg/dds099.
Sharif R, Mayes MD, Tan FK, Gorlova OY, Hummers LK, Shah AA, Furst DE, Khanna D, Martin J, Bossini-Castillo L, Gonzalez EB, Ying J, Draeger HT, Agarwal SK, Reveille JD, Arnett FC, Wigley FM, Assassi S: IRF5 polymorphism predicts prognosis in patients with systemic sclerosis. Ann Rheum Dis. 2012, 71: 1197-1202. 10.1136/annrheumdis-2011-200901.
Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, Lees CW, Balschun T, Lee J, Roberts R, Anderson CA, Bis JC, Bumpstead S, Ellinghaus D, Festen EM, Georges M, Green T, Haritunians T, Jostins L, Latiano A, Mathew CG, Montgomery GW, Prescott NJ, Raychaudhuri S, Rotter JI, Schumm P, Sharma Y, Simms LA, Taylor KD, Whiteman D: Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010, 42: 1118-1125. 10.1038/ng.717.
Anderson CA, Boucher G, Lees CW, Franke A, D’Amato M, Taylor KD, Lee JC, Goyette P, Imielinski M, Latiano A, Lagace C, Scott R, Amininejad L, Bumpstead S, Baidoo L, Baldassano RN, Barclay M, Bayless TM, Brand S, Buning C, Colombel JF, Denson LA, De Vos M, Dubinsky M, Edwards C, Ellinghaus D, Fehrmann RS, Floyd JA, Florin T, Franchimont D: Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet. 2011, 43: 246-252. 10.1038/ng.764.
Sestak AL, Furnrohr BG, Harley JB, Merrill JT, Namjou B: The genetics of systemic lupus erythematosus and implications for targeted therapy. Ann Rheum Dis. 2011, 70: i37-i43. 10.1136/ard.2010.138057.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC: PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007, 81: 559-575. 10.1086/519795.
Skol AD, Scott LJ, Abecasis GR, Boehnke M: Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat Genet. 2006, 38: 209-213. 10.1038/ng1706.
Ioannidis JP, Thomas G, Daly MJ: Validating, augmenting and refining genome-wide association signals. Nat Rev Genet. 2009, 10: 318-329. 10.1038/nrg2544.
Bossini-Castillo L, Martin JE, Broen J, Simeon CP, Beretta L, Gorlova OY, Vonk MC, Ortego-Centeno N, Espinosa G, Carreira P, Garcia De La Pena P, Oreiro N, Roman-Ivorra JA, Castillo MJ, Gonzalez-Gay MA, Saez-Comet L, Castellvi I, Schuerwegh AJ, Voskuyl AE, Hoffmann-Vold AM, Hesselstrand R, Nordin A, Lunardi C, Scorza R, Van Laar JM, Shiels PG, Herrick A, Worthington J, Fonseca C, Denton C, Denton C: Confirmation of TNIP1 but not RHOB and PSORS1C1 as systemic sclerosis risk factors in a large independent replication study. Ann Rheum Dis. 2013, 72: 602-607. 10.1136/annrheumdis-2012-201888.
Tontonoz P, Spiegelman BM: Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem. 2008, 77: 289-312. 10.1146/annurev.biochem.77.061307.091829.
Ghosh AK, Bhattacharyya S, Lakos G, Chen SJ, Mori Y, Varga J: Disruption of transforming growth factor beta signaling and profibrotic responses in normal skin fibroblasts by peroxisome proliferator-activated receptor gamma. Arthritis Rheum. 2004, 50: 1305-1318. 10.1002/art.20104.
Burgess HA, Daugherty LE, Thatcher TH, Lakatos HF, Ray DM, Redonnet M, Phipps RP, Sime PJ: PPARgamma agonists inhibit TGF-beta induced pulmonary myofibroblast differentiation and collagen production: implications for therapy of lung fibrosis. Am J Physiol Lung Cell Mol Physiol. 2005, 288: L1146-L1153. 10.1152/ajplung.00383.2004.
Kulkarni AA, Thatcher TH, Olsen KC, Maggirwar SB, Phipps RP, Sime PJ: PPAR-gamma ligands repress TGFbeta-induced myofibroblast differentiation by targeting the PI3K/Akt pathway: implications for therapy of fibrosis. PLoS One. 2011, 6: e15909-10.1371/journal.pone.0015909.
Wei J, Zhu H, Komura K, Lord G, Tomcik M, Wang W, Doniparthi S, Tamaki Z, Hinchcliff M, Distler JH, Varga J: A synthetic PPAR-gamma agonist triterpenoid ameliorates experimental fibrosis: PPAR-gamma-independent suppression of fibrotic responses. Ann Rheum Dis. 2013, Epub ahead of print
Wu M, Melichian DS, Chang E, Warner-Blankenship M, Ghosh AK, Varga J: Rosiglitazone abrogates bleomycin-induced scleroderma and blocks profibrotic responses through peroxisome proliferator-activated receptor-gamma. Am J Pathol. 2009, 174: 519-533. 10.2353/ajpath.2009.080574.
Wei J, Ghosh AK, Sargent JL, Komura K, Wu M, Huang QQ, Jain M, Whitfield ML, Feghali-Bostwick C, Varga J: PPARgamma downregulation by TGFss in fibroblast and impaired expression and function in systemic sclerosis: a novel mechanism for progressive fibrogenesis. PLoS One. 2010, 5: e13778-10.1371/journal.pone.0013778.
Glas J, Seiderer J, Markus C, Pfennig S, Wetzke M, Paschos E, Goke B, Ochsenkuhn T, Muller-Myhsok B, Diegelmann J, Roeske D, Brand S: Role of PPARG gene variants in inflammatory bowel disease. Inflamm Bowel Dis. 2011, 17: 1057-1058. 10.1002/ibd.21425.
Zhang ZF, Yang N, Zhao G, Zhu L, Wang LX: Association between the Pro12Ala polymorphism of peroxisome proliferator-activated receptor gamma 2 and inflammatory bowel disease: a meta-analysis. PLoS One. 2012, 7: e30551-10.1371/journal.pone.0030551.
Butt C, Gladman D, Rahman P: PPAR-gamma gene polymorphisms and psoriatic arthritis. J Rheumatol. 2006, 33: 1631-1633.
Lyssenko V, Jonsson A, Almgren P, Pulizzi N, Isomaa B, Tuomi T, Berglund G, Altshuler D, Nilsson P, Groop L: Clinical risk factors, DNA variants, and the development of type 2 diabetes. N Engl J Med. 2008, 359: 2220-2232. 10.1056/NEJMoa0801869.
Chu X, Pan CM, Zhao SX, Liang J, Gao GQ, Zhang XM, Yuan GY, Li CG, Xue LQ, Shen M, Liu W, Xie F, Yang SY, Wang HF, Shi JY, Sun WW, Du WH, Zuo CL, Shi JX, Liu BL, Guo CC, Zhan M, Gu ZH, Zhang XN, Sun F, Wang ZQ, Song ZY, Zou CY, Sun WH, Guo T: A genome-wide association study identifies two new risk loci for Graves’ disease. Nat Genet. 2011, 43: 897-901. 10.1038/ng.898.
Szymanski K, Bednarczuk T, Krajewski P, Ploski R: The replication of the association of the rs6832151 within chromosomal band 4p14 with Graves’ disease in a Polish Caucasian population. Tissue Antigens. 2012, 79: 380-383. 10.1111/j.1399-0039.2012.01854.x.
Acknowledgements
We thank Sofia Vargas, Sonia García, and Gema Robledo for their excellent technical assistance, and all the patients and control donors for their essential collaboration. We thank Banco Nacional de ADN (University of Salamanca, Spain), who supplied part of the control DNA samples.
This work was supported by the following grants: JM was funded by GEN-FER from the Spanish Society of Rheumatology, SAF2009-11110 and SAF2012-34435 from the Spanish Ministry of Economy and Competitiveness, CTS-4977, and CTS-180 from Junta de Andalucía, and is sponsored by the Orphan Disease Program grant from the European League Against Rheumatism (EULAR). This study was also funded by PI-0590-2010, from Consejería de Salud y Bienestar Social, Junta de Andalucía, Spain. JLCR and JM are funded by Consejería de Salud, Junta de Andalucía, through PI-0590-2010. ELI was supported by Ministerio de Educación, Cultura y Deporte through the program FPU. TRDJR was funded by the VIDI laureate from the Dutch Association of Research (NWO) and Dutch Arthritis Foundation (National Reumafonds). TW was granted by DFG WI 1031/6.1. Study on USA samples were supported by US National Institutes of Health and National Institute of Arthritis and Musculoskeletal Diseases (NIH-NIAMS) R01-AR-055258, Two-Stage Genome Wide Association Study in Systemic Sclerosis (MDM) and by the NIH-NIAMS Center of Research Translation (CORT) in SSc (P50AR054144) (MDM, FCA, FKT), the NIH-NIAMS SSc Family Registry and DNA Repository (N01-AR-0-2251) (MDM), NIH-KL2RR024149 (SA), K23AR061436 (SA), and the Department of Defense Congressionally Directed Medical Research Programs (W81XWH-07-01-0111) (MDM).
Spanish Scleroderma Group
Spanish Scleroderma Group: Norberto Ortego-Centeno and Raquel Ríos, Unidad de Enfermedades Sistémicas Autoinmunes, Department of Internal Medicine, Hospital Clínico Universitario San Cecilio, Granada; Nuria Navarrete, Department of Internal Medicine, Hospital Virgen de las Nieves, Granada; Rosa García Portales, Department of Rheumatology, Hospital Virgen de la Victoria, Málaga; María Teresa Camps, Department of Internal Medicine, Hospital Carlos Haya, Málaga; Antonio Fernández-Nebro, Department of Rheumatology, Hospital Carlos Haya, Málaga; María F. González-Escribano, Department of Immunology, Hospital Virgen del Rocío, Sevilla; Julio Sánchez-Román, Francisco José García-Hernández and Mª Jesús Castillo, Department of Internal Medicine, Hospital Virgen del Rocío, Sevilla; Mª Ángeles Aguirre and Inmaculada Gómez-Gracia, Department of Rheumatology, Hospital Reina Sofía/IMIBIC, Córdoba; Patricia Carreira, Department of Rheumatology. Hospital Universitario 12 de Octubre, Madrid; Benjamín Fernández-Gutiérrez and Luis Rodríguez-Rodríguez, Department of Rheumatology, Hospital Clínico San Carlos, Madrid; Esther Vicente, Department of Rheumatology, Hospital La Princesa, Madrid; José Luis Andreu and Mónica Fernández de Castro, Department of Rheumatology, Hospital Puerta de Hierro Majadahonda, Madrid; Paloma García de la Peña, Department of Rheumatology, Hospital Madrid Norte Sanchinarro, Madrid; Francisco Javier López-Longo and Lina Martínez, Department of Rheumatology, Hospital General Universitario Gregorio Marañón, Madrid; Vicente Fonollosa and Carmen Pilar Simeón, Department of Internal Medicine, Hospital Valle de Hebrón, Barcelona; Gerard Espinosa, Department of Internal Medicine, Hospital Clinic, Barcelona; Iván Castellví, Department of Rheumatology, Hospital de la Santa Creu i Sant Pau, Barcelona; Carlos Tolosa, Department of Internal Medicine, Hospital Parc Tauli, Sabadell; Anna Pros, Department of Rheumatology, Hospital Del Mar, Barcelona; Mónica Rodríguez Carballeira, Department of Internal Medicine, Hospital Universitari Mútua Terrasa, Barcelona; Francisco Javier Narváez, Department of Rheumatology, Hospital Universitari de Bellvitge, Barcelona; Manel Rubio Rivas, Department of Internal Medicine, Hospital Universitari de Bellvitge, Barcelona; Vera Ortiz Santamaría, Department of Rheumatology, Hospital General de Granollers, Granollers; Miguel Ángel González-Gay, Department of Rheumatology, Hospital Universitario Marqués de Valdecilla, IFIMAV, Santander; Bernardino Díaz and Luis Trapiella, Department of Internal Medicine, Hospital Central de Asturias, Oviedo; Adrián Sousa, Unidad de Trombosis y Vasculitis, Department of Internal Medicine, Hospital Xeral-Complexo Hospitalario Universitario de Vigo, Vigo; Patricia Fanlo Mateo, Department of Internal Medicine Hospital Virgen del Camino, Pamplona; Luis Sáez-Comet, Unidad de Enfermedades Autoinmunes Sistémicas, Department of Internal Medicine, Hospital Universitario Miguel Servet, Zaragoza; Federico Díaz and Vanesa Hernández, Department of Rheumatology, Hospital Universitario de Canarias, Tenerife; Emma Beltrán, Department of Rheumatology, Hospital General Universitario de Valencia, Valencia; and Francisco J. Blanco García and Natividad Oreiro, Department of Rheumatology, INIBIC-Hospital Universitario A Coruña, La Coruña.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
ELI and LBC contributed to the analysis and interpretation of data and the drafting the manuscript. CPS and AH participated in the acquisition of data and the drafting of the manuscript. JM contributed to the conception and design of the study and critically revised the manuscript. MVE, JJAS, JLC, JARI, MF, LB, AS, PA, CL, NH, GR, TW, AK, JHWD, AJS, MCV, AEV, PGS, JMvL, CF, CD, JW, SA, BPK, MDM, TRDJR, and the Spanish Scleroderma Group were involved in the acquisition of data and the revision of the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
13075_2013_4099_MOESM1_ESM.docx
Additional file 1: GWASs results from Allanore et al . [[6]] and Radstake et al . [[5]], and combined meta-analysis. Description: this file contains Additional file 1: Table S1 showing the results for the 66 selected SNPs in Allanore et al. and Radstake et al. GWASs, followed by the results of the combined meta-analysis performed in the present study. (DOCX 35 KB)
13075_2013_4099_MOESM2_ESM.docx
Additional file 2: Genotype and minor allele frequencies of rs310746 and rs6832151 SNPs in two European cohorts (Replication-step). Description: this file contains: Additional file 2: Table S2 showing the genotype and allele distributions of rs310746 and rs6832151 genetic variants in two European cohorts (1032 SSc cases and 6700 controls). (DOCX 14 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
López-Isac, E., Bossini-Castillo, L., Simeon, C.P. et al. A genome-wide association study follow-up suggests a possible role for PPARG in systemic sclerosis susceptibility. Arthritis Res Ther 16, R6 (2014). https://doi.org/10.1186/ar4432
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/ar4432