
Abstract
An adequate supply of oxygen and nutrients is essential for survival and metabolism of cells, and consequentially for normal homeostasis. Alterations in tissue oxygen tension have been postulated to contribute to a number of pathologies, including rheumatoid arthritis (RA), in which the characteristic synovial expansion is thought to outstrip the oxygen supply, leading to areas of synovial hypoxia and hypoperfusion. Indeed, the idea of a therapeutic modality aimed at 'starving' tissue of blood vessels was born from the concept that blood vessel formation (angiogenesis) is central to efficient delivery of oxygen to cells and tissues, and has underpinned the development of anti-angiogenic therapies for a range of cancers. An important and well characterized 'master regulator' of the adaptive response to alterations in oxygen tension is hypoxia-inducible factor (HIF), which is exquisitely sensitive to changes in oxygen tension. Activation of the HIF transcription factor signalling cascade leads to extensive changes in gene expression, which allow cells, tissues and organisms to adapt to reduced oxygenation. One of the best characterized hypoxia-responsive genes is the angiogenic stimulus vascular endothelial growth factor, expression of which is dramatically upregulated by hypoxia in many cells types, including RA synovial membrane cells. This leads to an apparent paradox, with the abundant synovial vasculature (which might be expected to restore oxygen levels to normal) occurring nonetheless together with regions of synovial hypoxia. It has been shown in a number of studies that vascular endothelial growth factor blockade is effective in animal models of arthritis; these findings suggest that hypoxia may activate the angiogenic cascade, thereby contributing to RA development. Recent data also suggest that, as well as activating angiogenesis, hypoxia may regulate many other features that are important in RA, such as cell trafficking and matrix degradation. An understanding of the biology of the HIF transcription family may eventually lead to the development of therapies that are aimed at interfering with this key signalling pathway, and hence to modulation of hypoxia-dependent pathologies such as RA.
Similar content being viewed by others


Introduction
Alterations in oxygen tension have been postulated to contribute to a number of pathologies, including rheumatoid arthritis (RA). Hypoxia refers to subnormal levels of oxygen in air, blood and tissue. Tissue hypoxia leads to cellular dysfunction and ultimately can lead to cell death, and the ability of cells to adapt to periods of hypoxia is therefore important for their survival. An important and well characterized 'master regulator' of the adaptive response to alterations in oxygen tension is hypoxia-inducible factor (HIF). Activation of the HIF signalling cascade leads to extensive changes in gene expression, which allow cells, tissues and organisms to adapt to reduced oxygenation. These changes include enhanced glucose uptake, increased expression of glycolytic enzymes and increased expression of angiogenic factors [1].
RA is a chronic systemic inflammatory disease, which affects approximately 1% of the population worldwide. The aetiology of RA is still not fully understood, but data suggest an interplay between environmental and genetic factors. The financial impact of RA is considerable because of the high level of functional impairment it causes; up to 30% of people with RA become permanently work disabled within 3 years of diagnosis if they do not receive medical treatment [2]. There is now considerable evidence that hypoxia is a feature of RA. Recent studies have also identified many parallels between hypoxia and acute infection and/or inflammation, such as that which is seen in RA. For example, HIF-1 is essential for myeloid cell-mediated inflammation and bactericidal capacity of phagocytes, suggesting crosstalk between angiogenesis and inflammation.
This review series examines the evidence for hypoxia in inflammatory and destructive joint disease, and discusses the interplay between alterations in oxygen tension, vascularity and inflammatory signalling pathways. In the present review we focus on current knowledge of the regulation of the HIF pathway, and then consider the potential role of hypoxia in the pathogenesis of RA.
Why is hypoxia a feature of rheumatoid arthritis?
Tissue hypoxia results from an inadequate supply of oxygen, with resultant effects on biological functions. Within the context of tumours, hypoxia is a well described phenomenon, arising from a hyperplastic response by the tumour cells that leads to an increased distance from pre-existing blood vessels. Because arthritic synovium is also characterized by an altered proliferative response, it is not surprising that hypoxia is also thought to contribute to RA development. At this point, it is worth noting that there is little agreement about what constitutes 'hypoxia'. Oxygen tensions under physiological conditions range from arterial blood levels to much lower tissue levels. Many studies ex vivo consider oxygen tension in relation to atmospheric oxygen levels, namely 20% to 21% oxygen, which is higher than in vivo oxygen levels. Moreover, some authors' definition of 'hypoxia' may actually be more analogous to physiological 'normoxia', with studies performed at 5% to 7% oxygen. The studies described in this review all utilized levels of oxygen below 5% when describing the effects of 'hypoxia'.
With regard to RA, the environment in the inflamed joint is characterized by a low partial pressure of oxygen. The first study demonstrating the hypoxic nature of rheumatoid synovium was carried out more than 30 years ago. Mean synovial fluid oxygen in RA knee joints was reported to be lower than in osteoarthritis (OA) patients or in traumatic effusions in otherwise healthy control individuals [3]. An interesting study also reported an inverse relationship between synovial fluid oxygen values and synovial fluid volume [4]. Despite these intriguing observations, it was only recently that we were able to measure synovial oxygen tension in RA patients directly using a highly sensitive gold microelectrode [5]. We observed that synovial tissue in RA patients was indeed hypoxic, with oxygen lower than in noninflamed synovium in patients without RA. The median oxygen in patients with RA was 26 mmHg (range 18 to 33 mmHg, equivalent to 2% to 4%), as compared with 74 mmHg in patients without RA (range 69 to 89 mmHg, equivalent to 9% to 12%). Furthermore, in a number of RA patients we were able to obtain matched measurements from invasive and encapsulating tenosynovium and from joint synovium, and we found that oxygen in invasive tenosynovium was 43% lower than in matched joint synovium, and 28% lower than in matched encapsulating tenosynovium. This suggests the existence of hypoxic gradients within RA synovium, and provides a potential mechanism for tendon rupture in RA patients, which could be driven by hypoxia-mediated upregulation of angiogenic and matrix-degrading factors.
A number of factors are believed to interplay to produce the hypoxic environment. As mentioned above, the formation of a hyperplastic inflammatory mass increases the distance between proliferating cells and their nearest blood vessels [6]. Several studies have demonstrated that the oxygen consumption of the RA synovium is elevated, possibly because of the increased proliferative activity of synovial cells, and that glucose is oxidized via an anaerobic, rather than aerobic, pathway [7, 8]. A recent study assessed whether synovial proliferation (assessed by ultrasonography as visible synovial thickening and nodular or villous appearance) differentially affects hypoxia in RA and OA. No difference was found between the OA patients with and without synovial proliferation in terms of synovial fluid oxygen, whereas RA patients had both increased proliferation and significantly lower synovial fluid oxygen levels, suggesting that the proliferative response may have different impacts on synovial oxygenation in RA and OA [9]. These findings of an anaerobic and acidic microenvironment have been supported by nuclear magnetic resonance spectroscopy, confirming the presence of low molecular weight metabolites, consistent with hypoxia [8]. Movement has been also proposed to result in intra-articular pressure exceeding synovial capillary perfusion pressure [10]. The raised pressure further compromises the vasculature and exacerbates the already ischaemic environment. Furthermore, these cycles of hypoxia-reperfusion are likely to generate reactive oxygen species, as has been demonstrated using electron spin resonance spectroscopy [11]. The data in RA patients are supported by findings showing reduced oxygen levels in the joints of arthritic mice [12, 13].
The HIF transcription factor signalling pathway
The alterations in synovial oxygen tension that are observed in RA synovium are likely to exert effects on the HIF transcription factors, which are considered to be the 'master regulators' of cellular responses to changes in oxygen tension. The HIF family was first analyzed and defined through studies of the glycoprotein hormone erythropoietin [14], which regulates red blood cell production. To date, it has been established that approximately 1% of all human genes are regulated by HIF, including genes that are involved in angiogenesis (in particular vascular endothelial growth factor [VEGF]), as well as apoptosis, vasomotor control, erythropoiesis and energy metabolism. HIF is a heterodimeric transcription factor that is composed of two different subunits: HIF-α, which is oxygen regulated, and HIF-β, which is expressed constitutively in the nucleus [15]. There are at least two α subunits, termed HIF-1α and HIF-2α. Regulation of HIF-dependent gene expression requires α subunit accumulation in the cytoplasm and translocation into the nucleus, which enables it to dimerize with β subunits of HIF. The HIF heterodimers are then recognized by co-activators and bind to the hypoxia-response elements (HREs) in the target gene to initiate transcription.
HIF: regulation by prolyl hydroxylases
In 1996, Jiang and coworkers [16] described that maximal levels of HIF-1α protein in HeLa cells exposed in vitro to different oxygen concentrations were observed at 0.5% oxygen, suggesting that HIF was possibly a cellular oxygen sensor. The main regulators of HIF-α post-translational modifications were subsequently characterized as oxygenases governed by oxygen, 2-oxoglutarate (2-OG), iron (Fe2+) and ascorbic acid (collectively termed HIF prolyl hydroxylase domain [PHD]-containing enzymes), and factor inhibiting HIF (FIH). HIF-α subunits encompass an oxygen-dependent degradation domain, responsible for hypoxic stabilization of α-subunits, and two transactivating domains, namely the amino-terminal transactivating domain and the carboxyl-terminal transactivating domain (C-TAD). The C-TAD has been shown to interact with co-activators such as p300 to activate transcription. Further upstream of the transactivation domains, a contiguous basic helix-loop-helix and Per-Arnt-Sim domain creates a functional interface for dimerization of HIF-α with HIF-β and binding to HRE. The PHD enzymes hydroxylate proline residues in the oxygen-dependent degradation domain, thus making HIF-α recognizable by the von Hippel Lindau (vHL) tumour suppressor protein/E3 ubiquitin ligase [17], which leads to polyubiquitination and proteolytic destruction of α subunits by the 26S proteasome. Thus, under conditions in which oxygen is limiting, HIF-α subunits accumulate and activate transcription of HRE-containing genes.
The PHD enzymes were first described by Epstein and coworkers [18] through a forward genetic approach to screening candidate 2-OG dependent dioxygenases in Caenorhabditis elegans and termed PHD-1, PHD-2 and PHD-3. The enzymes were also identified and described by other groups on the basis of similarity to mammalian procollagen prolyl-4-hydroxylase. The expression of PHD isoforms is highly variable between tissues, and they are also partitioned differently between nuclear and cytoplasmic compartments [19]. There is also substantial variation in the relative expression of the PHD isoforms in different cells, with PHD-2 being the most abundant HIF prolyl hydroxylase. Specific 'silencing' of all three enzymes using short interfering RNA has shown that PHD-2 is the major player in stabilizing HIF in normoxia in most, but not all, cell lines. Although PHD enzymes regulate stability of HIF and thereby induce cellular adaptations in response to hypoxia, it remains largely unknown how these enzymes are regulated. PHD-2 and PHD-3, and to a lesser extent PHD-1, are strongly induced by hypoxia in many cell types, thus resulting in the increased oxygen-mediated HIF-α degradation that is observed after long periods of hypoxia [20, 21].
The recent generation of mice with specific global or conditional inactivation of each of the three PHD enzymes is very promising and will promote better understanding of the functions of the enzymes. Mice homozygous for targeted disruptions in PHD-1 and PHD-3 genes are viable and appear normal. In contrast, targeted disruption of PHD-2 in mice led to embryonic lethality between embryonic days 12.5 and 14.5, caused by severe cardiac and placental defects, suggesting an important role of PHD-2 in development of the heart and placenta [22]. Because of the embryonic lethality after global deletion of PHD-2, Takeda and coworkers [23] conditionally inactivated lox P-flanked PHD-2 in adult mice using tamoxifen inducible Cre under the control of the ubiquitously expressed Rosa26 locus. This resulted in hyperactive angiogenesis and angiectasia in multiple organs, suggesting an essential role for PHD-2 in oxygen homeostasis of the adult vascular system. Another study from the same group showed that blood homeostasis in adult mice is mostly maintained by PHD-2 but can be further modulated by the combined actions of PHD-1 and PHD-3 [24]. Because hypoxia and HIF activation and angiogenesis are features of RA, it could be suggested that PHD enzymes are in some way downregulated in RA, and such conditional PHD knockout mice could in the future shed light on this hypothesis.
Finally, genetic studies have shown that loss of PHD-1, but not PHD-2 or PHD-3, selectively induced hypoxia tolerance in skeletal muscle. This indicates that even though all PHD enzymes are expressed in muscle, they are likely to play specific physiological roles. In PHD-1-deficient myofibres, oxygen consumption was reduced, leading to protection of the cells against the lethal effects of acute severe hypoxia [25]. In the same study it was shown that HIF-2α was a downstream mediator of PHD-1 in hypoxia tolerance. HIF-1α also appears to be involved in the PHD-1 pathway, although less prominently. These findings are of significant importance to our understanding of the molecular basis of hypoxia tolerance, not only in muscle but also in numerous other diseases (including cancer and RA) and in settings where induction of hypoxia tolerance might be of therapeutic value, such as organ preservation for transplantation.
There are nonetheless a number of questions that remain to be answered, including the existence of new targets other than HIF for prolyl hydroxylation and regulation. A recent report has revealed inhibitor of nuclear factor-κB (IκB) kinase (IKK)-2 to be a target for prolyl hydroxylation [26]. IKK-2 is a significant component of the nuclear factor-κB (NF-κB) signalling pathway, and it was shown that within its activation loop IKK-2 contains an evolutionarily conserved LxxLAP consensus motif for hydroxylation by PHD, thus linking two major human signalling systems, namely NF-κB and HIF. Mimicking hypoxia by treatment of cells with small interfering RNA against PHD-1 or PHD-2 or the pan-hydroxylase inhibitor dimethyloxalylglycine (a 2-OG analogue, and an inhibitor of both PHD and FIH) resulted in NF-κB activation via serine phosphorylation-dependent degradation of IκBα. The investigators suggested that in HeLa cells increased NF-κB activity during hypoxia was through decreased PHD activity, and that PHD-1 negatively regulated IKK-2 via prolyl hydroxylation. Again, if PHD enzymes are in some way downregulated in RA, then this might lead to activation of the NF-κB signalling cascade. However, there is hardly any evidence in the current literature of expression of the HIF regulating PHD enzymes in the RA synovium. Therefore, in the future it will important to study the expression and regulation of these enzymes in RA.
HIF: role of FIH
FIH is an asparaginyl β-hydroxylase, which belongs to the same superfamily of 2-OG and Fe2+-dependent dioxygenases as the PHD. As opposed to proteolytic regulation of HIF-α subunits through proline hydroxylation, FIH regulates HIF function by deactivating the C-TAD, using oxygen as a co-substrate, thereby preventing HIF-α heterodimerization with HIF-β and co-factors and preventing HIF transactivation in normoxia [27]. The C-TAD of HIF-α contains asparagine residues (Asn803 in HIF-1α and Asn851 in HIF-2α) targeted by FIH hydroxylation [28]. Hydroxylation occurs at the β-carbon of the asparagine residue, consequently (by way of steric hindrance) preventing the interaction of the HIF-α C-TAD with the cysteine/histidine-rich 1 domain of p300, a co-activator required for the heterodimerization and transcriptional activity of HIF [27]. The crystal structure of FIH reveals it to be a homodimeric protein [29], and disruption of dimerization of FIH, by use of site-directed mutagenesis, has shown the importance of the dimeric state for its function in recognizing HIF-α as a substrate [30]. Substrates other than HIF have been identified as targets for asparaginyl hydroxylation by FIH. These include proteins such as ankyrin repeat and SOCS box protein 4 (ASB4), thought to mediate ubiquitination of various target proteins, and the intracellular domain of the Notch receptor (involved in the maintenance of cells in an undifferentiated state), both of which notably contain ankyrin repeat motifs containing the asparagine residue hydroxylated by FIH [31]. Another target, identified only recently as an FIH substrate and also possessing an ankyrin repeat motif, is the IκB family of inhibitory proteins [32], providing further evidence that FIH-dependent aspara-ginyl hydroxylation is not restricted to HIF-α subunits. Although the functional result of asparaginyl hydroxylation of these proteins remains unclear (because the downstream effects are small), there is a suggestion that it may in fact involve HIF regulation, by the sequestering of FIH away from HIF, particularly in hypoxia.
Microenvironmental conditions in RA joints are characterized by low oxygen levels [3]. One property of FIH that contrasts with that of the PHD is its ability to function even in severe hypoxia [33]. In other words, when the availability of oxygen is low and PHD enzymes can no longer function (through lack of oxygen substrate), FIH is potentially still able to deactivate HIF that has escaped proteosomal degradation. It is unclear at present whether FIH is still active in RA synovium. As recently as 2005, a small molecule inhibitor was developed to inhibit FIH specifically and upregulate a host of bona fide HIF target genes such as erythropoietin and VEGF [34]. This selective inhibition could therefore be of future benefit for therapeutic strategies requiring upregulated HIF activity.
HIF regulation by inflammatory stimuli
In parallel to the oxygen dependent pathway, HIF-1α is also regulated by receptor-mediated signals under normoxic conditions [35–39], although the molecular pathways underlying these more subtle changes in HIF gene/protein expression have not been fully characterized. As is the case under hypoxic conditions, upregulation of HIF-1α by inflammatory cytokines such as tumour necrosis factor (TNF)-α and IL-1β is thought to involve at least in part stabilization of protein [35, 40, 41]. For example, TNF-α was shown to upregulate HIF-1α protein levels whereas the HIF-1α mRNA levels remained unchanged [35, 38, 42]. IL-1β has also been shown to induce HIF-1α protein in a lung epithelial cell line A549 through an NF-κB dependent pathway but did not alter the steady-state level of HIF-1α mRNA in these cells [42]. However, transcriptional effects have also been reported. Interestingly, in the context of RA, both IL-1β and TNF-α have been shown to increase mRNA for HIF-1α in RA fibroblasts [43, 44]. IL-1β could also induce HIF-1 DNA binding activity in these cells. Bacterial lipopolysaccharide has also been reported to upregulate HIF-1α transcription and/or protein levels [45, 46].
Several regulatory pathways have been reported to be involved in the control of HIF-α, in particular phosphatidyl-inositol 3-kinase [47–52], p42/p44 mitogen-activated protein kinase [53], p38 mitogen-activated protein kinase and protein kinase Cδ [54]. The NF-κB pathway has also been implicated. Recently, using IKK-2 deficient mice, it was shown that NF-κB is required for HIF-1α protein accumulation, and that absence of IKK-2 results in defective induction of HIF targets such as VEGF [55, 56]. Conversely, hypoxia itself has been identified as an activator of NF-κB [57, 58]. Given the importance of the HIF and NF-κB signalling cascades in the regulation of inflammation, further work to clarify the importance of crosstalk between these pathways is clearly required.
RA synovium is both hypoxic and expresses elevated levels of inflammatory cytokines. The HIF transcription factor family may thus represent an important convergence point in RA, integrating cellular responses to low oxygen tension and to inflammatory cytokines.
HIF and rheumatoid arthritis: regulation of angiogenesis and inflammation
What might be the consequence of the hypoxic milieu in terms of RA pathogenesis? The classic hypoxia-responsive gene is VEGF, which has been detected at higher levels in the serum and synovial fluid of RA patients [59, 60]. We have shown in several studies that hypoxia is a potent stimulus for VEGF induction in RA synovial membrane cell cultures, which contain lymphocytes, as well as macrophages and fibroblasts [60].
Besides VEGF, many other genes have been reported to be regulated by hypoxia in fibroblasts, including a variety of angiogenic and inflammatory mediators. Hypoxia has been reported to cause a general downregulation of gene expression in microarray studies in murine fibroblasts. Greijer and coworkers [61] observed a significant upregulation or down-regulation of 159 genes by hypoxia; of these 45 were up-regulated and 112 were downregulated. Using HIF-1α null mouse fibroblasts, these authors were able to establish that, of the genes that were upregulated in their study, 89% were dependent on HIF-1, as opposed to only 17% of the down-regulated genes. This supports a role for HIF-1 in up-regulating genes necessary for cell survival and adaptation to stress. Chemokines play a key role in regulating cell trafficking to RA synovium. Stromal cell-derived factor-1 is a chemo-kine of the C-X-C family that is involved in inflammation and angiogenesis. RA fibroblasts are capable of secreting large amounts of stromal cell-derived factor-1 in response to treatment with hypoxia (1% oxygen) for 24 hours [62]. Monocyte chemoattractant protein-1 is elevated in RA synovium. Interestingly, we and others have reported a suppressive effect of hypoxia on monocyte chemoattractant protein-1 in RA synovial cells [5, 63].
It is also becoming apparent that matrix metalloprotease (MMP) enzymes and their tissue inhibitors (TIMPs) are a further subset of molecules that may be regulated by hypoxia. The balance between MMPs and TIMPs is likely to influence cell invasion, within the context of angiogenesis (via degradation of extracellular matrix) and/or in terms of invasion by synovium of underlying tissue such cartilage, bone and tendon. A variety of MMPs have been shown to be regulated by hypoxia. When exposed to hypoxia, RA synovial fibroblasts exhibit increased protein levels of MMP-1 and MMP-3 [64]. Conversely, hypoxic RA synovial fibroblasts have been shown to decrease expression of TIMP-1 at both protein and mRNA levels [64]. TNF-α converting enzyme was also recently shown to be HIF-1 dependent [65], which could be important in regulating TNF-α levels in RA.
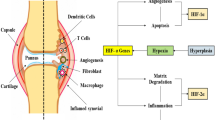
In summary, hypoxia may affect a host of genes that are involved in angiogenesis, apoptosis, cellular metabolism, matrix degradation and inflammation, thus perpetuating the cycle of reactions involved in RA development (Figure 1).

Role of hypoxia-regulated HIF transcription factors in RA. In the context of RA pathogenesis, hypoxia-induced stabilization of HIF-α protein can potentially modulate genes that are involved in angiogenesis (for example, VEGF), matrix degradation, apoptosis (for instance, BNIP-3), cellular metabolism (GLUT-1) and inflammation (cytokines and chemokines), thus perpetuating the destructive cascade of reactions. Furthermore, cytokines relevant to RA (IL-1 and TNF) can themselves modulate HIF levels. A schematic representation of a normal and RA joint is shown. Representative sections (×100 magnification, with bars indicating 20 μm) of RA tissue stained for HIF-1α and HIF-2α are shown, taken from two different RA patients. HIF-1α expression appears to be predominantly vascular associated, in areas of diffuse cellular infiltration, unlike HIF-2α, which was frequently associated with infiltrating cells distant form visible blood vessels. BNIP, BCL2/adenovirus E1B 19 kDa-interacting protein; COX, cyclo-oxygenase; GLUT, glucose transporter; HIF, hypoxia-inducible factor; IL, interleukin; MMP, matrix metalloprotease; RA, rheumatoid arthritis; TNF, tumour necrosis factor; VEGF, vascular endothelial growth factor.
Are there distinct roles for HIF isoforms?
The past decade has yielded striking evidence that HIF may become a key target in RA therapy. Hypoxia is known to influence cellular responses relevant to RA pathogenesis, and thus by specific HIF inhibition it should be possible to modulate the activity of cells. The question that should be answered first is, what are the individual roles of HIF-1α and HIF-2α, and which isoform should be blocked or activated?
A considerable amount of research has been carried out on HIF-1α and HIF-2α since the mid-1990s, showing their fundamental roles as mediators of transcriptional responses to hypoxia. A number of similarities have been shown, such as structure, regulation of activation and degradation via the vHL ubiquitin E3 ligase [17], as well as the mechanism of action, namely dimerization with HIF-1β, recognition and binding to HRE in the promoters of target genes [15]. Moreover, both isoforms are modified at the post-translational level by oxygen-dependent PHD and FIH-1 enzymes [18].
However, although there are many similarities between HIF-1α and HIF-2α, there is growing evidence revealing differences, implying that they play distinct biological roles in different cell types. The differences include presence in animals, with HIF-1α being older evolutionally, existing from C. elegans to humans, whereas HIF-2α is present only in complicated vertebrates, namely chickens, quails, and mammals. HIF-1α appears to be ubiquitously expressed, whereas HIF-2α is more tissue restricted, being primarily expressed in the embryo vasculature and subsequently in the lung, kidney and liver. This is mirrored in the number of regulated genes. It was reported using short interfering RNA and Affymetrix gene chip analysis of hepatoma cells that 3% of all genes were regulated by hypoxia, with HIF-2α regulating approximately 13% (36/271) of upregulated genes and 17% of downregulated genes (37/217) [66]. The vast majority of genes were HIF-1α dependent (75% of upregulated genes and 62% of downregulated genes), with the remainder apparently requiring both HIF-1α and HIF-2α. However, this study used human hepatoma Hep3B cell line, and it is not yet clear whether this might be true for cells in RA synovium.
Because of their structural similarities, it was believed that HIF-1α and HIF-2α were responsible for analogous responses to hypoxia. However, differences in RNA and protein stability (with HIF-1α being transiently expressed and HIF-2α expression being sustained in prolonged hypoxia) coupled with differences in co-factors engaged in regulation (such as NEMO, CITED-2 and ELK-1, which selectively cooperate with HIF-2α [67]) suggested that the two isoforms differ not only in terms of the number of HIF-regulated genes but also, and most importantly, in the pattern of gene expression. This is supported by evidence for a HIF-1α specific feedback loop mechanism that involves natural anti-sense HIF [68], PHD-2 and HIF-3α [69], and differences in expression upon cytokine stimulation. For example, IL-1 and TNF-α induce HIF-1α, but not HIF-2α, in RA synovial fibroblasts [70]. HIF-1α regulates genes involved in metabolism, regulating glycolysis and glucose uptake by glucose transporter-1 and glyceraldehyde 3-phosphate dehydrogenase [67, 71]. In addition HIF-1α activates angiogenesis, survival and invasion, most importantly in normal development and in response to stress. Conversely, HIF-2α regulates a small group of genes, and is involved specifically in renal tumourigenesis and regulation of genes with special functions. These biological functions ascribe to HIF-2α a unique role, in comparison with the broader and more general role played by HIF-1α.
The aforementioned studies indicate that HIF-1α and HIF-2α play different roles. However, some findings imply that they play completely opposing roles. In their 2005 study, Raval, Lau and their coworkers [72] observed that HIF-2α steers the anti-apoptotic response, because BCL2/adenovirus E1B 19 kDa-interacting protein (BNIP)3 (a pro-apoptotic factor) was downregulated by HIF-2α. In contrast, HIF-1α has pro-apoptotic properties because of upregulation of BNIP3. Indeed, BNIP3 has been reported to be upregulated by hypoxia in RA fibroblasts [73]. This is somewhat counterintuitive, because RA fibroblasts exhibit, if anything, reduced apoptosis. Additional striking evidence has been discovered in tumour development, showing that HIF-1α and HIF-2α display disparate effects on tumour growth [67]. It has become evident that α subunits can act in completely opposite ways in endothelial and breast cancer cells, in which hypoxia-responsive genes were HIF-1α dependent, and in renal carcinoma cells, which seem to be critically dependent on HIF-2α [67]. Raval and coworkers [72] have shown that in some cases over-expression of HIF-2α promotes tumour growth, whereas HIF-1α inhibits tumour growth, in contrast to breast cancer cells, in which proliferation was retarded by HIF-2α over-expression [74]. It has thus become clear that, by having contrasting effects on regulation of HIF-target genes, HIF-1α and HIF-2α may contribute to progression or regression of disease.
In RA synovium, HIF-1α and HIF-2α are expressed in the synovial lining and stromal cells [75]. In adjuvant-induced arthritis, HIF-1α has been localized to synovium of inflamed joints [12]. Conversely, targeted deletion of HIF-1α in cells of the myeloid lineage resulted in reduced arthritis in mice [76]. In RA synovium, we have also demonstrated that VEGF expression appears to closely resemble those of HIF-1α and HIF-2α [5]. VEGF was previously demonstrated to be regulated by HIF-1α in many cells. However, in cells with defective vHL and expressing only HIF-2α [67, 72] and in chondrocytes [77], VEGF was reduced by HIF-2α knockdown and not by HIF-1α. In summary, although both HIF-1α and HIF-2α have been shown to be expressed in RA synovium, it could be hypothesized that a switch might occur from a HIF-1α dependent pro-apoptotic phenotype to a more HIF-2α dependent 'tumour-like' proliferative phenotype, leading to synovial hyperplasia.
Conclusion
There is an emerging link between altered oxygen tension, angiogenesis, synovial invasion and disease progression in RA. The relative contributions of HIF-1α and HIF-2α in hypoxia-triggered cellular responses are subject to ongoing investigation. There are number of genes altered by hypoxia, among which some are HIF-1α dependent, some HIF-2α dependent and some respond equally to both isoforms. Many of these genes, such as VEGF, are critically involved in RA progression. Interestingly, HIF-2α is gaining more interest because studies have revealed that in some cell lines this isoform may be as important as HIF-1α. Based on the assumption that there are genes that are regulated by HIF-1α, HIF-2α or both, an understanding of the biology of the HIF transcription family may eventually lead to the development of therapies aimed at interfering with this key signalling pathway, and hence to modulation of hypoxia-dependent pathologies such as RA. Of relevance, the inhibitor 2-methoxyestradiol has been suggested to suppress HIF-1α and its downstream target genes such as VEGF and glucose transporter-1, and has also been shown to suppress arthritis in vivo in animal models. A clinical trial of 2-methoxyestradiol is planned in RA, and this may yield further insight into the links between hypoxia, angiogenesis, inflammatory cell trafficking and matrix breakdown in RA.
Note
This review is part of a series on Hypoxia edited by Ewa Paleolog.
Other articles in this series can be found at http://arthritis-research.com/series/ar_Hypoxia
Abbreviations
- BNIP:
-
BCL2/adenovirus E1B 19 kDa-interacting protein
- C-TAD:
-
carboxyl-terminal transactivating domain
- FIH:
-
factor inhibiting HIF
- HIF:
-
hypoxia-inducible factor
- HRE:
-
hypoxia-response element
- IκB:
-
inhibitor of nuclear factor-κB
- IKK:
-
inhibitor of nuclear factor-κB kinase
- IL:
-
interleukin
- MMP:
-
matrix metalloprotease
- NF-κB:
-
nuclear factor-κB
- OA:
-
osteoarthritis
- 2-OG:
-
2-oxoglutarate
- PHD:
-
prolyl hydroxylase domain
- RA:
-
rheumatoid arthritis
- TIMP:
-
tissue inhibitor of matrix metalloprotease
- TNF:
-
tumour necrosis factor
- VEGF:
-
vascular endothelial growth factor
- vHL:
-
von Hippel Lindau.
References
Wiesener MS, Maxwell PH: HIF and oxygen sensing; as important to life as the air we breathe?. Ann Med. 2003, 35: 183-190. 10.1080/0785389031000458233.
Sokka T: Work disability in early rheumatoid arthritis. Clin Exp Rheumatol. 2003, 21: S71-74.
Lund-Olesen K: Oxygen tension in synovial fluids. Arthritis Rheum. 1970, 13: 769-776. 10.1002/art.1780130606.
Richman AI, Su EY, Ho G: Reciprocal relationship of synovial fluid volume and oxygen tension. Arthritis Rheum. 1981, 24: 701-705. 10.1002/art.1780240512.
Sivakumar B, Akhavani MA, Winlove CP, Taylor PC, Paleolog EM, Kang N: Synovial hypoxia as a cause of tendon rupture in rheumatoid arthritis. J Hand Surg [Am]. 2008, 33: 49-58. 10.1016/j.jhsa.2007.09.002.
Larsen H, Akhavani MA, Raatz Y, Paleolog EM: Gene expression studies to investigate disease mechanisms in rheumatoid arthritis: does angiogenesis play a role?. Curr Rheumatol Rev. 2007, 3: 243-251. 10.2174/157339707782408991.
Blake DR, Merry P, Unsworth J, Kidd BL, Outhwaite JM, Ballard R, Morris CJ, Gray L, Lunec J: Hypoxic-reperfusion injury in the inflamed human joint. Lancet. 1989, 1: 289-293. 10.1016/S0140-6736(89)91305-6.
Naughton D, Whelan M, Smith EC, Williams R, Blake DR, Grootveld M: An investigation of the abnormal metabolic status of synovial fluid from patients with rheumatoid arthritis by high field proton nuclear magnetic resonance spectroscopy. FEBS Lett. 1993, 317: 135-138. 10.1016/0014-5793(93)81508-W.
Lee YA, Kim JY, Hong SJ, Lee SH, Yoo MC, Kim KS, Yang HI: Synovial proliferation differentially affects hypoxia in the joint cavities of rheumatoid arthritis and osteoarthritis patients. Clin Rheumatol. 2007, 26: 2023-2029. 10.1007/s10067-007-0605-2.
Jawed S, Gaffney K, Blake DR: Intra-articular pressure profile of the knee joint in a spectrum of inflammatory arthropathies. Ann Rheum Dis. 1997, 56: 686-689. 10.1136/ard.56.11.686.
Singh D, Nazhat NB, Fairburn K, Sahinoglu T, Blake DR, Jones P: Electron spin resonance spectroscopic demonstration of the generation of reactive oxygen species by diseased human synovial tissue following ex vivo hypoxia-reoxygenation. Ann Rheum Dis. 1995, 54: 94-99. 10.1136/ard.54.2.94.
Peters CL, Morris CJ, Mapp PI, Blake DR, Lewis CE, Winrow VR: The transcription factors hypoxia-inducible factor 1alpha and Ets-1 colocalize in the hypoxic synovium of inflamed joints in adjuvant-induced arthritis. Arthritis Rheum. 2004, 50: 291-296. 10.1002/art.11473.
Etherington PJ, Winlove P, Taylor P, Paleolog E, Miotla JM: VEGF release is associated with reduced oxygen tensions in experimental inflammatory arthritis. Clin Exp Rheumatol. 2002, 20: 799-805.
Semenza GL, Wang GL: A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992, 12: 5447-5454.
Wang GL, Jiang BH, Rue EA, Semenza GL: Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA. 1995, 92: 5510-5514. 10.1073/pnas.92.12.5510.
Jiang BH, Semenza GL, Bauer C, Marti HH: Hypoxia-inducible factor 1 levels vary exponentially over a physiologically relevant range of O2 tension. Am J Physiol. 1996, 271: C1172-C1180.
Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ: The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999, 399: 271-275. 10.1038/20459.
Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O'Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, Jaakkola P, Barstead R, Hodgkin J, Maxwell PH, Pugh CW, Schofield CJ, Ratcliffe PJ: C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001, 107: 43-54. 10.1016/S0092-8674(01)00507-4.
Metzen E, Berchner-Pfannschmidt U, Stengel P, Marxsen JH, Stolze I, Klinger M, Huang WQ, Wotzlaw C, Hellwig-Burgel T, Jelkmann W, Acker H, Fandrey J: Intracellular localisation of human HIF-1 alpha hydroxylases: implications for oxygen sensing. J Cell Sci. 2003, 116: 1319-1326. 10.1242/jcs.00318.
Marxsen JH, Stengel P, Doege K, Heikkinen P, Jokilehto T, Wagner T, Jelkmann W, Jaakkola P, Metzen E: Hypoxia-inducible factor-1 (HIF-1) promotes its degradation by induction of HIF-alpha-prolyl-4-hydroxylases. Biochem J. 2004, 381: 761-767. 10.1042/BJ20040620.
Pescador N, Cuevas Y, Naranjo S, Alcaide M, Villar D, Landazuri MO, Del Peso L: Identification of a functional hypoxia-responsive element that regulates the expression of the egl nine homologue 3 (egln3/phd3) gene. Biochem J. 2005, 390: 189-197. 10.1042/BJ20042121.
Takeda K, Ho VC, Takeda H, Duan LJ, Nagy A, Fong GH: Placental but not heart defects are associated with elevated hypoxia-inducible factor alpha levels in mice lacking prolyl hydroxylase domain protein 2. Mol Cell Biol. 2006, 26: 8336-8346. 10.1128/MCB.00425-06.
Takeda K, Cowan A, Fong GH: Essential role for prolyl hydroxylase domain protein 2 in oxygen homeostasis of the adult vascular system. Circulation. 2007, 116: 774-781. 10.1161/CIRCULATIONAHA.107.701516.
Takeda K, Aguila HL, Parikh NS, Li X, Lamothe K, Duan LJ, Takeda H, Lee FS, Fong GH: Regulation of adult erythropoiesis by prolyl hydroxylase domain proteins. Blood. 2008, 111: 3229-3235. 10.1182/blood-2007-09-114561.
Aragones J, Schneider M, Van Geyte K, Fraisl P, Dresselaers T, Mazzone M, Dirkx R, Zacchigna S, Lemieux H, Jeoung NH, Lambrechts D, Bishop T, Lafuste P, Diez-Juan A, Harten SK, Van Noten P, De Bock K, Willam C, Tjwa M, Grosfeld A, Navet R, Moons L, Vandendriessche T, Deroose C, Wijeyekoon B, Nuyts J, Jordan B, Silasi-Mansat R, Lupu F, Dewerchin M, Pugh C, Salmon P, Mortelmans L, Gallez B, Gorus F, Buyse J, Sluse F, Harris RA, Gnaiger E, Hespel P, Van Hecke P, Schuit F, Van Veldhoven P, Ratcliffe P, Baes M, Maxwell P, Carmeliet P: Deficiency or inhibition of oxygen sensor Phd1 induces hypoxia tolerance by reprogramming basal metabolism. Nat Genet. 2008, 40: 170-180. 10.1038/ng.2007.62.
Cummins EP, Berra E, Comerford KM, Ginouves A, Fitzgerald KT, Seeballuck F, Godson C, Nielsen JE, Moynagh P, Pouyssegur J, Taylor CT: Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc Natl Acad Sci USA. 2006, 103: 18154-18159. 10.1073/pnas.0602235103.
Ema M, Hirota K, Mimura J, Abe H, Yodoi J, Sogawa K, Poellinger L, Fujii-Kuriyama Y: Molecular mechanisms of transcription activation by HLF and HIF1alpha in response to hypoxia: their stabilization and redox signal-induced interaction with CBP/p300. EMBO J. 1999, 18: 1905-1914. 10.1093/emboj/18.7.1905.
Hewitson KS, McNeill LA, Riordan MV, Tian YM, Bullock AN, Welford RW, Elkins JM, Oldham NJ, Bhattacharya S, Gleadle JM, Ratcliffe PJ, Pugh CW, Schofield CJ: Hypoxia-inducible factor (HIF) asparagine hydroxylase is identical to factor inhibiting HIF (FIH) and is related to the cupin structural family. J Biol Chem. 2002, 277: 26351-26355. 10.1074/jbc.C200273200.
Elkins JM, Hewitson KS, McNeill LA, Seibel JF, Schlemminger I, Pugh CW, Ratcliffe PJ, Schofield CJ: Structure of factor-inhibiting hypoxia-inducible factor (HIF) reveals mechanism of oxidative modification of HIF-1 alpha. J Biol Chem. 2003, 278: 1802-1806. 10.1074/jbc.C200644200.
Lancaster DE, McNeill LA, McDonough MA, Aplin RT, Hewitson KS, Pugh CW, Ratcliffe PJ, Schofield CJ: Disruption of dimerization and substrate phosphorylation inhibit factor inhibiting hypoxia-inducible factor (FIH) activity. Biochem J. 2004, 383: 429-437. 10.1042/BJ20040735.
Ferguson JE, Wu Y, Smith K, Charles P, Powers K, Wang H, Patterson C: ASB4 is a hydroxylation substrate of FIH and promotes vascular differentiation via an oxygen-dependent mechanism. Mol Cell Biol. 2007, 27: 6407-6419. 10.1128/MCB.00511-07.
Cockman ME, Lancaster DE, Stolze IP, Hewitson KS, McDonough MA, Coleman ML, Coles CH, Yu X, Hay RT, Ley SC, Pugh CW, Oldham NJ, Masson N, Schofield CJ, Ratcliffe PJ: Post-translational hydroxylation of ankyrin repeats in IkappaB proteins by the hypoxia-inducible factor (HIF) asparaginyl hydroxylase, factor inhibiting HIF (FIH). Proc Natl Acad Sci USA. 2006, 103: 14767-14772. 10.1073/pnas.0606877103.
Koivunen P, Hirsila M, Gunzler V, Kivirikko KI, Myllyharju J: Catalytic properties of the asparaginyl hydroxylase (FIH) in the oxygen sensing pathway are distinct from those of its prolyl 4-hydroxylases. J Biol Chem. 2004, 279: 9899-9904. 10.1074/jbc.M312254200.
McDonough MA, McNeill LA, Tilliet M, Papamicael CA, Chen QY, Banerji B, Hewitson KS, Schofield CJ: Selective inhibition of factor inhibiting hypoxia-inducible factor. J Am Chem Soc. 2005, 127: 7680-7681. 10.1021/ja050841b.
Zhou J, Schmid T, Brune B: Tumor necrosis factor-alpha causes accumulation of a ubiquitinated form of hypoxia inducible factor-1alpha through a nuclear factor-kappaB-dependent pathway. Mol Biol Cell. 2003, 14: 2216-2225. 10.1091/mbc.E02-09-0598.
Scharte M, Han X, Bertges DJ, Fink MP, Delude RL: Cytokines induce HIF-1 DNA binding and the expression of HIF-1-dependent genes in cultured rat enterocytes. Am J Physiol Gastrointest Liver Physiol. 2003, 284: G373-G384.
Hellwig-Burgel T, Rutkowski K, Metzen E, Fandrey J, Jelkmann W: Interleukin-1beta and tumor necrosis factor-alpha stimulate DNA binding of hypoxia-inducible factor-1. Blood. 1999, 94: 1561-1567.
Albina JE, Mastrofrancesco B, Vessella JA, Louis CA, Henry WL, Reichner JS: HIF-1 expression in healing wounds: HIF-1alpha induction in primary inflammatory cells by TNF-alpha. Am J Physiol Cell Physiol. 2001, 281: C1971-1977.
Bilton RL, Booker GW: The subtle side to hypoxia inducible factor (HIFalpha) regulation. Eur J Biochem. 2003, 270: 791-798. 10.1046/j.1432-1033.2003.03446.x.
Metzen E, Zhou J, Jelkmann W, Fandrey J, Brune B: Nitric oxide impairs normoxic degradation of HIF-1alpha by inhibition of prolyl hydroxylases. Mol Biol Cell. 2003, 14: 3470-3481. 10.1091/mbc.E02-12-0791.
McMahon S, Charbonneau M, Grandmont S, Richard DE, Dubois CM: Transforming growth factor beta1 induces hypoxia-inducible factor-1 stabilization through selective inhibition of PHD2 expression. J Biol Chem. 2006, 281: 24171-24181. 10.1074/jbc.M604507200.
Jung Y, Isaacs JS, Lee S, Trepel J, Liu ZG, Neckers L: Hypoxia-inducible factor induction by tumour necrosis factor in normoxic cells requires receptor-interacting protein-dependent nuclear factor kappa B activation. Biochem J. 2003, 370: 1011-1017. 10.1042/BJ20021279.
Thornton RD, Lane P, Borghaei RC, Pease EA, Caro J, Mochan E: Interleukin 1 induces hypoxia-inducible factor 1 in human gingival and synovial fibroblasts. Biochem J. 2000, 350 (Pt 1): 307-312. 10.1042/0264-6021:3500307.
Westra J, Brouwer E, Bos R, Posthumus MD, Doornbos-van der Meer B, Kallenberg CG, Limburg PC: Regulation of cytokine-induced HIF-1alpha expression in rheumatoid synovial fibroblasts. Ann N Y Acad Sci. 2007, 1108: 340-348. 10.1196/annals.1422.035.
Frede S, Stockmann C, Freitag P, Fandrey J: Bacterial lipopolysaccharide induces HIF-1 activation in human monocytes via p44/42 MAPK and NF-kappaB. Biochem J. 2006, 396: 517-527. 10.1042/BJ20051839.
Nishi K, Oda T, Takabuchi S, Oda S, Fukuda K, Adachi T, Semenza GL, Shingu K, Hirota K: LPS induces hypoxia-inducible factor 1 activation in macrophage-differentiated cells in a reactive oxygen species-dependent manner. Antioxid Redox Signal. 2008, 10: 983-995. 10.1089/ars.2007.1825.
Mazure NM, Chen EY, Laderoute KR, Giaccia AJ: Induction of vascular endothelial growth factor by hypoxia is modulated by a phosphatidylinositol 3-kinase/Akt signaling pathway in Ha-ras-transformed cells through a hypoxia inducible factor-1 transcriptional element. Blood. 1997, 90: 3322-3331.
Alvarez-Tejado M, Alfranca A, Aragones J, Vara A, Landazuri MO, del Peso L: Lack of evidence for the involvement of the phosphoinositide 3-kinase/Akt pathway in the activation of hypoxia-inducible factors by low oxygen tension. J Biol Chem. 2002, 277: 13508-13517. 10.1074/jbc.M200017200.
Jiang BH, Jiang G, Zheng JZ, Lu Z, Hunter T, Vogt PK: Phosphatidylinositol 3-kinase signaling controls levels of hypoxia-inducible factor 1. Cell Growth Differ. 2001, 12: 363-369.
Stiehl DP, Jelkmann W, Wenger RH, Hellwig-Burgel T: Normoxic induction of the hypoxia-inducible factor 1alpha by insulin and interleukin-1beta involves the phosphatidylinositol 3-kinase pathway. FEBS Lett. 2002, 512: 157-162. 10.1016/S0014-5793(02)02247-0.
Treins C, Giorgetti-Peraldi S, Murdaca J, Semenza GL, Van Obberghen E: Insulin stimulates hypoxia-inducible factor 1 through a phosphatidylinositol 3-kinase/target of rapamycin-dependent signaling pathway. J Biol Chem. 2002, 277: 27975-27981. 10.1074/jbc.M204152200.
Belaiba RS, Bonello S, Zahringer C, Schmidt S, Hess J, Kietzmann T, Gorlach A: Hypoxia up-regulates hypoxia-inducible factor-1alpha transcription by involving phosphatidylinositol 3-kinase and nuclear factor kappaB in pulmonary artery smooth muscle cells. Mol Biol Cell. 2007, 18: 4691-4697. 10.1091/mbc.E07-04-0391.
Richard DE, Berra E, Gothie E, Roux D, Pouyssegur J: p42/p44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1alpha (HIF-1alpha) and enhance the transcriptional activity of HIF-1. J Biol Chem. 1999, 274: 32631-32637. 10.1074/jbc.274.46.32631.
Baek SH, Lee UY, Park EM, Han MY, Lee YS, Park YM: Role of protein kinase Cdelta in transmitting hypoxia signal to HSF and HIF-1. J Cell Physiol. 2001, 188: 223-235. 10.1002/jcp.1117.
Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, Johnson RS, Haddad GG, Karin M: NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008, 453: 807-811. 10.1038/nature06905.
van Uden P, Kenneth NS, Rocha S: Regulation of hypoxia-inducible factor-1alpha by NF-kappaB. Biochem J. 2008, 412: 477-484. 10.1042/BJ20080476.
Walmsley SR, McGovern NN, Whyte MK, Chilvers ER: The HIF/VHL pathway: from oxygen sensing to innate immunity. Am J Respir Cell Mol Biol. 2008, 38: 251-255. 10.1165/rcmb.2007-0331TR.
Walmsley SR, Print C, Farahi N, Peyssonnaux C, Johnson RS, Cramer T, Sobolewski A, Condliffe AM, Cowburn AS, Johnson N, Chilvers ER: Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J Exp Med. 2005, 201: 105-115. 10.1084/jem.20040624.
Koch AE, Harlow LA, Haines GK, Amento EP, Unemori EN, Wong WL, Pope RM, Ferrara N: Vascular endothelial growth factor. A cytokine modulating endothelial function in rheumatoid arthritis. J Immunol. 1994, 152: 4149-4156.
Paleolog EM, Young S, Stark AC, McCloskey RV, Feldmann M, Maini RN: Modulation of angiogenic vascular endothelial growth factor by tumor necrosis factor alpha and interleukin-1 in rheumatoid arthritis. Arthritis Rheum. 1998, 41: 1258-1265. 10.1002/1529-0131(199807)41:7<1258::AID-ART17>3.0.CO;2-1.
Greijer AE, Groep van der P, Kemming D, Shvarts A, Semenza GL, Meijer GA, Wiel van de MA, Belien JA, van Diest PJ, Wall van der E: Up-regulation of gene expression by hypoxia is mediated predominantly by hypoxia-inducible factor 1 (HIF-1). J Pathol. 2005, 206: 291-304. 10.1002/path.1778.
Hitchon C, Wong K, Ma G, Reed J, Lyttle D, El-Gabalawy H: Hypoxia-induced production of stromal cell-derived factor 1 (CXCL12) and vascular endothelial growth factor by synovial fibroblasts. Arthritis Rheum. 2002, 46: 2587-2597. 10.1002/art.10520.
Safronova O, Nakahama K, Onodera M, Muneta T, Morita I: Effect of hypoxia on monocyte chemotactic protein-1 (MCP-1) gene expression induced by Interleukin-1beta in human synovial fibroblasts. Inflamm Res. 2003, 52: 480-486. 10.1007/s00011-003-1205-5.
Cha HS, Ahn KS, Jeon CH, Kim J, Song YW, Koh EM: Influence of hypoxia on the expression of matrix metalloproteinase-1, -3 and tissue inhibitor of metalloproteinase-1 in rheumatoid synovial fibroblasts. Clin Exp Rheumatol. 2003, 21: 593-598.
Charbonneau M, Harper K, Grondin F, Pelmus M, McDonald PP, Dubois CM: Hypoxia-inducible factor mediates hypoxic and tumor necrosis factor α-induced Increases in tumor necrosis factor-α converting enzyme/ADAM17 expression by synovial cells. J Biol Chem. 2007, 282: 33714-33724. 10.1074/jbc.M704041200.
Warnecke C, Weidemann A, Volke M, Schietke R, Wu X, Knaup KX, Hackenbeck T, Bernhardt W, Willam C, Eckardt KU, Wiesener MS: The specific contribution of hypoxia-inducible factor-2alpha to hypoxic gene expression in vitro is limited and modulated by cell type-specific and exogenous factors. Exp Cell Res. 2008, 314: 2016-2027. 10.1016/j.yexcr.2008.03.003.
Sowter HM, Raval RR, Moore JW, Ratcliffe PJ, Harris AL: Predominant role of hypoxia-inducible transcription factor (Hif)-1alpha versus Hif-2alpha in regulation of the transcriptional response to hypoxia. Cancer Res. 2003, 63: 6130-6134.
Thrash-Bingham CA, Tartof KD: aHIF: a natural antisense transcript overexpressed in human renal cancer and during hypoxia. J Natl Cancer Inst. 1999, 91: 143-151. 10.1093/jnci/91.2.143.
Maynard MA, Evans AJ, Hosomi T, Hara S, Jewett MA, Ohh M: Human HIF-3alpha4 is a dominant-negative regulator of HIF-1 and is down-regulated in renal cell carcinoma. FASEB J. 2005, 19: 1396-1406. 10.1096/fj.05-3788com.
Larsen H, Feldmann M, Paleolog EM: Synovial fibroblasts are important mediators of synovial angiogenesis in the hypoxic rheumatoid joint. Vascular Pharmacology. 2006, 45: e118-10.1016/j.vph.2006.08.317.
Carroll VA, Ashcroft M: Role of hypoxia-inducible factor (HIF)-1alpha versus HIF-2alpha in the regulation of HIF target genes in response to hypoxia, insulin-like growth factor-I, or loss of von Hippel-Lindau function: implications for targeting the HIF pathway. Cancer Res. 2006, 66: 6264-6270. 10.1158/0008-5472.CAN-05-2519.
Raval RR, Lau KW, Tran MG, Sowter HM, Mandriota SJ, Li JL, Pugh CW, Maxwell PH, Harris AL, Ratcliffe PJ: Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol Cell Biol. 2005, 25: 5675-5686. 10.1128/MCB.25.13.5675-5686.2005.
Kammouni W, Wong K, Ma G, Firestein GS, Gibson SB, El-Gabalawy HS: Regulation of apoptosis in fibroblast-like syn-oviocytes by the hypoxia-induced Bcl-2 family member Bcl-2/adenovirus E1B 19-kd protein-interacting protein 3. Arthritis Rheum. 2007, 56: 2854-2863. 10.1002/art.22853.
Blancher C, Moore JW, Talks KL, Houlbrook S, Harris AL: Relationship of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha expression to vascular endothelial growth factor induction and hypoxia survival in human breast cancer cell lines. Cancer Res. 2000, 60: 7106-7113.
Giatromanolaki A, Sivridis E, Maltezos E, Athanassou N, Papa-zoglou D, Gatter KC, Harris AL, Koukourakis MI: Upregulated hypoxia inducible factor-1alpha and -2alpha pathway in rheumatoid arthritis and osteoarthritis. Arthritis Res Ther. 2003, 5: R193-201. 10.1186/ar756.
Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, Firestein GS, Gerber HP, Ferrara N, Johnson RS: HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003, 112: 645-657. 10.1016/S0092-8674(03)00154-5.
Lafont JE, Talma S, Murphy CL: Hypoxia-inducible factor 2alpha is essential for hypoxic induction of the human articular chondrocyte phenotype. Arthritis Rheum. 2007, 56: 3297-3306. 10.1002/art.22878.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Muz, B., Khan, M.N., Kiriakidis, S. et al. Hypoxia. The role of hypoxia and HIF-dependent signalling events in rheumatoid arthritis. Arthritis Res Ther 11, 201 (2009). https://doi.org/10.1186/ar2568
Published:
DOI: https://doi.org/10.1186/ar2568