
Abstract
Osteoarthritis (OA) is a degenerative joint disease characterized by progressive loss of articular cartilage, subchondral bone sclerosis, osteophyte formation, and synovial inflammation, causing substantial physical disability, impaired quality of life, and significant health care utilization. Traditionally, non-steroidal anti-inflammatory drugs (NSAIDs), including selective cyclooxygenase (COX)-2 inhibitors, have been used to treat pain and inflammation in OA. Besides its anti-inflammatory properties, evidence is accumulating that celecoxib, one of the selective COX-2 inhibitors, has additional disease-modifying effects. Celecoxib was shown to affect all structures involved in OA pathogenesis: cartilage, bone, and synovium. As well as COX-2 inhibition, evidence indicates that celecoxib also modulates COX-2-independent signal transduction pathways. These findings raise the question of whether celecoxib, and potentially other coxibs, is more than just an anti-inflammatory and analgesic drug. Can celecoxib be considered a disease-modifying osteoarthritic drug? In this review, these direct effects of celecoxib on cartilage, bone, and synoviocytes in OA treatment are discussed.
Similar content being viewed by others


Introduction
Osteoarthritis (OA) is the most common joint disorder in western countries, affecting over 70% of adults aged 55 to 70 years [1, 2]. It is characterized by progressive loss of articular cartilage, subchondral bone sclerosis, osteophyte formation, and synovial inflammation, causing substantial physical disability, impaired quality of life, and significant health care utilization. As OA incidence increases with age, OA will become a major health issue and socio-economic problem in the coming decades [3]. Historically, OA was seen as a degenerative disease caused solely by the 'wear and tear' process of ageing cartilage. Now it is recognized as a more dynamic, complex disease involving numerous factors affecting the whole joint [4]. Various risk factors for development of OA have been identified - age, sex, and genetic and bio-mechanical factors - contributing to degeneration of articular cartilage and changes in bone and synovium.
Traditionally, non-steroidal anti-inflammatory drugs (NSAIDs) have been used to treat pain and inflammation in OA [5]. The anti-inflammatory effects of NSAIDs are mainly due to their ability to inhibit cyclooxygenase (COX), impairing production of prostaglandins, which are important mediators of the inflammatory response and pain. COX enzymes metabolize arachidonic acid, forming prostaglandin H2, which is subsequently metabolized by prostaglandin E synthase into prostaglandin E2 (PGE2) [6]. Two isoforms of the COX enzyme exist: constitutively expressed homeostatic COX-1 found in most tissues, and COX-2, which is not expressed in normal healthy tissues and cells but is induced by various pro-inflammatory, catabolic, and stress mediators, such as cytokines, growth factors, and increased loading [7]. Beneficial effects of NSAIDs are thought to be mediated by COX-2 inhibition, whereas unwanted gastrointestinal effects are caused by inhibitory effects on COX-1 [8]. This led to the development of selective COX-2 inhibitors. Celecoxib (SC-58635; 4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfon-amide) was the first US Food and Drug Administration-approved selective COX-2 inhibitor and is now widely used in OA treatment [9].
Besides its anti-inflammatory properties, evidence is accumulating that celecoxib has additional disease modifying effects. Celecoxib has been shown to affect all structures involved in OA pathogenesis: cartilage, bone, and synovium [10–12]. As well as COX-2 inhibition, evidence indicates that celecoxib also modulates COX-2-independent signal transduction pathways [13]. These findings raise the question of whether celecoxib is more than just an anti-inflammatory and analgesic drug - does celecoxib also slow down OA disease progression and can it be viewed as a disease-modifying osteoarthritic drug? In this review, the direct effects of celecoxib on cartilage, bone, and synoviocytes in OA treatment are discussed.
It is important to note that some of the effects described may be related to the coxib class of drugs as a whole, some may be specific to celecoxib, and some may result from a general COX-inhibiting effect. This review does not intend to distinguish between these but focuses on the properties of celecoxib specifically. Only when celecoxib has been compared to other treatments have such comparisons been taken into account. Moreover, this review does not discuss the issue of side effects and clinical efficacy of celecoxib, but focuses on its potential tissue structure-modifying, mostly chondroprotective, effects.
Methods
Two electronic databases were searched for relevant publications: PubMed (1990 to March 2010) and EMBASE (1990 to March 2010). Key words used were: celecoxib/Celebrex/SC-58635, osteoarthritis/arthrosis/OA, cartilage/chondrocytes, synovium/synovial/synoviocytes, and bone. Celecoxib studies regarding its effects on cartilage, bone, and synovium were selected by screening title and abstract. Publications not written in English or not containing original data were excluded. Reviews concerning subjects like the cost-effectiveness and cardiovascular/gastrointestinal side effects of celecoxib and the use of celecoxib in cancer treatment have been published and are therefore not covered in this review.
Celecoxib: direct effects on cartilage
In OA, chondrocytes fail to maintain the equilibrium between synthesis and degradation of the extracellular matrix, resulting in progressive disruption of the structural integrity of cartilage. Initially, chondrocytes compensate for the enhanced catabolic processes by increasing synthesis of collagens and proteoglycans. However, as OA progresses, the increasing catabolic enzyme activity can no longer be counterbalanced [14]. IL-1β and TNF-α play key roles in the destructive process by stimulating expression and release of proteases, such as collagenases and aggrecanases, including matrix metalloproteinases (MMPs) and a disintegrin and metalloproteinase with trombospondin repeats (ADAMTS), which degrade collagen and aggrecan. These pro-inflammatory cytokines stimulate synthesis and release of nitric oxide (NO) and PGE2 [15]. Chondrocytes from OA patients show elevated COX-2 expression, and its product PGE2 is increased in OA cartilage [16]. The function of PGE2 in OA is not exactly clear as it has both catabolic and anabolic effects in cartilage [17, 18]. NSAIDs could potentially affect cartilage through their inhibition of PGE2 production.
Proteoglycan turnover
Celecoxib dose-dependently inhibits glycosaminoglycan release and stimulates proteoglycan synthesis in healthy human articular cartilage explants when exposed to peripheral blood mononuclear cells from rheumatoid arthritis patients or IL-1β and TNF-α [19]. The fact that the decreased proteoglycan synthesis induced by IL-1β and TNF-α is reversed by celecoxib indicates that this drug can also exert its effects directly on activated cartilage. Furthermore, in OA cartilage explants, celecoxib stimulated proteoglycan synthesis and retention of newly formed proteoglycans [20–22]. The non-selective COX inhibitors diclofenac and naproxen did not affect proteoglycan turnover in OA cartilage, and indomethacin and an experimental COX-1 selective inhibitor (SC-560) had adverse effects [20, 21]. This difference in NSAID effects supports COX-2 involvement in catabolic activity regulation in cartilage, whereas COX-1 activity might have a more physiological role in chondrocytes.
No effect of celecoxib on proteoglycan turnover was observed in healthy cartilage [19, 22]. This is in contrast to the protective in vitro effect of celecoxib on end-stage OA cartilage obtained at joint replacement surgery. For the treatment of OA in clinical practice, it would be beneficial if celecoxib could influence proteoglycan turnover in earlier stages of disease. It was shown that in both degenerated (pre-clinical) and late-stage OA cartilage, celecoxib not only stimulated proteoglycan synthesis and retention of newly formed proteoglycans, but also had favorable effects on proteoglycan content. Importantly, proteoglycan content in degenerated cartilage normalized in vitro during celecoxib treatment, suggesting celecoxib treatment in the early stages of OA could slow down or even reverse the destructive process [22].
Whereas the in vitro effects of celecoxib on OA cartilage are beneficial, results obtained with isolated chondrocytes are not consistent. In a mechanically stretched monolayer of chondrocytes, celecoxib had a positive effect on aggrecan expression and reduced the release of chondroitin sulfate [23]. In contrast, celecoxib had no positive effect on proteoglycan turnover of osteoarthritic chondrocytes cultured in alginate beads [24], of a monolayer of chondrocytes [25], nor in an in vitro model of post-traumatic OA [26]. This variation in the effects of celecoxib could potentially be due to differences in chondrocyte culture models, whereas cartilage explants probably better reflect the in vivo situation.
Prostaglandin E2-induced catabolism
A possible way in which celecoxib exerts its effect on proteoglycan turnover is inhibition of PGE2 production. PGE2 is highly expressed in OA cartilage and studies indicate a pivotal role for PGE2 in OA cartilage metabolism [27]. Expression of PGE2 and COX-2 in OA cartilage is strongly inhibited by celecoxib [10, 21, 22, 28, 29]. PGE2 enhances IL-1β-/TNF-α-induced proteoglycan release, resulting in decreased proteoglycan content in cartilage explants [28]. The effect of PGE2 on the synthesis of proteoglycans remains controversial; in OA cartilage, proteoglycan synthesis is inhibited by PGE2 [17], whereas PGE2 does not affect proteoglycan synthesis rate in healthy cartilage [28]. This discrepancy could be due to differences in expression levels of individual members of the EP receptor family (EP1 to EP4) through which PGE2 exerts its effects. EP4 has been implicated in mediating catabolic effects because it is highly expressed in OA cartilage [17]. IL-1-induced expression of EP4 in cultured OA chondrocytes is decreased by celecoxib [29], but not consistently [17]. The overall negative effect of PGE2 on proteoglycan turnover in cartilage might be mediated through the EP4 receptor (Figure 1).

Schematic representation of the in vitro effects of celecoxib (CBX) on cartilage degeneration. Cyclooxygenase (COX)-2 expression in the chondrocyte is induced by inflammatory mediators such as IL-1β and TNF-α (1). Subsequently, the prostanoid receptor EP4 is up-regulated via a COX-2-dependent mechanism (2). Increased COX-2 activity results in large concentrations of prostaglandin E2 (PGE2) (3). PGE2 exerts its effects through the prostanoid receptor EP4 (4), resulting in the increased expression of matrix metalloproteinases (MMPs) and a disintegrin and metalloproteinase with thrombospondin repeats (ADAMTS)-5. Furthermore, PGE2 augments the release of newly formed proteoglycans from cartilage and reduces the synthesis of proteoglycans (5). IL-1β and TNF-α also activate the transcription factors NF-κB and JNK (6), which stimulate the expression of inducible nitric oxide synthase (iNOS) (7), resulting in the formation of nitric oxide (NO) (8). NO has a potential role in inducing chondrocyte apoptosis, inhibiting proteoglycan synthesis and stimulating MMP activity (9). Together, the effects of NO and PGE2 result in cartilage degeneration. Celecoxib prevents the negative effects of PGE2 and NO on cartilage destruction by inhibiting both COX-2 and NF-κB/JNK, thereby potentially slowing cartilage degradation in osteoarthritis.
PGE2 inhibits collagen synthesis and stimulates expression of MMP and ADAMTS-5, proteolytic enzymes involved in the degradation of collagens and proteoglycans [17, 30, 31]. Theoretically, celecoxib could also prevent cartilage destruction by inhibiting induction of MMP expression in OA cartilage. Both inhibitory and stimulatory effects of celecoxib on IL-1-induced expression of MMP-13 in OA chondrocytes have been reported [10, 17]. Also, there is no agreement on the effect of celecoxib on MMP-1 expression in cartilage [10, 25, 32]. Celecoxib reverses IL-1β-induced ADAMTS-5 expression in OA cartilage explants [17]. As such, it could prevent enhanced proteoglycan turnover in OA by affecting both MMP and ADAMTS-5 expression. But our understanding of the influence of celecoxib on PGE2-induced cartilage catabolism is clearly far from complete and it would be worthwhile to explore this role in more detail.
Nitric oxide, NF-κB, and chondrocyte apoptosis
NO plays an important role in cartilage destruction in OA - for example, by inhibiting matrix synthesis, activating MMPs, and inducing chondrocyte apoptosis [33–35]. Because NO is an attractive target in OA treatment, several studies have addressed the question of whether celecoxib influences NO production, although little agreement has been reached. Several studies found inhibitory effects of celecoxib on NO production in chondrocytes [25, 32, 36], whereas others did not [28, 37]. These contradictory effects are potentially due to differences in culture models, treatment duration, and celecoxib concentration used.
In articular chondrocytes, NO production is regulated by NF-κB, JunNH2-terminal kinase (JNK) and p38 [32, 38]. Celecoxib was shown to suppress NO production by inactivating JNK and NF-κB [32]. An inhibitory effect of celecoxib on NF-κB signaling in OA chondrocytes was reported previously [10]. NF-κB has an essential role in OA pathogenesis, being involved in cytokine stimulation, MMP and ADAMTS expression, and diminished secretion of extracellular matrix proteins by chondrocytes. Inhibition of NF-κB could potentially be beneficial in OA treatment. Interestingly, it was reported that celecoxib reduces expression of IL-1 [37] and IL-6 [24], both inflammatory cytokines involved in OA pathogenesis [39]. It is currently unknown how celecoxib mediates its effects on cytokine expression and NF-κB activity.
Celecoxib induced apoptosis in a dose-dependent manner in chondrocytes derived from cartilage from patients with OA [25], although reduced apoptosis via COX inhibition by celecoxib has also been reported [26].
In general, celecoxib has favorable effects on cartilage destruction in vitro, thereby theoretically slowing down disease progress in vivo (Figure 1).
Celecoxib: effects on synoviocytes
Inhibition of signal transduction and pro-inflammatory mediators
Although originally viewed as a non-inflammatory arthropathy, a pivotal role of synovial inflammation in OA progression is now recognized. Imaging studies have shown synovium changes in early and late OA [40]. Histologically, synovium from OA patients shows hyperplasia, increased lining layer thickness, blood vessel formation and mononuclear cell infiltration, mainly consisting of macrophage like cells. IL-1β and TNF-α levels are increased in OA synoviocytes, potentially contributing to disease progression by activating chondrocytes and synovial fibroblasts [41, 42]. Enhanced PGE2 and COX-2 expression in synovial fluid and synovial membrane have been observed [43, 44]. Several effects of celecoxib on synovium, with a focus on fibroblasts, have been described. Celecoxib reversed IL-1β-induced PGE2 and COX-2 protein expression in synovial fibroblasts. Furthermore, celecoxib inhibited IL-1β-induced activation of NF-κB in synovial fibroblasts from OA patients [10]. NF-κB induces expression of large numbers of inflammatory mediators and plays a major role in the initiation and maintenance of synovitis, synovial hyperplasia, and inhibition of synovial apoptosis in rheumatoid arthritis. Although less is known concerning the function of NF-κB in osteoarthritic synovium, it is clear that celecoxib could reduce expression of various inflammatory mediators by down-regulation of NF-κB [45].
Proteolysis
Among the downstream factors of NF-κB are MMPs, which play a crucial role in cartilage degradation in OA. Both MMP-1 and MMP-13 levels are enhanced in OA; MMP-1 is predominantly released by synovial cells, and MMP-13 is highly expressed by chondrocytes [46]. MMP-2 and MMP-9 are also elevated in the osteoarthritic joint. MMP-2 expression is regulated by COX-2. Several NSAIDs, including celecoxib, inhibit MMP-2 secretion in OA synovial fibroblast cultures [47]. Furthermore, celecoxib can decrease the expression of MMP-9 and urokinase-type plasminogen activator (u-PA) and its inhibitor PAI [47]. Alterations in u-PA and PAI expression have been found in osteoarthritic tissue and contribute to a disturbed proteolytic balance [48].
It was shown that celecoxib, but no other selective COX-2 inhibitors, enhances MMP-1 and MMP-13 protein expression in IL-1β-stimulated synoviocytes [10, 49]. This observation does not corroborate the inhibitory effect of celecoxib on MMP-1 expression in rheumatoid arthritis synoviocytes [50]. This discrepancy could be due to different concentrations used, celecoxib being stimulatory at low concentrations (0.5 to 1 μM) and inhibitory at higher concentrations (5 to 10 μM). Evidently, a stimulatory effect of celecoxib on synovial MMP-1 and MMP-13 expression could be detrimental in OA treatment [10]. In conclusion, celecoxib influences the balance of proteolytic enzymes in OA synovium, and although this appears to be generally beneficial (reducing expression of MMP-2, MMP-9, and uPA), adverse effects have been reported as well (increasing expression of MMP-1 and MMP-13).
Apoptosis
Recently, it was shown that celecoxib dose-dependently inhibits proliferation and induces apoptosis in synovial fibroblasts obtained from OA patients [51, 52]. This is in agreement with findings in rheumatoid arthritis [53–55]. Remarkably, various other COX-2 selective inhibitors, including nimesulide and rofecoxib, did not induce apoptosis of synovial fibroblasts, indicating that celecoxib stimulates apoptosis in a COX-2-independent manner [51]. In cancer cells celecoxib has been shown to modulate apoptosis pathways by inhibiting anti-apoptotic proteins, elevating Ca2+ concentration and altering NF-kB signaling (reviewed in [56]). Although the exact pro-apoptotic mechanism of celecoxib in synovial tissue remains to be established, it is evident that anti-proliferative and pro-apoptotic effects of celecoxib on synovium are beneficial in reducing synovial hyperplasia and potentially slow down synovitis-mediated OA disease progress.
Taken together, celecoxib modulates several pathogenic mechanisms of synovial cells that are not always affected by other NSAIDs, suggesting that celecoxib may have additional, COX-2-independent value in the treatment of OA (Figure 2).

Schematic representation of the major actions of celecoxib (CBX) in synovium. In osteoarthritic synovium, increased levels of IL-1β and TNF-α stimulate the expression of cyclooxygenase (COX)-2 and the ensuing production of prostaglandin E2 (PGE2). PGE2 augments the expression of proteolytic enzymes, including matrix metalloproteinases (MMPs) and urokinase-type plasminogen activator (uPA), thereby contributing to the destruction of the articular joint. Celecoxib has a beneficial effect on synovium through its inhibition of COX-2 (1). In addition, celecoxib inhibits IL-1β-induced activation of NF-κB, and hence diminishes the expression of various inflammatory mediators (2). A third potential favorable effect of celecoxib on synovium is its induction of apoptosis in synovial fibroblasts (3). This would theoretically contribute to decreased synovial hyperplasia and reduced expression of inflammatory mediators. PAI, plasminogen activator inhibitor.
Celecoxib: effects on subchondral bone
Subchondral bone in osteoarthritis
Subchondral bone sclerosis and osteophyte formation are radiographic hallmarks of end-stage OA. Several studies suggest that bone remodeling in OA is biphasic: an early decrease in trabecular bone formation, followed by an increase in subchondral bone density and stiffness [57, 58]. The initial thinning of the subchondral plate coincides with changes in articular cartilage, suggesting a pivotal role for the cartilage and subchondral bone interaction in OA progression. In established OA, the increased subchondral bone stiffness probably contributes to further cartilage degeneration [59].
Osteoclastogenesis
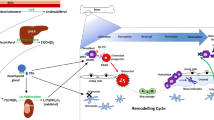
Osteoclasts play a pivotal role in the destruction of subchondral bone [4, 14, 59]. Osteoclastogenesis and activation of mature osteoclasts are critically regulated by the receptor activator of NF-κB ligand (RANKL). RANKL mediates its function by binding to its cell-surface receptor RANK on osteoclast precursor cells and osteoclasts, thus stimulating differentiation and activation of osteoclasts. It is mainly expressed by osteoblasts and stromal cells, where expression of RANKL is COX-2-dependent [60]. During inflammation RANKL is also produced by T lymphocytes and fibroblast-like synoviocytes. Osteoprotegerin (OPG), a soluble decoy receptor for RANKL, can prevent the biological effects of RANKL, and the ratio between OPG and RANKL determines whether the balance is in favor of bone resorption or bone formation [61, 62]. Interestingly, two osteoblast subpopulations were identified in OA, one with a low OPG/RANKL ratio that favors bone resorption, and one with a high OPG/RANKL ratio that promotes bone formation [61, 63]. Inhibition of COX-2 by NSAIDs diminishes RANKL production by osteoblasts, and since RANKL is an important inducer of osteoclastogenesis, celecoxib inhibited osteoclast differentiation in co-cultures of osteoblasts and bone marrow-derived cells [12, 64]. Besides affecting osteoclastogenesis indirectly through its effect on osteoblasts, celecoxib also directly influenced osteoclast precursor cells by inhibiting COX-2 expression. Adding celecoxib to bone marrow-derived monocyte/macrophage cells, in the absence of stromal cells, suppresses RANKL-induced osteoclast differentiation [65, 66]. This celecoxib effect was reversed by PGE2, indicating that RANKL-induced COX-2 and PGE2 expression in osteoclast precursors is critically involved in osteoclastogenesis [65] (Figure 3).

Schematic representation of the main effects of celecoxib (CBX) on subchondral bone. IL-1β induces the expression of cyclooxygenase (COX)-2 in chondrocytes (1) and osteoblasts (2), leading to production of receptor activator of NF-κB ligand (RANKL). RANKL stimulates the differentiation of osteoclast precursor cells into quiescent osteoclasts (3). Furthermore, it induces the expression of COX-2 and prostaglandin E2 (PGE2) in quiescent osteoclasts, and subsequently PGE2 activates osteoclasts in both an autocrine and paracrine manner (4). Celecoxib inhibits the COX-2-dependent RANKL production by chondrocytes and osteoblasts, thereby avoiding osteoclastogenesis and osteoblast activity. Furthermore, celecoxib directly inhibits the differentiation of precursor cells, independent of RANKL production by stromal cells. Celecoxib can also directly affect the osteoclasts themselves by decreasing RANKL-induced PGE2 expression and inhibiting carbonic anhydrase. Decreased carbonic anhydrase activity will diminish acidification of the resorption pit, and hence decrease osteoclast activity (5).
Osteoclast activity
Besides inhibiting osteoclast differentiation, celecoxib is able to almost completely inhibit the activity of human osteoclasts [66]. Slightly lesser effects were observed with indomethacin, and no effects were seen with a selective COX-1 inhibitor, suggesting a COX-2-dependent pathway is involved [66]. However, other mechanisms might be involved in inhibiting osteoclast activity as well. Celecoxib, as well as other sulfonamide-type COX-2 inhibitors, contain an aryl sulfonamide moiety that inhibits carbonic anhydrase II [67]. Abundantly expressed on the inner surface of osteoclasts, carbonic anhydrase II catalyzes conversion of CO2 and H2O into bicarbonate and H+. Acidification in the resorption pit is required for dissolution of the inorganic matrix of bone [68]. Treatment with celecoxib reduced carbonic anhydrase activity and thereby inhibited osteoclast activity, an effect not observed for COX-inhibitors without this sulfonamide moiety [12].
Osteoprotegerin/RANKL in chondrocytes
Recently, it was found that human chondrocytes express OPG, RANKL and RANK [61, 69]. Interestingly, the OPG/RANKL ratio is significantly lower in OA chondrocytes compared to healthy chondrocytes [70]. This shift in OPG/RANKL ratio is mediated by PGE2 [69, 71], and inhibition of PGE2 production by celecoxib resulted in a higher OPG/RANKL ratio [71, 72]. It was shown that RANKL produced by chondrocytes can stimulate osteoclastogenesis [73, 74] and, furthermore, as a chemoattractant for peripheral blood monocytes, it could attract osteoclast precursor cells to the joint [75]. Inhibition of chondrocyte RANKL expression by celecoxib might thus prevent subchondral bone loss (Figure 3).
Celecoxib: effects in vivo
Cartilage
In vitro experiments have shown a cartilage-sparing effect of celecoxib in OA cartilage; however, in vivo data, from either human or animals, are scarce. Contrary to its positive effects on cartilage degeneration in vitro, no chondroprotective effect of celecoxib in the canine groove model of OA was observed [76]. Although PGE2 levels in the joint were inhibited, celecoxib did not improve cartilage histopathology or proteoglycan turnover. This lack of chondroprotective effect might have been due to increased loading of the joint in the celecoxib-treated group compared to the placebo-treated group, where no analgesics were given [76]. Conversely, celecoxib was shown to reduce cartilage damage in collagen-induced osteoarthritis in rabbits; histopathological evaluation showed less cartilage erosion, reduced cartilage fibrillation and decreased loss of chondrocytes. Proteoglycan content, determined by Safranin-O staining intensity, was higher than in the placebo-treated group [77]. Next to the direct effects of celecoxib, the anti-inflammatory effects of celecoxib may have caused this chondroprotective effect as the model depends on inflammation and the number of inflammatory cells and the PGE2 concentration in synovial fluid was significantly reduced by celecoxib.
Few studies have described the in vivo effects of celecoxib on cartilage destruction in OA patients [37, 44, 78–80]. However, these studies generally have limitations with respect to their small size and short duration. A way to study drug effects is to treat patients with severe knee OA waiting for joint replacement surgery and analyze the cartilage ex vivo. In this manner, a beneficial effect of celecoxib on cartilage degradation after 4 weeks of treatment was observed [78]. Although no differences in the histopathological Mankin score were observed, proteoglycan synthesis rate and retention of newly formed proteoglycans was significantly increased in celecoxib-treated OA patients compared to indomethacin-treated or untreated patients. The expression of key players in the destructive process, NO and PGE2, was inhibited by both celecoxib and indomethacin [78]. Hence, differences in cartilage proteoglycan turnover between celecoxib- and indomethacin-treated patients could result from specific effects of indomethacin-induced COX-1 inhibition on cartilage [20, 21], or from COX-2-independent actions of celecoxib [13]. Using a similar approach, long-term (3 months) effects of celecoxib and aceclofenac were studied in OA patients [37]. It was demonstrated that expression of COX-2, microsomal prostaglandin E synthase-1 (mPGES-1) and inducible NO synthase, an enzyme involved in NO generation, was strongly reduced in both celecoxib- and aceclofenac-treated patients. Only celecoxib was shown to inhibit expression of the PGE2 receptors EP2 and EP4, as well as TNF-α and IL-1β, in articular cartilage. A positive correlation exists between TNF-α/IL-1β levels and cartilage damage [81], suggesting a chondroprotective effect of celecoxib in vivo.
The effects of celecoxib treatment on disease progression are more ambiguous [79, 80, 82]. In an observational study, conventional NSAID use was associated with enhanced cartilage destruction compared to selective COX-2 inhibitors. Furthermore, the COX-2 inhibitors rofecoxib and celecoxib showed beneficial effects on tibial cartilage defects in knee OA compared to no medication [82]. Recently, the effect of celecoxib treatment (200 mg daily, 12 months) on cartilage volume loss was studied compared to a historical cohort of patients receiving standard care [79]. Using quantitative magnetic resonance imaging, no protective celecoxib effect on knee cartilage was found. Only one randomized controlled trial has addressed the effects of celecoxib on cartilage degeneration [80]. Patients who met radio-graphic criteria grade 2 and 3 (Kellgren and Lawrence) were blinded and given celecoxib, chondroitin sulfate, glucosamine or placebo. Unexpectedly, no differences in joint space narrowing (measured radiographically) or disease progression between celecoxib- and placebo-treated groups were observed after 2 years follow-up [80]. Less than anticipated loss of joint space width in the placebo-treated group hampered the study and prevented a strong conclusion. Moreover, the results found in these studies were obtained in an un-controlled trial set-up and, as such, could be affected by the selection of patients. Also, the numbers of patients used in most studies is rather limited.
Figure 4 summarizes the suggested in vivo effects of celecoxib. The beneficial in vitro effects and the some-what controversial in vivo effects on cartilage, mostly based upon weak evidence, clearly indicate the requirement for properly designed randomized controlled trials on the potential disease-modifying osteoarthritic drug effects of celecoxib.

Summary of the effects of celecoxib on osteoarthritis in vivo (animal and human). Celecoxib has different in vivo effects in the different tissues of the osteoarthritic knee joint. In cartilage celecoxib has a positive effect on proteoglycan content, as well as an inhibiting effect on several mediators of inflammatory and cartilage destruction processes. In synovial tissue celecoxib shows inhibiting effects on cytokines and the number of synovial macrophages. Celecoxib shows a positive effect on trabecular bone volume and a negative effect on bone resorption and the number of osteoclasts in subchondral bone. COX, cyclooxygenase; iNOS, inducible nitric oxide synthase; MMP, matrix metalloproteinase; NO, nitric oxide; PGE2, prostaglandin E2.
Synovium
Celecoxib has been shown to reduce synovitis, leukocyte infiltration and synovial hyperplasia in different arthritis animal models [83–85]. In the synovium of severe knee OA patients, inhibitory effects of celecoxib on IL-1β and TNF-α expression have been demonstrated [44, 78]. Furthermore, celecoxib reduced IL-6 concentrations in the synovial fluid of patients with moderately severe OA after 2 weeks of treatment [86]. Interestingly, aceclofenac and indomethacin had no or only moderate effects on cytokine expression in these studies [44, 78].
Reduction of pro-inflammatory cytokines in synovial fluid by celecoxib could be the result of decreased production by chondrocytes, as has been shown in vitro [24]. However, synovial macrophages are also an important source of pro-inflammatory cytokines [42]. Ex vivo analysis of OA synovium after in vivo celecoxib treatment showed a significant reduction in synovial macrophage numbers, which was not observed for aceclofenac [44]. This macrophage depletion might be due to enhanced apoptosis in response to celecoxib, which has a pro-apoptotic effect on synoviocytes and macrophages [51, 53, 55]. Decreasing macrophage numbers would result in lower pro-inflammatory mediator levels in synovial fluid. Only one study has addressed the influence of celecoxib on MMP activity in synovial tissue; despite controversial results on MMP activity in synoviocytes in vitro, no celecoxib effect on MMP activity was demonstrated in vivo [78].
In conclusion, under certain conditions pro-inflammatory cytokines play a crucial role in OA pathogenesis by inhibiting proteoglycan synthesis, inducing chondrocyte apoptosis and activating other cells. Preventing enhanced production of these inflammatory mediators by celecoxib will likely slow disease processes. Several lines of evidence indicate that synovial changes can be among the first to occur in OA (reviewed in [87]), suggesting early treatment could slow or maybe prevent joint damage. As little research has focused on the effects of celecoxib on synovial tissue, further research should elucidate the effects of celecoxib in disease progression.
Bone
Various studies have shown a beneficial effect of celecoxib on bone in vivo [12, 88–90]. Celecoxib, but not other NSAIDs, reduced bone mineral density loss [12, 88, 90] and enhanced trabecular bone volume in adjuvant- and collagen-induced arthritis in rats [88–90]. The increased trabecular bone volume correlated with reduced serum type I collagen C-telopeptide, a bone resorption marker representing osteoclast activity [12, 88], and other bone resorption parameters [89]. Whereas celecoxib did not affect bone formation, it suppressed osteoclast numbers in tibia of arthritic animals [89, 90]. These celecoxib effects were partly mediated by RANKL, as celecoxib decreased expression of RANKL in synovial tissue, bone marrow cells and cartilage in vivo [71, 89]. As shown in vitro, celecoxib inhibited both osteoclastogenesis and osteoclast activation, thereby directly diminishing bone destruction.
Despite celecoxib being used for treatment of OA for many years, no effects of it on serum markers of bone resorption and formation or on structural changes in bone have been reported. As celecoxib has beneficial effects on bone resorption in vitro and in vivo in animal models, it would be interesting to explore these effects on bone metabolism in OA patients in more detail.
Conclusion
Despite celecoxib being approved for OA treatment for over a decade, few studies have addressed the disease-modifying properties of this selective COX-2 inhibitor, specifically in vivo. This review does not address the clinical risk and side effects related to the clinical benefits of celecoxib but focuses on the disease-modifying properties of this compound. However, the increased risk of myocardial infarction and worsening of high blood pressure can not be ignored when prescribing celecoxib. These issues have been extensively described in other reviews and are still under discussion at present. Also, it is not the intention of this review to compare in a systematic way the disease-modifying effects of celecoxib with other coxibs and conventional NSAIDs. As such, all effects described might be partly class-specific and partly celecoxib-specific. Nonetheless, celecoxib's chondroprotective effects - prevention of synovial hyperplasia, and inhibition of bone destruction in vitro and in vivo specifically in animal models - suggest that it and maybe other coxibs could potentially slow OA disease progression in humans. At present, however, good quality randomized controlled trials examining the disease-modifying effects of celecoxib are lacking. Future studies should elucidate the actual role of celecoxib and other selective coxibs as disease-modifying osteoarthritic drugs.
Abbreviations
- ADAMTS:
-
a disintegrin and metalloproteinase with thrombospondin repeats
- COX:
-
cyclooxygenase
- IL:
-
interleukin
- JNK:
-
JunNH2-terminal kinase
- MMP:
-
matrix metalloproteinase
- NF:
-
nuclear factor
- NO:
-
nitric oxide
- NSAID:
-
non-steroidal anti-inflammatory drug
- OA:
-
osteoarthritis
- OPG:
-
osteoprotegerin
- PAI:
-
plasminogen activator inhibitor
- PGE2:
-
prostaglandin E2
- RANK(L):
-
receptor activator of NF-κB (ligand)
- TNF:
-
tumor necrosis factor
- u-PA:
-
urokinase-type plasminogen activator.
References
Bijlsma JW, Knahr K: Strategies for the prevention and management of osteoarthritis of the hip and knee. Best Pract Res Clin Rheumatol. 2007, 21: 59-76. 10.1016/j.berh.2006.08.013.
Dillon CF, Rasch EK, Gu Q, Hirsch R: Prevalence of knee osteoarthritis in the United States: arthritis data from the Third National Health and Nutrition Examination Survey 1991-94. J Rheumatol. 2006, 33: 2271-2279.
Sangha O: Epidemiology of rheumatic diseases. Rheumatology (Oxford). 2000, 39 (Suppl 2): 3-12.
Clouet J, Vinatier C, Merceron C, Pot-vaucel M, Maugars Y, Weiss P, Grimandi G, Guicheux J: From osteoarthritis treatments to future regenerative therapies for cartilage. Drug Discov Today. 2009, 14: 913-925. 10.1016/j.drudis.2009.07.012.
Crofford LJ, Lipsky PE, Brooks P, Abramson SB, Simon LS, van de Putte LB: Basic biology and clinical application of specific cyclooxygenase-2 inhibitors. Arthritis Rheum. 2000, 43: 4-13. 10.1002/1529-0131(200001)43:1<4::AID-ANR2>3.0.CO;2-V.
Vane JR: Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol. 1971, 231: 232-235.
Martel-Pelletier J, Pelletier JP, Fahmi H: Cyclooxygenase-2 and prostaglandins in articular tissues. Semin Arthritis Rheum. 2003, 33: 155-167. 10.1016/S0049-0172(03)00134-3.
Hawkey CJ: COX-2 inhibitors. Lancet. 1999, 353: 307-314. 10.1016/S0140-6736(98)12154-2.
Bingham CO: Development and clinical application of COX-2-selective inhibitors for the treatment of osteoarthritis and rheumatoid arthritis. Cleve Clin J Med. 2002, 69 (Suppl 1): SI5-12.
Alvarez-Soria MA, Herrero-Beaumont G, Sanchez-Pernaute O, Bellido M, Largo R: Diacerein has a weak effect on the catabolic pathway of human osteoarthritis synovial fibroblast--comparison to its effects on osteoarthritic chondrocytes. Rheumatology (Oxford). 2008, 47: 627-633. 10.1093/rheumatology/ken116.
Mastbergen SC, Bijlsma JW, Lafeber FP: Selective COX-2 inhibition is favorable to human early and late-stage osteoarthritic cartilage: a human in vitro study. Osteoarthritis Cartilage. 2005, 13: 519-526. 10.1016/j.joca.2005.02.004.
Katagiri M, Ogasawara T, Hoshi K, Chikazu D, Kimoto A, Noguchi M, Sasamata M, Harada S, Akama H, Tazaki H, Chung UI, Takato T, Nakamura K, Kawaguchi H: Suppression of adjuvant-induced arthritic bone destruction by cyclooxygenase-2 selective agents with and without inhibitory potency against carbonic anhydrase II. J Bone Miner Res. 2006, 21: 219-227.
Tegeder I, Pfeilschifter J, Geisslinger G: Cyclooxygenase-independent actions of cyclooxygenase inhibitors. FASEB J. 2001, 15: 2057-2072. 10.1096/fj.01-0390rev.
Abramson SB, Attur M: Developments in the scientific understanding of osteoarthritis. Arthritis Res Ther. 2009, 11: 227-10.1186/ar2655.
Goldring MB: Osteoarthritis and cartilage: the role of cytokines. Curr Rheumatol Rep. 2000, 2: 459-465. 10.1007/s11926-000-0021-y.
Hardy MM, Seibert K, Manning PT, Currie MG, Woerner BM, Edwards D, Koki A, Tripp CS: Cyclooxygenase 2-dependent prostaglandin E2 modulates cartilage proteoglycan degradation in human osteoarthritis explants. Arthritis Rheum. 2002, 46: 1789-1083. 10.1002/art.10356.
Attur M, Al-Mussawir HE, Patel J, Kitay A, Dave M, Palmer G, Pillinger MH, Abramson SB: Prostaglandin E2 exerts catabolic effects in osteoarthritis cartilage: evidence for signaling via the EP4 receptor. J Immunol. 2008, 181: 5082-5088.
Tchetina EV, Di Battista JA, Zukor DJ, Antoniou J, Poole AR: Prostaglandin PGE2 at very low concentrations suppresses collagen cleavage in cultured human osteoarthritic articular cartilage: this involves a decrease in expression of pro-inflammatory genes, collagenases and COL10A1, a gene linked to chondrocyte hypertrophy. Arthritis Res Ther. 2007, 9: R75-10.1186/ar2273.
Mastbergen SC, Lafeber FP, Bijlsma JW: Selective COX-2 inhibition prevents pro-inflammatory cytokine-induced cartilage damage. Rheumatology (Oxford). 2002, 41: 801-808. 10.1093/rheumatology/41.7.801.
El Hajjaji H, Marcelis A, Devogelaer JP, Manicourt DH: Celecoxib has a positive effect on the overall metabolism of hyaluronan and proteoglycans in human osteoarthritic cartilage. J Rheumatol. 2003, 30: 2444-2451.
Mastbergen SC, Jansen NW, Bijlsma JW, Lafeber FP: Differential direct effects of cyclo-oxygenase-1/2 inhibition on proteoglycan turnover of human osteoarthritic cartilage: an in vitro study. Arthritis Res Ther. 2006, 8: R2-10.1186/ar1846.
Mastbergen SC, Bijlsma JW, Lafeber FP: Selective COX-2 inhibition is favorable to human early and late-stage osteoarthritic cartilage: a human in vitro study. Osteoarthritis Cartilage. 2005, 13: 519-526. 10.1016/j.joca.2005.02.004.
Iimoto S, Watanabe S, Takahashi T, Shimizu A, Yamamoto H: The influence of Celecoxib on matrix synthesis by chondrocytes under mechanical stress in vitro. IntJ Mol Med. 2005, 16: 1083-1088.
Sanchez C, Mateus MM, Defresne MP, Crielaard JM, Reginster JY, Henrotin YE: Metabolism of human articular chondrocytes cultured in alginate beads. Longterm effects of interleukin 1beta and nonsteroidal anti-inflammatory drugs. J Rheumatol. 2002, 29: 772-782.
Nakamura H, Masuko K, Yudoh K, Kato T, Nishioka K: Effects of celecoxib on human chondrocytes--enhanced production of chemokines. Clin Exp Rheumatol. 2007, 25: 11-16.
Jeffrey JE, Aspden RM: Cyclooxygenase inhibition lowers prostaglandin E2 release from articular cartilage and reduces apoptosis but not proteoglycan degradation following an impact load in vitro. Arthritis Res Ther. 2007, 9: R129-10.1186/ar2346.
Amin AR, Attur M, Patel RN, Thakker GD, Marshall PJ, Rediske J, Stuchin SA, Patel IR, Abramson SB: Superinduction of cyclooxygenase-2 activity in human osteoarthritis-affected cartilage. Influence of nitric oxide. J Clin Invest. 1997, 99: 1231-1237. 10.1172/JCI119280.
Mastbergen SC, Bijlsma JW, Lafeber FP: Synthesis and release of human cartilage matrix proteoglycans are differently regulated by nitric oxide and prostaglandin-E2. Ann Rheum Dis. 2008, 67: 52-58. 10.1136/ard.2006.065946.
Watanabe Y, Namba A, Honda K, Aida Y, Matsumura H, Shimizu O, Suzuki N, Tanabe N, Maeno M: IL-1beta stimulates the expression of prostaglandin receptor EP4 in human chondrocytes by increasing production of prostaglandin E2. Connect Tissue Res. 2009, 50: 186-193. 10.1080/03008200802588451.
Goldring MB, Birkhead J, Sandell LJ, Kimura T, Krane SM: Interleukin 1 suppresses expression of cartilage-specific types II and IX collagens and increases types I and III collagens in human chondrocytes. J Clin Invest. 1988, 82: 2026-2037. 10.1172/JCI113823.
Nagase H, Kashiwagi M: Aggrecanases and cartilage matrix degradation. Arthritis Res Ther. 2003, 5: 94-103.
Tsutsumi R, Ito H, Hiramitsu T, Nishitani K, Akiyoshi M, Kitaori T, Yasuda T, Nakamura T: Celecoxib inhibits production of MMP and NO via down-regulation of NF-kappaB and JNK in a PGE2 independent manner in human articular chondrocytes. Rheumatol Int. 2008, 28: 727-736. 10.1007/s00296-007-0511-6.
Pelletier JP, Jovanovic DV, Lascau-Coman V, Fernandes JC, Manning PT, Connor JR, Currie MG, Martel-Pelletier J: Selective inhibition of inducible nitric oxide synthase reduces progression of experimental osteoarthritis in vivo: possible link with the reduction in chondrocyte apoptosis and caspase 3 level. Arthritis Rheum. 2000, 43: 1290-1299. 10.1002/1529-0131(200006)43:6<1290::AID-ANR11>3.0.CO;2-R.
Blanco FJ, Ochs RL, Schwarz H, Lotz M: Chondrocyte apoptosis induced by nitric oxide. Am J Pathol. 1995, 146: 75-85.
Cao M, Westerhausen-Larson A, Niyibizi C, Kavalkovich K, Georgescu HI, Rizzo CF, Hebda PA, Stefanovic-Racic M, Evans CH: Nitric oxide inhibits the synthesis of type-II collagen without altering Col2A1 mRNA abundance: prolyl hydroxylase as a possible target. Biochem J. 1997, 324 (Pt 1): 305-310.
Matsuda K, Nakamura S, Matsushita T: Celecoxib inhibits nitric oxide production in chondrocytes of ligament-damaged osteoarthritic rat joints. Rheumatol Int. 2006, 26: 991-995. 10.1007/s00296-006-0107-6.
Alvarez-Soria MA, Herrero-Beaumont G, Moreno-Rubio J, Calvo E, Santillana J, Egido J, Largo R: Long-term NSAID treatment directly decreases COX-2 and mPGES-1 production in the articular cartilage of patients with osteoarthritis. Osteoarthritis Cartilage. 2008, 16: 1484-1493. 10.1016/j.joca.2008.04.022.
Schmidt N, Pautz A, Art J, Rauschkolb P, Jung M, Erkel G, Goldring MB, Kleinert H: Transcriptional and post-transcriptional regulation of iNOS expression in human chondrocytes. Biochem Pharmacol. 2010, 79: 722-732. 10.1016/j.bcp.2009.10.012.
Wilbrink B, Nietfeld JJ, Den OW, van Roy JL, Bijlsma JW, Huber-Bruning O: Role of TNF alpha, in relation to IL-1 and IL-6 in the proteoglycan turnover of human articular cartilage. Br J Rheumatol. 1991, 30: 265-271. 10.1093/rheumatology/30.4.265.
Loeuille D, Chary-Valckenaere I, Champigneulle J, Rat AC, Toussaint F, Pinzano-Watrin A, Goebel JC, Mainard D, Blum A, Pourel J, Netter P, Gillet P: Macroscopic and microscopic features of synovial membrane inflammation in the osteoarthritic knee: correlating magnetic resonance imaging findings with disease severity. Arthritis Rheum. 2005, 52: 3492-3501. 10.1002/art.21373.
Benito MJ, Veale DJ, FitzGerald O, van den Berg WB, Bresnihan B: Synovial tissue inflammation in early and late osteoarthritis. Ann Rheum Dis. 2005, 64: 1263-1267. 10.1136/ard.2004.025270.
Bondeson J, Blom AB, Wainwright S, Hughes C, Caterson B, van den Berg WB: The role of synovial macrophages and macrophage-produced mediators in driving inflammatory and destructive responses in osteoarthritis. Arthritis Rheum. 2010, 62: 647-657.
Sahap AO: Leukotriene B4 and prostaglandin E2-like activity in synovial fluid in osteoarthritis. Prostaglandins Leukot Essent Fatty Acids. 1990, 39: 253-254. 10.1016/0952-3278(90)90002-3.
Alvarez-Soria MA, Largo R, Santillana J, Sanchez-Pernaute O, Calvo E, Hernandez M, Egido J, Herrero-Beaumont G: Long term NSAID treatment inhibits COX-2 synthesis in the knee synovial membrane of patients with osteoarthritis: differential proinflammatory cytokine profile between celecoxib and aceclofenac. Ann Rheum Dis. 2006, 65: 998-1005. 10.1136/ard.2005.046920.
Berenbaum F: Signaling transduction: target in osteoarthritis. Curr Opin Rheumatol. 2004, 16: 616-622. 10.1097/01.bor.0000133663.37352.4a.
Tetlow LC, Adlam DJ, Woolley DE: Matrix metalloproteinase and proinflammatory cytokine production by chondrocytes of human osteoarthritic cartilage: associations with degenerative changes. Arthritis Rheum. 2001, 44: 585-594. 10.1002/1529-0131(200103)44:3<585::AID-ANR107>3.0.CO;2-C.
Yang SF, Hsieh YS, Lue KH, Chu SC, Chang IC, Lu KH: Effects of nonsteroidal anti-inflammatory drugs on the expression of urokinase plasminogen activator and inhibitor and gelatinases in the early osteoarthritic knee of humans. Clin Biochem. 2008, 41: 109-116. 10.1016/j.clinbiochem.2007.10.011.
Busso N, Peclat V, So A, Sappino AP: Plasminogen activation in synovial tissues: differences between normal, osteoarthritis, and rheumatoid arthritis joints. Ann Rheum Dis. 1997, 56: 550-557. 10.1136/ard.56.9.550.
Pillinger MH, Rosenthal PB, Tolani SN, Apsel B, Dinsell V, Greenberg J, Chan ES, Gomez PF, Abramson SB: Cyclooxygenase-2-derived E prostaglandins down-regulate matrix metalloproteinase-1 expression in fibroblast-like synoviocytes via inhibition of extracellular signal-regulated kinase activation. J Immunol. 2003, 171: 6080-6089.
Cha HS, Ahn KS, Jeon CH, Kim J, Koh EM: Inhibitory effect of cyclooxygenase-2 inhibitor on the production of matrix metalloproteinases in rheumatoid fibroblast-like synoviocytes. Rheumatol Int. 2004, 24: 207-211.
Kusunoki N, Ito T, Sakurai N, Suguro T, Handa H, Kawai S: A novel celecoxib derivative potently induces apoptosis of human synovial fibroblasts. J Pharmacol Exp Ther. 2005, 314: 796-803. 10.1124/jpet.105.086116.
Lev-Ari S, Strier L, Kazanov D, Elkayam O, Lichtenberg D, Caspi D, Arber N: Curcumin synergistically potentiates the growth-inhibitory and pro-apoptotic effects of celecoxib in osteoarthritis synovial adherent cells. Rheumatology (Oxford). 2006, 45: 171-177.
Kusunoki N, Yamazaki R, Kawai S: Pro-apoptotic effect of nonsteroidal anti-inflammatory drugs on synovial fibroblasts. Mod Rheumatol. 2008, 18: 542-551. 10.1007/s10165-008-0102-8.
Parada-Turska J, Rzeski W, Majdan M, Kandefer-Szerszen M, Turski WA: Effect of glutamate receptor antagonists and antirheumatic drugs on proliferation of synoviocytes in vitro. Eur J Pharmacol. 2006, 535: 95-97. 10.1016/j.ejphar.2006.02.009.
Audo R, Deschamps V, Hahne M, Combe B, Morel J: Apoptosis is not the major death mechanism induced by celecoxib on rheumatoid arthritis synovial fibroblasts. Arthritis Res Ther. 2007, 9: R128-10.1186/ar2342.
Grosch S, Maier TJ, Schiffmann S, Geisslinger G: Cyclooxygenase-2 (COX-2)-independent anticarcinogenic effects of selective COX-2 inhibitors. J Natl Cancer Inst. 2006, 98: 736-747. 10.1093/jnci/djj206.
Muraoka T, Hagino H, Okano T, Enokida M, Teshima R: Role of subchondral bone in osteoarthritis development: A comparative study of two strains of guinea pigs with and without spontaneously occurring osteoarthritis. Arthritis Rheum. 2007, 56: 3366-3374. 10.1002/art.22921.
Bolbos RI, Zuo J, Banerjee S, Link TM, Ma CB, Li X, Majumdar S: Relationship between trabecular bone structure and articular cartilage morphology and relaxation times in early OA of the knee joint using parallel MRI at 3 T. Osteoarthritis Cartilage. 2008, 16: 1150-1159. 10.1016/j.joca.2008.02.018.
Karsdal MA, Leeming DJ, Dam EB, Henriksen K, Alexandersen P, Pastoureau P, Altman RD, Christiansen C: Should subchondral bone turnover be targeted when treating osteoarthritis?. Osteoarthritis Cartilage. 2008, 16: 638-646. 10.1016/j.joca.2008.01.014.
Chikazu D, Katagiri M, Ogasawara T, Ogata N, Shimoaka T, Takato T, Nakamura K, Kawaguchi H: Regulation of osteoclast differentiation by fibroblast growth factor 2: stimulation of receptor activator of nuclear factor kappaB ligand/osteoclast differentiation factor expression in osteoblasts and inhibition of macrophage colony-stimulating factor function in osteoclast precursors. J Bone Miner Res. 2001, 16: 2074-2081. 10.1359/jbmr.2001.16.11.2074.
Tat SK, Pelletier JP, Velasco CR, Padrines M, Martel-Pelletier J: New perspective in osteoarthritis: the OPG and RANKL system as a potential therapeutic target?. Keio J Med. 2009, 58: 29-40. 10.2302/kjm.58.29.
Yavropoulou MP, Yovos JG: Osteoclastogenesis--current knowledge and future perspectives. J Musculoskelet Neuronal Interact. 2008, 8: 204-216.
Tat SK, Pelletier JP, Lajeunesse D, Fahmi H, Duval N, Martel-Pelletier J: Differential modulation of RANKL isoforms by human osteoarthritic subchondral bone osteoblasts: influence of osteotropic factors. Bone. 2008, 43: 284-291. 10.1016/j.bone.2008.04.006.
Igarashi K, Woo JT, Stern PH: Effects of a selective cyclooxygenase-2 inhibitor, celecoxib, on bone resorption and osteoclastogenesis in vitro. Biochem Pharmacol. 2002, 63: 523-532. 10.1016/S0006-2952(01)00861-9.
Han SY, Lee NK, Kim KH, Jang IW, Yim M, Kim JH, Lee WJ, Lee SY: Transcriptional induction of cyclooxygenase-2 in osteoclast precursors is involved in RANKL-induced osteoclastogenesis. Blood. 2005, 106: 1240-1245. 10.1182/blood-2004-12-4975.
Kawashima M, Fujikawa Y, Itonaga I, Takita C, Tsumura H: The effect of selective cyclooxygenase-2 inhibitor on human osteoclast precursors to influence osteoclastogenesis in vitro. Mod Rheumatol. 2009, 19: 192-198. 10.1007/s10165-008-0149-6.
Weber A, Casini A, Heine A, Kuhn D, Supuran CT, Scozzafava A, Klebe G: Unexpected nanomolar inhibition of carbonic anhydrase by COX-2-selective celecoxib: new pharmacological opportunities due to related binding site recognition. J Med Chem. 2004, 47: 550-5578. 10.1021/jm030912m.
Segovia-Silvestre T, Neutzsky-Wulff AV, Sorensen MG, Christiansen C, Bollerslev J, Karsdal MA, Henriksen K: Advances in osteoclast biology resulting from the study of osteopetrotic mutations. Hum Genet. 2009, 124: 561-577. 10.1007/s00439-008-0583-8.
Komuro H, Olee T, Kuhn K, Quach J, Brinson DC, Shikhman A, Valbracht J, Creighton-Achermann L, Lotz M: The osteoprotegerin/receptor activator of nuclear factor kappaB/receptor activator of nuclear factor kappaB ligand system in cartilage. Arthritis Rheum. 2001, 44: 2768-2776. 10.1002/1529-0131(200112)44:12<2768::AID-ART464>3.0.CO;2-I.
Kwan TS, Amiable N, Pelletier JP, Boileau C, Lajeunesse D, Duval N, Martel-Pelletier J: Modulation of OPG, RANK and RANKL by human chondrocytes and their implication during osteoarthritis. Rheumatology (Oxford). 2009, 48: 1482-1490. 10.1093/rheumatology/kep300.
Moreno-Rubio J, Herrero-Beaumont G, Tardio L, Alvarez-Soria MA, Largo R: Nonsteroidal antiinflammatory drugs and prostaglandin E(2) modulate the synthesis of osteoprotegerin and RANKL in the cartilage of patients with severe knee osteoarthritis. Arthritis Rheum. 2010, 62: 478-488. 10.1002/art.27204.
Watanabe Y, Namba A, Aida Y, Honda K, Tanaka H, Suzuki N, Matsumura H, Maeno M: IL-1beta suppresses the formation of osteoclasts by increasing OPG production via an autocrine mechanism involving celecoxib-related prostaglandins in chondrocytes. Mediators Inflamm. 2009, 2009: 308596-
Usui M, Xing L, Drissi H, Zuscik M, O'Keefe R, Chen D, Boyce BF: Murine and chicken chondrocytes regulate osteoclastogenesis by producing RANKL in response to BMP2. J Bone Miner Res. 2008, 23: 314-325.
Masuyama R, Stockmans I, Torrekens S, Van LR, Maes C, Carmeliet P, Bouillon R, Carmeliet G: Vitamin D receptor in chondrocytes promotes osteoclastogenesis and regulates FGF23 production in osteoblasts. J Clin Invest. 2006, 116: 3150-3159. 10.1172/JCI29463.
Breuil V, Schmid-Antomarchi H, Schmid-Alliana A, Rezzonico R, Euller-Ziegler L, Rossi B: The receptor activator of nuclear factor (NF)-kappaB ligand (RANKL) is a new chemotactic factor for human monocytes. FASEB J. 2003, 17: 1751-1753.
Mastbergen SC, Marijnissen AC, Vianen ME, Zoer B, van Roermund PM, Bijlsma JW, Lafeber FP: Inhibition of COX-2 by celecoxib in the canine groove model of osteoarthritis. Rheumatology (Oxford). 2006, 45: 405-413. 10.1093/rheumatology/kei187.
Huh JE, Baek YH, Kim YJ, Lee JD, Choi DY, Park DS: Protective effects of butanol fraction from Betula platyphyla var. japonica on cartilage alterations in a rabbit collagenase-induced osteoarthritis. J Ethnopharmacol. 2009, 123: 515-521. 10.1016/j.jep.2008.08.028.
de Boer TN, Huisman AM, Polak AA, Niehoff AG, van Rinsum AC, Saris D, Bijlsma JW, Lafeber FJ, Mastbergen SC: The chondroprotective effect of selective COX-2 inhibition in osteoarthritis: ex vivo evaluation of human cartilage tissue after in vivo treatment. Osteoarthritis Cartilage. 2009, 17: 482-488. 10.1016/j.joca.2008.09.002.
Raynauld JP, Martel-Pelletier J, Beaulieu A, Bessette L, Morin F, Choquette D, Haraoui B, Abram F, Pelletier JP: An open-label pilot study evaluating by magnetic resonance imaging the potential for a disease-modifying effect of celecoxib compared to a modelized historical control cohort in the treatment of knee osteoarthritis. Semin Arthritis Rheum. 2010, 40: 185-192. 10.1016/j.semarthrit.2009.10.003.
Sawitzke AD, Shi H, Finco MF, Dunlop DD, Bingham CO, Harris CL, Singer NG, Bradley JD, Silver D, Jackson CG, Lane NE, Oddis CV, Wolfe F, Lisse J, Furst DE, Reda DJ, Moskowitz RW, Williams HJ, Clegg DO: The effect of glucosamine and/or chondroitin sulfate on the progression of knee osteoarthritis: a report from the glucosamine/chondroitin arthritis intervention trial. Arthritis Rheum. 2008, 58: 3183-3191. 10.1002/art.23973.
Marks PH, Donaldson ML: Inflammatory cytokine profiles associated with chondral damage in the anterior cruciate ligament-deficient knee. Arthroscopy. 2005, 21: 1342-1347. 10.1016/j.arthro.2005.08.034.
Ding C, Cicuttini F, Jones G: Do NSAIDs affect longitudinal changes in knee cartilage volume and knee cartilage defects in older adults?. Am J Med. 2009, 122: 836-842. 10.1016/j.amjmed.2009.03.022.
Thwin MM, Douni E, Arjunan P, Kollias G, Kumar PV, Gopalakrishnakone P: Suppressive effect of secretory phospholipase A2 inhibitory peptide on interleukin-1beta-induced matrix metalloproteinase production in rheumatoid synovial fibroblasts, and its antiarthritic activity in hTNFtg mice. Arthritis Res Ther. 2009, 11: R138-10.1186/ar2810.
Gebhard HH, Zysk SP, Schmitt-Sody M, Jansson V, Messmer K, Veihelmann A: The effects of Celecoxib on inflammation and synovial microcirculation in murine antigen-induced arthritis. Clin Exp Rheumatol. 2005, 23: 63-70.
El-Ghazaly MA, Nada AS, El-Hazek RM, Khayyal MT: Effect of selective COX-2 inhibitor, celecoxib on adjuvant-induced arthritis model in irradiated rats. Int J Radiat Biol. 2010, 86: 1079-1087. 10.3109/09553002.2010.501839.
Bianchi M, Broggini M, Balzarini P, Franchi S, Sacerdote P: Effects of nimesulide on pain and on synovial fluid concentrations of substance P, interleukin-6 and interleukin-8 in patients with knee osteoarthritis: comparison with celecoxib. Int J Clin Pract. 2007, 61: 1270-1277. 10.1111/j.1742-1241.2007.01453.x.
Sutton S, Clutterbuck A, Harris P, Gent T, Freeman S, Foster N, Barrett-Jolley R, Mobasheri A: The contribution of the synovium, synovial derived inflammatory cytokines and neuropeptides to the pathogenesis of osteoarthritis. Vet J. 2009, 179: 10-24. 10.1016/j.tvjl.2007.08.013.
Noguchi M, Kimoto A, Sasamata M, Miyata K: Micro-CT imaging analysis for the effect of celecoxib, a cyclooxygenase-2 inhibitor, on inflammatory bone destruction in adjuvant arthritis rats. J Bone Miner Metab. 2008, 26: 461-468.
Taketa T, Sakai A, Tanaka S, Nakai K, Menuki K, Yamane H, Tanaka K, Nakamura T: Selective cyclooxygenase-2 inhibitor prevents reduction of trabecular bone mass in collagen-induced arthritic mice in association with suppression of RANKL/OPG ratio and IL-6 mRNA expression in synovial tissues but not in bone marrow cells. J Bone Miner Metab. 2008, 26: 143-151. 10.1007/s00774-007-0808-2.
Tsuboi H, Nampei A, Matsui Y, Hashimoto J, Kawai S, Ochi T, Yoshikawa H: Celecoxib prevents juxta-articular osteopenia and growth plate destruction adjacent to inflamed joints in rats with collagen-induced arthritis. Mod Rheumatol. 2007, 17: 115-122. 10.1007/s10165-007-0552-4.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
MZ, JAGR, FPJGL and SM declare that they have no competing interests. The work of TNB is supported by an unrestricted grant from Pfizer. JWJB received a consultancy fee from Pfizer (<US$10,000).
Authors' contributions
All authors contributed to the design, drafting and revision of the manuscript, and all authors approved the final submission.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Zweers, M.C., de Boer, T.N., van Roon, J. et al. Celecoxib: considerations regarding its potential disease-modifying properties in osteoarthritis . Arthritis Res Ther 13, 239 (2011). https://doi.org/10.1186/ar3437
Published:
DOI: https://doi.org/10.1186/ar3437