
Abstract
Significant advances have been made in understanding the genetic basis of systemic sclerosis (scleroderma) in recent years. Can these discoveries lead to individualized monitoring and treatment? Besides robustly replicated genetic susceptibility loci, several genes have been recently linked to various systemic sclerosis disease manifestations. Furthermore, inclusion of genetic studies in design and analysis of drug trials could lead to development of genetic biomarkers that predict treatment response. Future genetic studies in well-characterized systemic sclerosis cohorts paired with advanced analytic approaches can lead to development of genetic biomarkers for targeted diagnostic and therapeutic interventions in systemic sclerosis.
Similar content being viewed by others
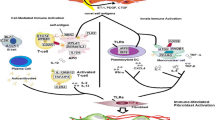


Background
Systemic sclerosis (SSc or scleroderma) is a multisystem, uncommon disease characterized by fibrosis in skin and internal organs, immune dysregulation, and vasculopathy. Its pathogenesis remains poorly understood but there is a growing body of evidence implicating in part genetic factors. However, the genetic basis for SSc is defined by multiple genes that have only modest effect on disease susceptibility [1, 2]. Moreover, the disease is thought to arise from an interaction between genetic factors and environmental triggers.
SSc is subdivided into limited and diffuse types based on the extent of skin involvement [3]. Furthermore, SSc can be sub-grouped based on the presence of non-overlapping autoantibodies that are associated with various disease manifestations [4]. The standardized mortality ratio of patients with SSc is 3.5 [5] which is higher than most other rheumatic diseases. Reliable predictors of disease course and therapeutic options are very limited. Genetic data are not time-dependent and do not change over the course of disease; thus they are attractive candidates for development of predictive biomarkers. In this review, we will examine the implication of recent discoveries in SSc genetics for drug development and identification of predictive biomarkers.
Recent advances in SSc genetics
Case-control candidate gene studies have identified several robust SSc susceptibility loci that have been confirmed in subsequent independent studies (reviewed in [1, 2]). The majority of these genes such as IRF5 [6], STAT4 [7], BANK1 [8] and BLK [9] belong to pathways involved in immune regulation. Furthermore, three genome wide association studies (GWAS) allowed unbiased genetic profiling of patients with SSc [10–12]. These studies have confirmed genes in the major histocompatibility complex (MHC) as the strongest susceptibility loci. Furthermore, a GWAS follow-up study confirmed that HLA-DQB1, HLA-DPA1/B1, and NOTCH4 associations with SSc are likely confined to SSc specific auto-antibodies [13].
Multiple non-MHC susceptibility loci also have been identified in the above-mentioned studies. As shown in Table 1, the most robust associations are in genes related to innate immunity, as well as B- and T-cell activation. For example, IRF5 belongs to a family of transcription factors in the type I interferon pathway which is an important component of the innate immunity, whereas CD247 encodes the T-cell receptor zeta subunit modulating T-cell activation. The majority of these gene variants are also risk loci for other autoimmune diseases, especially for systemic lupus erythematosus (SLE) [2, 14]. This indicates that SSc has a shared immune pathogenesis with other autoimmune diseases providing further support for the concept of quantitative thresholds in immune-cell signaling. In this concept, several genetic factors of relatively small effect may cumulatively create a state of susceptibility to autoimmune diseases (reviewed in [15]). Self-reactive B and T-cells are a normal component of the immune system. However, they are usually kept in check by regulatory mechanisms in the thymus/bone marrow or peripheral blood. In the concept of quantitative threshold, the implicated genetic variations lead cumulatively to an impairment of necessary biological processes for destruction of self-reactive immune cells and regulating auto-reactivity. Validity of this concept in SSc is supported by the fact that several SSc genetic susceptibility loci overlap not only with SLE but also with other autoimmune diseases. For example, STAT4 is also implicated in rheumatoid arthritis [16], and primary biliary cirrhosis [17]. Similarly, PTPN22 is a susceptibility locus in rheumatoid arthritis [18], type 1 diabetes mellitus [19], and also SSc [20].
Some of the confirmed SSc susceptibility loci show a stronger association with its serological or clinical (limited versus diffuse) [13] subtypes than the overall disease. Several genetic associations in the HLA [8, 21] or non-HLA regions, such as BANK1, IRF8, SOX5 and IRF7 are mainly with the SSc-related autoantibodies (e.g. anti-centromere or anti-topoisomerase I) or clinical subtypes of disease [1, 2, 8, 22]. Furthermore, many of the identified single nucleotide polymorphisms (SNPs) are merely a tag genetic variant for the yet to be identified causal allele. This is also applicable to GWA studies, because the utilized platforms provide more than 80% coverage for common polymorphisms in human genome by investigating SNPs that are in strong linkage disequilibrium with multiple other SNPs and serve as proxies for gene areas. Advances in gene sequencing techniques will permit large scale sequencing of these susceptibility genes to pinpoint the actual causal variant.
Some of the reported genetic associations in one ethnic group might not replicate in other ethnicities. The reported polymorphisms might not tag the causal locus in all ethnic groups because of the varying linkage disequilibrium structure among different ethnicities. Alternatively, the reported genetic associations might be truly an ethnic specific susceptibility locus for SSc.
It is noteworthy that the gene variants of interest do not operate in isolation as they are parts of intertwined biological pathways. Therefore, examination of gene-gene or gene-environment interactions can lead to better understanding of SSc pathogenesis. Lastly, mechanistic studies are needed to elucidate how these immune system gene variants contribute to the cross-talk among immune, vascular and fibrotic pathways leading to the unique phenotype of SSc.
Implication of SSc genetics for predicting disease severity and organ involvement
SSc is associated with high morbidity and mortality. The disease related mortality is mainly driven by internal organ involvement [23], especially severity of lung disease [24, 25]. As shown in Table 2, several studies have also investigated the association of MHC and non-MHC genetic loci with interstitial lung disease (ILD), pulmonary arterial hypertension (PAH), scleroderma renal crisis, and mortality. It is important to point out that the comparison of SSc patients with a particular disease manifestation with patients without that particular organ involvement (case-case analysis) is more relevant for biomarker development than comparison of patient with the disease manifestation to unaffected controls (case-control analysis). The main reason for this notion is that the prognostic biomarkers are useful if they can aid clinicians to subgroup patients (cases-case analysis) based on the expected disease progression. A case to control comparison does not occur in the clinical settings because the diagnosis of SSc is already established before clinicians become interested in predicting the disease course. IRF5 gene variants have been linked to overall mortality independent of disease type and serology [26]. CTGF [27], HGF [28], IRAK1 [29], IRF5 [6, 26, 30], MMP-12 [31], SP-B [32] polymorphisms are reported to be associated with ILD. The case definition for ILD varies considerably, some investigators have relied on the presence of reticular or ground glass opacities on high resolution chest computer tomography (HRCT) while others have focused on severity of ILD based on the pulmonary function results. The former approach does not differentiate between the mild stable ILD and its severe progressive forms. Furthermore, IL23R [33], KCNA5 [34], TLR2 [35], TNAIP3 [36], and UPAR [37] genes are reported to be associated with PAH while HLA-DRB1*04:07 and *13:04 were associated with scleroderma renal crisis [38].
However, the above findings need to be replicated in independent studies. Furthermore, the currently available cross-sectional patient populations for SSc genetic studies are most likely affected by survival bias, i.e. the examined prevalent cohorts with longstanding disease are depleted of patients with the most progressive and severe form of SSc. For example, SSc patients with rapidly progressive ILD have a higher mortality [39], therefore patient samples with long-standing disease (mean disease duration > 5 years) are depleted of the most severe form of ILD. This can lead to decreased frequency of genetic loci associated with more severe forms of disease in the investigated patient samples. Examination of incident cases with longitudinal follow-up can avoid problems arising from survival bias. Furthermore, the genetic severity loci might be different than the genes linked to SSc susceptibility. For example, HGF was not a susceptibility locus for SSc but was associated with end-stage lung disease among Japanese SSc patients [28]. A careful phenotypic characterization of patients examined in GWAS can permit an unbiased profiling of severity loci. This will also allow combination of genetic data with other clinical and serological markers of disease severity for risk prediction.
Risk prediction in genetically complex diseases like SSc requires statistical approaches that extend beyond separate odds ratios for each SNP of interest. Genotypes at multiple SNPs can be combined into cumulative scores calculated according to the number of severity alleles carried. Furthermore, risk reclassification statistics can be used to combine genetic and clinical data. In this approach, patients in the intermediate risk group based on clinical data are reassigned to low- or high-risk categories using the pertinent genetic information.
Implication of SSc genetics for treatment selection
The newly identified genetic susceptibility pathways can lead to identification of novel therapeutic targets and guide drug development. Indeed, some of the currently investigated biologic therapies for SSc match appropriately to these pathways. These include anti-interferon (e.g. sifalimumab) and anti-B-cell agents (e.g. rituximab) [40]. Furthermore, the SSc genetic data lend support to T-cell directed therapies (e.g. abatacept). However, there are no reported large-scale, randomized controlled studies of B-cell, T-cell, interferon directed therapies in patients with SSc.
Beyond identification of new therapeutic targets, the genetic information might be used to identify the high responsive group to a particular biologic treatment. There are no data on predictive significance of genetic information for response to treatment in SSc. This requires the collection of genetic material in drug trials and careful analysis of genetic information conditional on the study outcomes. Considering the modest effect of these gene variants on the disease susceptibility, we might be underpowered to examine the predictive significance of these factors in drug trials using traditional (frequentist) statistical methods (especially after sample partitioning into treatment and control arms). Bayesian analysis of trial results in uncommon diseases such as SSc [41] might lead to more flexible and clinically useful biomarker development.
Independent of disease susceptibility genes, the genetic information can be used to predict drug metabolism and development of adverse effects (pharmacogenetics). For example, polymorphism in the UGT1A9 affect metabolism of mycophenolate mofetil and predict acute rejection in renal transplant patients [42, 43]. Despite the widespread use of mycophenolate mofetil, the role of this polymorphism for response to treatment and development of adverse events has not been investigated in SSc patients.
In a recently published study, a polymorphism in the IL-6 gene predicted response to rituximab in a sample of patients with SLE and other rheumatic diseases that included patients with SSc [44].
Conclusion
The significant advances in SSc genetics represent an opportunity for biomarker development. Careful phenotypic characterization, independent confirmation of current findings, inclusion of genetic studies in drug trials, and utilization of novel analytic approaches paired with advanced high-throughput technologies can potentially lead to identification of genetic markers that predict disease severity and response to treatment in SSc.
Authors' information
SA is associate professor of medicine/rheumatology at the University of Texas-Houston (USA). His research focuses on correlation of genomic data with important clinical outcomes in systemic sclerosis and other rheumatic diseases.
TR is a professor of Rheumatology & Clinical Immunology at the University of Utrecht (The Netherlands). His area of research focuses on mechanistic and genetic translational studies in systemic sclerosis and other rheumatic diseases.
MM is a professor of medicine/rheumatology at the University of Texas-Houston (USA). Her research focuses on genetic and clinical studies in systemic sclerosis.
JM is a professor of genetics in Instituto de Parasitología y Biomedicina López-Neyra, Consejo Superior de Investigaciones Científicas (CSIC) in Granada (Spain). His research focuses on genetics of systemic sclerosis, as well as other rheumatic and autoimmune diseases.
Abbreviations
- GWAS:
-
Genome wide association studies
- HLA:
-
Human leukocyte antigen
- HRCT:
-
High resolution chest computer tomography
- ILD:
-
Interstitial Lung Disease
- MHC:
-
Major histocompatibility complex
- PAH:
-
Pulmonary arterial hypertension
- SNP:
-
Single nucleotide polymorphism
- SSc:
-
Systemic sclerosis.
References
Martin J, Fonseca C: The genetics of scleroderma. Curr Rheumatol Rep. 2011, 13: 13-20. 10.1007/s11926-010-0139-5.
Martin JE, Bossini-Castillo L, Martin J: Unraveling the genetic component of systemic sclerosis. Hum Genet. 2012, 131: 1023-1037. 10.1007/s00439-011-1137-z.
Leroy EC, Black C, Fleischmajer R, Jablonska S, Krieg T, Medsger TA, Rowell N, Wollheim F: Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol. 1988, 15: 202-205.
Steen VD: Autoantibodies in systemic sclerosis. Semin Arthritis Rheum. 2005, 35: 35-42. 10.1016/j.semarthrit.2005.03.005.
Elhai M, Meune C, Avouac J, Kahan A, Allanore Y: Trends in mortality in patients with systemic sclerosis over 40 years: a systematic review and meta-analysis of cohort studies. Rheumatology (Oxford). 2011, 51: 1017-1026.
Dieude P, Guedj M, Wipff J, Avouac J, Fajardy I, Diot E, Granel B, Sibilia J, Cabane J, Mouthon L, Cracowski JL, Carpentier PH, Hachulla E, Meyer O, Kahan A, Boileau C, Allanore Y: Association between the IRF5 rs2004640 functional polymorphism and systemic sclerosis: A new perspective for pulmonary fibrosis. Arthritis Rheum. 2009, 60: 225-233. 10.1002/art.24183.
Rueda B, Broen J, Simeon C, Hesselstrand R, Diaz B, Suarez H, Ortego-Centeno N, Riemekasten G, Fonollosa V, Vonk MC, van den Hoogen FH, Sanchez-Roman J, guirre-Zamorano MA, Garcia-Portales R, Pros A, Camps MT, Gonzalez-Gay MA, Coenen MJ, Airo P, Beretta L, Scorza R, van LJ, Gonzalez-Escribano MF, Nelson JL, Radstake TR, Martin J: The STAT4 gene influences the genetic predisposition to systemic sclerosis phenotype. Hum Mol Genet. 2009, 18: 2071-2077. 10.1093/hmg/ddp119.
Rueda B, Gourh P, Broen J, Agarwal SK, Simeon C, Ortego-Centeno N, Vonk MC, Coenen M, Riemekasten G, Hunzelmann N, Hesselstrand R, Tan FK, Reveille JD, Assassi S, Garcia-Hernandez FJ, Carreira P, Camps M, Fernandez-Nebro A, Garcia de la Peña P, Nearney T, Hilda D, Gónzalez-Gay MA, Airo P, Beretta L, Scorza R, Radstake TR, Mayes MD, Arnett FC, Martin J: BANK1 functional variants are associated with susceptibility to diffuse systemic sclerosis in Caucasians. Ann Rheum Dis. 2010, 69: 700-705. 10.1136/ard.2009.118174.
Gourh P, Agarwal SK, Martin E, Divecha D, Rueda B, Bunting H, Assassi S, Paz G, Shete S, McNearney T, Draeger H, Reveille JD, Radstake TR, Simeon CP, Rodriguez L, Vicente E, Gonzalez-Gay MA, Mayes MD, Tan FK, Martin J, Arnett FC: Association of the C8orf13-BLK region with systemic sclerosis in North-American and European populations. J Autoimmun. 2010, 34: 155-162. 10.1016/j.jaut.2009.08.014.
Radstake TR, Gorlova O, Rueda B, Martin JE, Alizadeh BZ, Palomino-Morales R, Coenen MJ, Vonk MC, Voskuyl AE, Schuerwegh AJ, Broen JC, van Riel PL, van 't SR, Italiaander A, Ophoff RA, Riemekasten G, Hunzelmann N, Simeon CP, Ortego-Centeno N, Gonzalez-Gay MA, Gonzalez-Escribano MF, Airo P, van LJ, Herrick A, Worthington J, Hesselstrand R, Smith V, De KF, Houssiau F, Chee MM, et al: Genome-wide association study of systemic sclerosis identifies CD247 as a new susceptibility locus. Nat Genet. 2010, 42: 426-429. 10.1038/ng.565.
Zhou X, Lee JE, Arnett FC, Xiong M, Park MY, Yoo YK, Shin ES, Reveille JD, Mayes MD, Kim JH, Song R, Choi JY, Park JA, Lee YJ, Lee EY, Song YW, Lee EB: HLA-DPB1 and DPB2 are genetic loci for systemic sclerosis: a genome-wide association study in Koreans with replication in North Americans. Arthritis Rheum. 2009, 60: 3807-3814. 10.1002/art.24982.
Allanore Y, Saad M, Dieude P, Avouac J, Distler JH, Amouyel P, Matucci-Cerinic M, Riemekasten G, Airo P, Melchers I, Hachulla E, Cusi D, Wichmann HE, Wipff J, Lambert JC, Hunzelmann N, Tiev K, Caramaschi P, Diot E, Kowal-Bielecka O, Valentini G, Mouthon L, Czirjak L, Damjanov N, Salvi E, Conti C, Muller M, Muller-Ladner U, Riccieri V, Ruiz B, et al: Genome-wide scan identifies TNIP1, PSORS1C1, and RHOB as novel risk loci for systemic sclerosis. PLoS Genet. 2011, 7: e1002091-10.1371/journal.pgen.1002091.
Gorlova O, Martin JE, Rueda B, Koeleman BP, Ying J, Teruel M, az-Gallo LM, Broen JC, Vonk MC, Simeon CP, Alizadeh BZ, Coenen MJ, Voskuyl AE, Schuerwegh AJ, van Riel PL, Vanthuyne M, van 't SR, Italiaander A, Ophoff RA, Hunzelmann N, Fonollosa V, Ortego-Centeno N, Gonzalez-Gay MA, Garcia-Hernandez FJ, Gonzalez-Escribano MF, Airo P, van LJ, Worthington J, Hesselstrand R, Smith V, et al: Identification of Novel Genetic Markers Associated with Clinical Phenotypes of Systemic Sclerosis through a Genome-Wide Association Strategy. PLoS Genet. 2011, 7: e1002178-10.1371/journal.pgen.1002178.
Guerra SG, Vyse TJ, Cunninghame Graham DS: The genetics of lupus: a functional perspective. Arthritis Res Ther. 2012, 14: 211-10.1186/ar3844.
Cho JH, Gregersen PK: Genomics and the multifactorial nature of human autoimmune disease. N Engl J Med. 2011, 365: 1612-1623. 10.1056/NEJMra1100030.
Remmers EF, Plenge RM, Lee AT, Graham RR, Hom G, Behrens TW, de Bakker PI, Le JM, Lee HS, Batliwalla F, Li W, Masters SL, Booty MG, Carulli JP, Padyukov L, Alfredsson L, Klareskog L, Chen WV, Amos CI, Criswell LA, Seldin MF, Kastner DL, Gregersen PK: STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med. 2007, 357: 977-986. 10.1056/NEJMoa073003.
Mells GF, Floyd JA, Morley KI, Cordell HJ, Franklin CS, Shin SY, Heneghan MA, Neuberger JM, Donaldson PT, Day DB, Ducker SJ, Muriithi AW, Wheater EF, Hammond CJ, Dawwas MF, UK PBC Consortium, Wellcome Trust Case Control Consortium, Jones DE, Peltonen L, Alexander GJ, Sandford RN, Anderson CA: Genome-wide association study identifies 12 new susceptibility loci for primary biliary cirrhosis. Nat Genet. 2011, 43: 329-332. 10.1038/ng.789.
Begovich AB, Carlton VE, Honigberg LA, Schrodi SJ, Chokkalingam AP, Alexander HC, Ardlie KG, Huang Q, Smith AM, Spoerke JM, Conn MT, Chang M, Chang SY, Saiki RK, Catanese JJ, Leong DU, Garcia VE, McAllister LB, Jeffery DA, Lee AT, Batliwalla F, Remmers E, Criswell LA, Seldin MF, Kastner DL, Amos CI, Sninsky JJ, Gregersen PK: A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet. 2004, 75: 330-337. 10.1086/422827.
Criswell LA, Pfeiffer KA, Lum RF, Gonzales B, Novitzke J, Kern M, Moser KL, Begovich AB, Carlton VE, Li W, Lee AT, Ortmann W, Behrens TW, Gregersen PK: Analysis of families in the multiple autoimmune disease genetics consortium (MADGC) collection: the PTPN22 620W allele associates with multiple autoimmune phenotypes. Am J Hum Genet. 2005, 76: 561-571. 10.1086/429096.
Diaz-Gallo LM, Gourh P, Broen J, Simeon C, Fonollosa V, Ortego-Centeno N, Agarwal S, Vonk MC, Coenen M, Riemekasten G, Hunzelmann N, Hesselstrand R, Tan FK, Reveille JD, Assassi S, García-Hernandez FJ, Carreira P, Camps MT, Fernandez-Nebro A, de la Peña PG, Nearney T, Hilda D, González-Gay MA, Airo P, Beretta L, Scorza R, Herrick A, Worthington J, Pros A, Gómez-Gracia I, Trapiella L, Espinosa G, Castellvi I, Witte T, de Keyser F, Vanthuyne M, Mayes MD, Radstake TR, Arnett FC, Martin J, Rueda B: Analysis of the influence of PTPN22 gene polymorphisms in systemic sclerosis. Ann Rheum Dis. 2011, 70: 454-462. 10.1136/ard.2010.130138.
Arnett FC, Gourh P, Shete S, Ahn CW, Honey R, Agarwal SK, Tan FK, McNearney T, Fischbach M, Fritzler MJ, Mayes MD, Reveille JD: Major Histocompatibility Complex (MHC) class II alleles, haplotypes, and epitopes which confer susceptibility or protection in the fibrosing autoimmune disease systemic sclerosis: analyses in 1300 Caucasian, African-American and Hispanic cases and 1000 controls. Ann Rheum Dis. 2009, 69: 822-827.
Carmona FD, Gutala R, Simeon CP, Carreira P, Ortego-Centeno N, Vicente-Rabaneda E, Garcia-Hernandez FJ, Garcia de la PP, Fernandez-Castro M, Martinez-Estupinan L, Egurbide MV, Tsao BP, Gourh P, Agarwal SK, Assassi S, Mayes MD, Arnett FC, Tan FK, Martin J, Spanish Scleroderma Group: Novel identification of the IRF7 region as an anticentromere autoantibody propensity locus in systemic sclerosis. Ann Rheum Dis. 2012, 71: 114-119. 10.1136/annrheumdis-2011-200275.
Ioannidis JP, Vlachoyiannopoulos PG, Haidich AB, Medsger TA, Lucas M, Michet CJ, Kuwana M, Yasuoka H, van den HF, Te BL, van Laar JM, Verbeet NL, Matucci-Cerinic M, Georgountzos A, Moutsopoulos HM: Mortality in systemic sclerosis: an international meta-analysis of individual patient data. Am J Med. 2005, 118: 2-10. 10.1016/j.amjmed.2004.04.031.
Steen VD, Medsger TA: Changes in causes of death in systemic sclerosis, 1972-2002. Ann Rheum Dis. 2007, 66: 940-944. 10.1136/ard.2006.066068.
Tyndall AJ, Bannert B, Vonk M, Airo P, Cozzi F, Carreira PE, Bancel DF, Allanore Y, Muller-Ladner U, Distler O, Iannone F, Pellerito R, Pileckyte M, Miniati I, Ananieva L, Gurman AB, Damjanov N, Mueller A, Valentini G, Riemekasten G, Tikly M, Hummers L, Henriques MJ, Caramaschi P, Scheja A, Rozman B, Ton E, Kumanovics G, Coleiro B, Feierl E, et al: Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis. 2010, 69: 1809-1815. 10.1136/ard.2009.114264.
Sharif R, Mayes MD, Tan FK, Gorlova OY, Hummers LK, Shah AA, Furst DE, Khanna D, Martin J, Bossini-Castillo L, Gonzalez EB, Ying J, Draeger HT, Agarwal SK, Reveille JD, Arnett FC, Wigley FM, Assassi S: IRF5 polymorphism predicts prognosis in patients with systemic sclerosis. Ann Rheum Dis. 2012, 71: 1197-1202. 10.1136/annrheumdis-2011-200901.
Fonseca C, Lindahl GE, Ponticos M, Sestini P, Renzoni EA, Holmes AM, Spagnolo P, Pantelidis P, Leoni P, McHugh N, Stock CJ, Shi-Wen X, Denton CP, Black CM, Welsh KI, du Bois RM, Abraham DJ: A polymorphism in the CTGF promoter region associated with systemic sclerosis. N Engl J Med. 2007, 357: 1210-1220. 10.1056/NEJMoa067655.
Hoshino K, Satoh T, Kawaguchi Y, Kuwana M: Association of hepatocyte growth factor promoter polymorphism with severity of interstitial lung disease in Japanese patients with systemic sclerosis. Arthritis Rheum. 2011, 63: 2465-2472. 10.1002/art.30415.
Dieude P, Bouaziz M, Guedj M, Riemekasten G, Airo P, Muller M, Cusi D, Matucci-Cerinic M, Melchers I, Koenig W, Salvi E, Wichmann HE, Cuomo G, Hachulla E, Diot E, Hunzelmann N, Caramaschi P, Mouthon L, Riccieri V, Distler J, Tarner I, Avouac J, Meyer O, Kahan A, Chiocchia G, Boileau C, Allanore Y: Evidence of the contribution of the X chromosome to systemic sclerosis susceptibility: association with the functional IRAK1 196Phe/532Ser haplotype. Arthritis Rheum. 2011, 63: 3979-3987. 10.1002/art.30640.
Dieude P, Dawidowicz K, Guedj M, Legrain Y, Wipff J, Hachulla E, Diot E, Sibilia J, Mouthon L, Cabane J, Amoura Z, Crakowski JL, Carpentier P, Avouac J, Meyer O, Kahan A, Boileau C, Allanore Y: Phenotype-haplotype correlation of IRF5 in systemic sclerosis: role of 2 haplotypes in disease severity. J Rheumatol. 2010, 37: 987-992. 10.3899/jrheum.091163.
Manetti M, Ibba-Manneschi L, Fatini C, Guiducci S, Cuomo G, Bonino C, Bazzichi L, Liakouli V, Giacomelli R, Abbate R, Bombardieri S, Montecucco C, Valentini G, Matucci-Cerinic M: Association of a functional polymorphism in the matrix metalloproteinase-12 promoter region with systemic sclerosis in an Italian population. J Rheumatol. 2010, 37: 1852-1857. 10.3899/jrheum.100237.
Sumita Y, Sugiura T, Kawaguchi Y, Baba S, Soejima M, Murakawa Y, Hara M, Kamatani N: Genetic polymorphisms in the surfactant proteins in systemic sclerosis in Japanese: T/T genotype at 1580 C/T (Thr131Ile) in the SP-B gene reduces the risk of interstitial lung disease. Rheumatology (Oxford). 2008, 47: 289-291.
Agarwal SK, Gourh P, Shete S, Paz G, Divecha D, Reveille JD, Assassi S, Tan FK, Mayes MD, Arnett FC: Association of interleukin 23 receptor polymorphisms with anti-topoisomerase-I positivity and pulmonary hypertension in systemic sclerosis. J Rheumatol. 2009, 36: 2715-2723. 10.3899/jrheum.090421.
Wipff J, Dieude P, Guedj M, Ruiz B, Riemekasten G, Cracowski JL, Matucci-Cerinic M, Melchers I, Humbert M, Hachulla E, Airo P, Diot E, Hunzelmann N, Caramaschi P, Sibilia J, Valentini G, Tiev K, Girerd B, Mouthon L, Riccieri V, Carpentier PH, Distler J, Amoura Z, Tarner I, Degano B, Avouac J, Meyer O, Kahan A, Boileau C, Allanore Y: Association of a KCNA5 gene polymorphism with systemic sclerosis-associated pulmonary arterial hypertension in the European Caucasian population. Arthritis Rheum. 2010, 62: 3093-3100. 10.1002/art.27607.
Broen JC, Bossini-Castillo L, van BL, Vonk MC, Knaapen H, Beretta L, Rueda B, Hesselstrand R, Herrick A, Worthington J, Hunzelman N, Denton CP, Fonseca C, Riemekasten G, Kiener HP, Scorza R, Simeon CP, Ortego-Centeno N, Gonzalez-Gay MA, Airo P, Coenen MJ, Martin J, Radstake TR, Spanish Systemic Sclerosis Group: A rare polymorphism in the gene for Toll-like receptor 2 is associated with systemic sclerosis phenotype and increases the production of inflammatory mediators. Arthritis Rheum. 2012, 64: 264-271. 10.1002/art.33325.
Dieude P, Guedj M, Wipff J, Ruiz B, Riemekasten G, Matucci-Cerinic M, Melchers I, Hachulla E, Airo P, Diot E, Hunzelmann N, Cabane J, Mouthon L, Cracowski JL, Riccieri V, Distler J, Meyer O, Kahan A, Boileau C, Allanore Y: Association of the TNFAIP3 rs5029939 variant with systemic sclerosis in the European Caucasian population. Ann Rheum Dis. 2010, 69: 1958-1964. 10.1136/ard.2009.127928.
Manetti M, Allanore Y, Revillod L, Fatini C, Guiducci S, Cuomo G, Bonino C, Riccieri V, Bazzichi L, Liakouli V, Cipriani P, Giacomelli R, Abbate R, Bombardieri S, Valesini G, Montecucco C, Valentini G, Ibba-Manneschi L, Matucci-Cerinic M: A genetic variation located in the promoter region of the UPAR (CD87) gene is associated with the vascular complications of systemic sclerosis. Arthritis Rheum. 2011, 63: 247-256.
Nguyen B, Mayes MD, Arnett FC, Del JD, Reveille JD, Gonzalez EB, Draeger HT, Perry M, Hendiani A, Anand KK, Assassi S: HLA-DRB1*0407 and *1304 are risk factors for scleroderma renal crisis. Arthritis Rheum. 2011, 63: 530-534.
Assassi S, Sharif R, Lasky RE, McNearney TA, Estrada YMR, Draeger HT, Nair DK, Fritzler MJ, Reveille JD, Arnett FC, Mayes MD, Genisos ST: Predictors of interstitial lung disease in early systemic sclerosis: a prospective longitudinal study of the GENISOS cohort. Arthritis Res Ther. 2010, 12: R166-10.1186/ar3125.
Daoussis D, Liossis SN, Tsamandas AC, Kalogeropoulou C, Kazantzi A, Sirinian C, Karampetsou M, Yiannopoulos G, Andonopoulos AP: Experience with rituximab in scleroderma: results from a 1-year, proof-of-principle study. Rheumatology (Oxford). 2010, 49: 271-280. 10.1093/rheumatology/kep093.
Johnson SR, Feldman BM, Pope JE, Tomlinson GA: Shifting our thinking about uncommon disease trials: the case of methotrexate in scleroderma. J Rheumatol. 2009, 36: 323-329.
van Schaik RH, van AM, de Fijter JW, Hartmann A, Schmidt J, Budde K, Kuypers D, Le MY, van der WM, Mamelok R, van GT: UGT1A9 -275T>A/-2152C>T polymorphisms correlate with low MPA exposure and acute rejection in MMF/tacrolimus-treated kidney transplant patients. Clin Pharmacol Ther. 2009, 86: 319-327. 10.1038/clpt.2009.83.
Baldelli S, Merlini S, Perico N, Nicastri A, Cortinovis M, Gotti E, Remuzzi G, Cattaneo D: C-440T/T-331C polymorphisms in the UGT1A9 gene affect the pharmacokinetics of mycophenolic acid in kidney transplantation. Pharmacogenomics. 2007, 8: 1127-1141. 10.2217/14622416.8.9.1127.
Robledo G, vila-Fajardo CL, Marquez A, Ortego-Centeno N, Callejas Rubio JL, de Ramon GE, Sanchez-Roman J, Garcia-Hernandez FJ, Rios-Fernandez R, Gonzalez-Escribano MF, Camps Garcia MT, Castillo Palma MJ, Ayala MD, Martin J: Association Between -174 Interleukin-6 Gene Polymorphism and Biological Response to Rituximab in Several Systemic Autoimmune Diseases. DNA Cell Biol. 2012, 31: 1486-91. 10.1089/dna.2012.1684.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1741-7015/11/9/prepub
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
SA, TR, MM, and JM were involved in drafting and revising of the manuscript and approved its final version. All authors meet the criteria for authorship.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Assassi, S., Radstake, T.R., Mayes, M.D. et al. Genetics of scleroderma: implications for personalized medicine?. BMC Med 11, 9 (2013). https://doi.org/10.1186/1741-7015-11-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1741-7015-11-9