
Abstract
Alzheimer’s disease (AD) is a devastating neurodegenerative disorder, afflicting more than one-third of people over the age of 85. While many therapies for AD are in late-stage clinical testing, rational drug design based on distinct signaling pathways in this disorder is only now emerging. Here we review the putative signaling pathway of amyloid-beta (Aβ), by which the tyrosine kinase Fyn is activated via cell surface binding of Aβ oligomers to cellular prion protein. Several lines of evidence implicate Fyn in the pathogenesis of AD, and its interaction with both Aβ and Tau renders Fyn a unique therapeutic target that addresses both of the major pathologic hallmarks of AD. We are currently enrolling patients in a phase Ib study of saracatinib (AZD0530), a small molecule inhibitor with high potency for Src and Fyn, for the treatment of AD. The results of this trial and a planned phase IIa multisite study will provide important data regarding the potential for this therapeutic strategy in AD.
Similar content being viewed by others


Review
Introduction
While the last few decades have produced significant advances in our understanding of Alzheimer’s disease (AD) pathogenesis, clinical therapeutic trials have yet to yield a disease-modifying intervention. AD is invariably associated with diffuse cortical deposition of amyloid-beta (Aβ) plaques and tau-positive neurofibrillary tangles, and considerable resources have therefore been committed to developing therapies to reduce Aβ burden. This approach is supported by a large body of literature suggesting that Aβ plays a critical role in AD. A limitation to this approach, however, has been the lack of a well-defined downstream pathway linking specific misfolded forms of Aβ to neurotoxicity. Many previously developed upstream approaches to reduce Aβ levels have effects on multiple cellular pathways, inadvertently targeting essential biological processes that may counter the intended therapeutic effects. For these reasons, the neuronal signaling pathway triggered by Aβ has been the subject of intense study.
The various elements now thought to comprise a distinct Aβ signaling cascade have come from many sources dating back at least several decades. In this review, we highlight the recent finding that extracellular oligomeric Aβ binds cellular prion protein (PrPC) with high affinity, activating an intracellular signaling cascade coupled to the protein tyrosine kinase Fyn [1–4]. While several elements of this proposed pathway might be targeted as a novel therapeutic strategy in AD, Fyn kinase is of particular interest given its widely accepted role in AD pathogenesis, and the clinically available drugs targeting this protein. A phase Ib study of saracatinib (AZD0530), a Src family kinase inhibitor with high potency for Fyn and Src kinase, is currently underway to treat patients with AD (NCT01864655), with a planned proof-of-concept phase IIa trial in 2014.
Fyn and Src family kinases
Fyn is a 59 kDa protein belonging to the Src family of nonreceptor tyrosine kinases (SFKs), which also includes Src, Yes, Yrk, Blk, Fgr, Hck, Lck, and Lyn [5]. The prototype member of the family is Src kinase, so named for the critical role of viral Src, contained in the Rous sarcoma virus genome, to transform chick fibroblasts infected with this virus into a sarcoma (src) [6]. The Src family kinases are grouped together based on a shared domain structure consisting of three Src homology domains (SH1 to SH3), a variable N-terminus region, and the SH4 domain that anchors the protein to the cytosolic plasma membrane [7]. Variations within the N-terminus region as well as the SH4 domain are thought to be largely responsible for the different physiologic functions among the Src family members [8–10].
The Fyn gene is located on chromosome 6q21, and has three known splice variants [11]. The known active forms are FynT and FynB, with the former expressed mostly in cells of hematopoietic origin while FynB is ubiquitously expressed, with particularly high levels in the brain [11, 12]. In addition to FynB, the other Src family members Src, Lck, Lyn, and Yes are also expressed to varying degrees in the central nervous system, with Fyn and Src being the most extensively studied with regard to brain function.
Fyn activity, like that of other Src family kinases, is regulated by intramolecular interactions that depend on equilibrium between tyrosine phosphorylation and dephosphorylation. In the basal state, catalytic activity is constrained by engagement of the SH2 domain by a phosphorylated C-terminal tyrosine 531 [13]. External protein interactions with the SH2 and SH3 domains by phosphorylation at Tyr 420 in the activation loop of the kinase domain, and/or by dephosphorylation of Tyr 531, result in Fyn activation [13]. Dephosphorylation at Tyr 420, such as that seen with striatal-enriched tyrosine phosphatase 61, significantly reduces Fyn activity [14].
Fyn and central nervous system functions
The biological functions of Fyn are diverse and include processes such as T-cell receptor signaling, cell division and adhesion, platelet function, synaptic function and plasticity, and central nervous system (CNS) myelination (Table 1) [5, 15]. Of particular interest for AD are multiple lines of evidence linking Fyn kinase to synaptic function. Fyn has been localized to the post-synaptic density (PSD) fraction of the brain, which is the primary post-synaptic site for signaling transduction and processing in neurons. Amongst its substrates are receptors for the major excitatory transmitter, glutamate [16], which plays a critical role in synaptic function. Fyn regulates glutamate receptor trafficking and synaptic plasticity in part by specifically phosphorylating the N-methyl-d-aspartate-type glutamate receptor subunits NR2A and NR2B [17–21]. These subunits are critical for both long-term potentiation and long-term depression, which are thought to model the underlying mechanisms of learning and memory [22, 23]. Brain slices from mice lacking Fyn expression have blunted long-term potentiation, and mice show impaired contextual fear memory function [19, 24], indicating that Fyn plays an important role in regulating synaptic plasticity. This is further corroborated by the finding that overexpression of a constitutively active Fyn transgene reduces the threshold for long-term potentiation induction in mouse brain hippocampal slices [25], and Fyn knockout mice have an age-dependent reduction in dendritic spines, the single contact points between an axon and a dendrite critical for synaptic function [26].
In summary, Fyn regulates key aspects of synaptic physiology. Activation of Fyn enhances synaptic function, which, as discussed in subsequent sections, can have the undesired effect of rendering neurons more vulnerable to synaptotoxicity. A reduction in Fyn activation has the opposite effect, and while this may be neuroprotective, excessive inhibition may lead to impairments in long-term potentiation, and hypothetically affect cognitive function in humans. A therapeutic approach aimed at maintaining the delicate balance between activation and inhibition of Fyn will thus probably optimize the function of individual synapses, as well as the broader neuronal network.
Beyond synaptic function, Fyn has been shown to have a unique role in CNS myelination. Fyn associates with the large myelin-associated glycoprotein, and this interaction plays an important role in the signaling cascade required for the initial stages of myelination [27]. Consistent with this function, mice lacking Fyn expression have significantly reduced brain myelination, a role not shared by other Src family kinases present in CNS oligodendrocytes, including Src and Lyn [27, 28].
Fyn kinase and Alzheimer’s disease
Fyn is elevated in Alzheimer’s disease brain and activated by amyloid-beta
An early focus in AD research was to identify proteins responsible for the pathologic phosphorylation of various tau residues. In 1993, Shirazi and Wood reported that a subset of neurons from AD brain exhibited strong Fyn immunoreactivity compared with control brains, and that these neurons were also positive for abnormally phosphorylated Tau protein [29]. The possible connection between Fyn and AD led to numerous studies focusing mainly on the downstream effects of Aβ on tyrosine kinases in cell culture. An early study suggested that Aβ25–35 and Aβ1–40 increased tyrosine phosphorylation in a dose-dependent manner, although the exact Aβ preparation was not well characterized [30]. Other synthetic preparations reported to consist of mostly fibrillar Aβ also showed an increase in tyrosine phosphorylated proteins in cell culture, suggesting the importance of tyrosine kinases in AD [31, 32]. While the majority of early studies in AD used fibrillary or mixed preparations of Aβ, it is now widely believed that soluble intermediates of this peptide, termed Aβ oligomer (Aβo), are the most relevant assemblies in AD pathogenesis [33]. These specific conformations of Aβ, initially named Aβ-derived diffusible ligands, were found to cause apoptosis in organotypic mouse brain slices, with full protection conferred by the lack of Fyn expression [34].
Amyloid-beta oligomers activate Fyn via cellular prion protein
A critical early step in AD pathophysiology is the process by which Aβo interacts with the neuronal surface to trigger downstream pathology, and studies of this pathway have further implicated Fyn in AD pathophysiology. In a unique genome-wide unbiased screen for cell surface proteins binding Aβo, we identified PrPC[1]. Aβ binding to PrPC has consistently been shown to be of high affinity and is exquisitely specific for the oligomeric form, with little or no affinity for fibrillary or monomeric Aβ peptide [1, 35–37]. PrPC is not essential for all Aβ-related phenotypes [36–39]. However, it is required for cell death in vitro, for reduced survival of APP/PS1 transgenic mice, for epileptiform discharges, for synapse loss, for serotonin fiber degeneration and for spatial learning and memory deficits [2, 40–46]. The ability of human AD brain-derived Aβ species to suppress hippocampal synaptic plasticity depends upon PrPC, and human AD brain extracts contain Aβo species capable of interacting with PrPC as well as Aβ–PrPC complexes [2, 47–49].
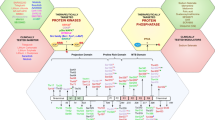
Once Aβo forms and binds to PrPC at the cell surface, changes in neuronal biochemistry occur (Figure 1). Both PrPC and Fyn were found to be enriched in the PSD, and exposure of cultured mouse hippocampal neurons to Aβo activated Fyn by phosphorylating the Tyr 420 residue [2]. This interaction depends entirely on the normal expression of PrPC, and in Prnp−/− cultures activation of Fyn by Aβo is eliminated [2]. Brain extracts from patients with AD also stimulate Fyn activation in mouse cortical cultures, but control brain extracts do not, further suggesting that the Aβ–PrPC–Fyn pathway is relevant in human disease [2].

mGluR5 couples amyloid-beta oligomer–cellular prion protein to post-synaptic signaling. Schematic illustrating a central role of Fyn in amyloid-beta oligomer (Aβo) signaling. Binding of Aβo to cellular prion protein (PrPC) triggers mGluR5-dependent signaling events. Proteins are clustered in the post-synaptic density (PSD) and alter N-methyl-d-aspartate receptor (NMDA-R) function, calcium and protein translation. Tau plays a role in localizing Fyn and is a Fyn substrate. The net result of aberrant PrPC–mGluR5–Fyn signaling is synaptic malfunction and loss. Aβ, amyloid-beta; APP, amyloid precursor protein; PrP, prion protein.
The connection between Aβo–PrPC complexes at the cell surface and intracellular Fyn kinase has recently been shown to require the metabotropic glutamate receptor, mGluR5 [3]. This transmembrane protein has a physiological role in linking extracellular glutamate levels to calcium mobilization, to protein translation in dendrites and to tyrosine kinase signaling. For Aβo signaling, we screened transmembrane PSD proteins for the ability to couple Aβo–PrPC complexes to Fyn activation in transfected non-neuronal cells, and only mGluR5 was positive. In neurons and in mice, mGluR5 is required for downstream neuronal dysfunction caused by Aβ. Extracellular Aβ oligomers thus trigger neuronal signal transduction from PrPC to mGluR5 to Fyn kinase.
Fyn activation by amyloid-beta oligomer–cellular prion protein activates N-methyl-d-aspartate receptors and induces dendritic spine loss
N-methyl-d-aspartate receptors play a key role in synaptic plasticity and AD, and intracellular segments of the NR2A and NR2B subunits contain tyrosine residues phosphorylated by SFKs [50]. We showed that Aβo induces a dose-dependent increase in the phosphorylation of N-methyl-d-aspartate receptors, specifically the Fyn-specific phosphorylation of NR2B at Y-1472 [2]. Aβo-induced NR2B phosphorylation at this site is not detected in Prnp−/− or Fyn−/− cultures. Moreover, the role of Fyn is gene-dose dependent, being reduced in heterozygous neurons [2]. Synaptic loss is a hallmark of AD, and several in vitro studies have described dendritic spine loss after acute Aβo exposure [51–54]. We found that in vitro dendritic spine destabilization by Aβo is eliminated in embryonic hippocampal Fyn−/− neurons, providing further evidence that Fyn plays a critical role in Aβ-induced synaptotoxicity [2].
Fyn is implicated in Alzheimer’s disease by interacting with Tau
In addition to its role in Aβ signaling, Fyn is also involved in Tau phosphorylation. It is well known that, in addition to Aβ plaques, AD brain contains diffuse deposits of neurofibrillary tangles, composed of hyperphosphorylated Tau.
Tau may play a critical role in mediating downstream neurodegeneration in AD, and a recent report showed that a 50% reduction in tau expression reversed cognitive impairments in a transgenic AD model [55]. Similar to studies of Aβo, studies of Tau have implicated Fyn mechanistically in AD. Fyn physically associates with Tau, and can phosphorylate tyrosine residues, including Tyr18, near the amino terminus [56–59]. Tyr18 is also phosphorylated in neurofibrillary tangles in human AD brain, suggesting a possible clinical relevance [58]. Activation of Fyn by the Aβo–PrPC complex also leads to downstream Tau phosphorylation [4].
Critically, Fyn and Tau interact genetically to modulate synapse loss, behavioral deficits and electroencephalographic abnormalities in amyloid precursor protein (APP) transgenic mice [60]. Tau was recently reported to have a novel dendritic function to target Fyn to the PSD [61]. Without functional Tau, Fyn is uncoupled from N-methyl-d-aspartate receptors, and Aβ toxicity is rescued [61]. As expected, postsynaptic Fyn levels were reduced in mice with either absent Tau expression or the expression of a truncated Tau transgene, and were increased in mice overexpressing full-length Tau [61]. In both cases, total Fyn levels and activity were normal compared with wild-type littermates [61]. Two studies have reported a genetic association between single nucleotide polymorphisms at the FYN locus and cerebrospinal fluid Tau levels in AD, although this was not observed in all cohorts [62, 63]. PrPC-Fyn signaling thus appears to couple Aβ and Tau pathologies, and Fyn can be considered a lynchpin of AD pathophysiology (Table 2).
In vivo studies of the cellular prion protein–mGluR5–Fyn pathway in Alzheimer’s disease mouse models
Several groups, including ours, have studied the PrPC–mGluR5–Fyn pathway in mouse models overexpressing an APP transgene known to cause autosomal dominant AD. Genetic removal or pharmacologic blockade of PrPC reverses memory impairments and synapse loss in transgenic AD mice [43, 44], although these findings have not been successfully replicated in all models [39]. PrPC does not span the plasma membrane, and thus needs a transmembrane partner to initiate intracellular signaling. This partner is mGluR5, and its blockage with the antagonist MTEP reverses both spatial memory impairments and hippocampal synapse loss in AD mice [3]. Fyn is the third step in the PrPC–mGluR5–Fyn pathway, and several preclinical studies in AD mice have been reported.
Overexpressing Fyn was found to accelerate synapse loss and the onset of cognitive impairment in the J9 (APPswe/Ind) transgenic AD mouse model, while removing Fyn expression rescued synapse loss in the J20 (APPswe/Ind) transgenic AD model [64, 65]. Because J20 mice manifest PrPC-independent dysfunction [39], Fyn represents a common downstream effector required in both PrPC-dependent and PrPC-independent pathways. This further supports Fyn as an attractive target in AD therapy. In the APP23 (APPswe) transgenic model, overexpression of a truncated Tau protein, which competes with full-length Tau for the Fyn binding site, rescued cognitive impairments, and was associated with a reduction in PSD Fyn levels [61]. The findings from these two transgenic lines were not replicated when reducing Fyn expression in a different AD model expressing a tau transgene, in addition to APP and Presenilin 1 (3xTg-AD) [66]. However, the negative study focused on subtle and sex-specific deficits. Interestingly, inhibiting striatal-enriched tyrosine phosphatase 61, which results in Fyn activation, reversed impairments in spatial memory in the 3xTg-AD model [67].
While these results differ from the majority of studies to date, they highlight the complexity of Fyn homeostatic regulation, and the importance of ongoing efforts to characterize these important signaling pathways in humans. While a specific Fyn inhibitor has not yet been studied in an AD model, the SFK inhibitor dasatinib was administered to APP/PS1 transgenic mice in one study [68]. The authors focused on Aβ-induced SFK signaling in microglial cells and reported that dasatinib reduced microgliosis, lowered tumor necrosis factor-alpha levels in the brain and improved behavior in a T-maze test. These data further support the use of SFK inhibitors in AD, and suggest that the benefits may derive from modifying microglial toxicity as well as altering Aβo synaptotoxic signaling in neurons.
Targeting Fyn as a novel therapy in Alzheimer’s disease
Potential adverse effects of Fyn inhibition
Taken together, the preclinical evidence strongly supports targeting Fyn as a promising therapeutic intervention in AD. However, Fyn is involved in a diverse set of physiologic processes important to normal function, and disrupting some of these processes could potentially have unintended consequences. Most important is the role of Fyn in synaptic function and plasticity. While the relationship between long-term potentiation and human memory function continues to be debated, excessive inhibition of Fyn could have an adverse effect on memory and cognitive function. These effects have not been reported in clinical studies of SFK inhibitors for cancer, but this will need to be monitored closely in a more vulnerable population with little cognitive reserve, such as patients with AD. Preclinical data strongly suggest that Fyn is activated by Aβ, and thus a therapeutic strategy aimed at lowering this activity to a physiologic level is likely to represent the most effective approach, with the least serious adverse effects.
While Fyn also regulates CNS myelination, this function may not be as relevant in the fully mature CNS, where myelination is complete. Fyn has been shown to be part of a signaling pathway important for myelin regeneration in chronic demyelinating diseases [69]. While partial inhibition of Fyn seems unlikely to have a clinically meaningful effect on myelination in AD, the possible side effects of excessive Fyn inhibition on CNS myelination in humans must be considered when evaluating new therapies aimed at reducing Fyn activity.
FynT is part of the T-cell receptor, and is an important mediator of both T-cell signaling and differentiation of CD4+ cells into T-helper type 2 cells in rodents, the latter cells being critical for the humoral immune response [70, 71]. Both processes are essential to normal immune function, and while they are not exclusively regulated by Fyn, monitoring immune function in patients receiving SFK inhibitors is critical.
Finally, Fyn also affects the hematologic system, and associates with glycoprotein IIb–IIIa on platelets, a well-known integrin that has been targeted in cardiovascular disease and stroke [72]. Removal of Fyn expression in mice decreases platelet function and increases the bleeding time [72]. A similar function in humans is not known, and the same effects on platelets have not been demonstrated when Fyn is partially removed. Nevertheless, these findings must be considered when inhibiting Fyn in human subjects.
Pharmacologic compounds targeting Fyn in Alzheimer’s disease
Masitinib – a tyrosine kinase inhibitor selective for c-Kit, platelet-derived growth factor receptor and, to a lesser degree, Fyn and Lyn – was recently studied in a 24-week, phase II dose-ranging trial in France, involving 34 patients with mild-to-moderate AD [73]. Masitinib was reasonably well tolerated, and was associated with improvements in cognition and daily function at 12 and 24 weeks, thus supporting tyrosine kinase inhibition as a treatment strategy in AD. A large international phase III trial was launched earlier this year to evaluate the efficacy and safety of two doses of masitinib compared with placebo (ClinicalTrials.gov: NCT01872598).
Saracatinib (AZD0530) is a small molecule inhibitor of Src family kinases, blocking Src, Fyn, Yes and Lyn, with 2 to 10 nM potency [74]. At 10-fold to 100-fold higher concentrations the compound also inhibits Abl family kinases, but there is no detectable activity in this concentration range against other kinase families, including c-kit, Csk and platelet-derived growth factor receptor. AZD0530’s specific inhibition of Fyn and SFKs has led to its development as therapy for solid tumors, because Src family kinases regulate tumor cell adhesion, migration and invasion, and also regulate proliferation [74]. Clinical tolerability and oral bioavailability have been demonstrated, but phase II studies have shown limited benefit as a single agent in specific oncological indications [75–79]. For tumor cell migration and for oncological applications, it is estimated that >98% kinase inhibition is required so clinical doses have targeted concentrations >20-fold above the kinase half maximal inhibitory concentration, in the 200 to 1,000 nM concentration range.
Preliminary studies indicate that the dose needed to disrupt the Aβ–PrPC–Fyn signaling cascade is significantly lower than that needed for cancer therapy. We recently launched a phase Ib study of saracatinib in patients with AD (ClinicalTrials.gov: NCT01864655). The goal of the study is to establish safety, tolerability, and CNS penetration in patients with AD, in preparation for a phase IIa proof-of-concept clinical trial beginning in 2014. Our data will provide important data on the effectiveness of Fyn and Src family kinases inhibition for the treatment of AD. Future approaches may target more specific aspects of the Aβ-PrPC–mGluR5–Fyn pathway, particularly addressing mechanisms of Fyn activation unique to this signaling cascade.
Conclusions
Despite the enormous and growing burden of AD, there remains no effective disease-modifying therapy today. The approaches now in clinical trials are limited in their mechanisms of action, with most centering on efforts to alter Aβ itself, or its production, clearance or aggregation. No major trial has centered on the signal transduction downstream of toxic Aβ species, and while there are efforts to develop approaches targeting Tau and Tau kinases in AD, none provides a unified approach to Aβ and Tau. Fyn represents a unique therapeutic target in AD as it is central to Aβ signal transduction, and has major functional interactions with Tau, thereby unifying the two major pathologies in AD. Developing a disease-modifying therapy in AD would revolutionize the care of millions of patients worldwide, with a major impact on rapidly escalating healthcare costs. The hope is that of the numerous discoveries that are being made in models of AD, such as the Aβ–PrPC–Fyn signaling pathway, some will reach patients with AD over the next decade.
Abbreviations
- AD:
-
Alzheimer’s disease
- APP:
-
Amyloid precursor protein
- Aβ:
-
Amyloid-beta
- Aβo:
-
Amyloid-beta oligomer
- CNS:
-
Central nervous system
- PrPC:
-
Cellular prion protein
- PSD:
-
Post-synaptic density
- SFK:
-
Src family of nonreceptor tyrosine kinase.
References
Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM: Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009, 457: 1128-1132. 10.1038/nature07761.
Um JW, Nygaard HB, Heiss JK, Kostylev MA, Stagi M, Vortmeyer A, Wisniewski T, Gunther EC, Strittmatter SM: Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat Neurosci. 2012, 15: 1227-1235. 10.1038/nn.3178.
Um JW, Kaufman AC, Kostylev M, Heiss JK, Stagi M, Takahashi H, Kerrisk ME, Vortmeyer A, Wisniewski T, Koleske AJ, Gunther EC, Nygaard HB, Strittmatter SM: Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer Aβ oligomer bound to cellular prion protein. Neuron. 2013, 79: 887-902. 10.1016/j.neuron.2013.06.036.
Larson M, Sherman MA, Amar F, Nuvolone M, Schneider JA, Bennett DA, Aguzzi A, Lesne SE: The complex PrP(c)–Fyn couples human oligomeric Aβ with pathological tau changes in Alzheimer’s disease. J Neurosci. 2012, 32: 16857a-16871a. 10.1523/JNEUROSCI.1858-12.2012.
Thomas SM, Brugge JS: Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol. 1997, 13: 513-609. 10.1146/annurev.cellbio.13.1.513.
Martin GS: The hunting of the Src. Nat Rev Mol Cell Biol. 2001, 2: 467-475. 10.1038/35073094.
Boggon TJ, Eck MJ: Structure and regulation of Src family kinases. Oncogene. 2004, 23: 7918-7927. 10.1038/sj.onc.1208081.
Sicheri F, Kuriyan J: Structures of Src-family tyrosine kinases. Curr Opin Struct Biol. 1997, 7: 777-785. 10.1016/S0959-440X(97)80146-7.
Jelic D, Mildner B, Kostrun S, Nujic K, Verbanac D, Culic O, Antolovic R, Brandt W: Homology modeling of human Fyn kinase structure: discovery of rosmarinic acid as a new Fyn kinase inhibitor and in silico study of its possible binding modes. J Med Chem. 2007, 50: 1090-1100. 10.1021/jm0607202.
Yang K, Belrose J, Trepanier CH, Lei G, Jackson MF, MacDonald JF: Fyn, a potential target for Alzheimer’s disease. J Alzheimers Dis. 2011, 27: 243-252.
Goldsmith JF, Hall CG, Atkinson TP: Identification of an alternatively spliced isoform of the fyn tyrosine kinase. Biochem Biophys Res Commun. 2002, 298: 501-504. 10.1016/S0006-291X(02)02510-X.
Cooke MP, Perlmutter RM: Expression of a novel form of the fyn proto-oncogene in hematopoietic cells. New Biol. 1989, 1: 66-74.
Kramer-Albers EM, White R: From axon-glial signalling to myelination: the integrating role of oligodendroglial Fyn kinase. Cell Mol Life Sci. 2011, 68: 2003-2012. 10.1007/s00018-010-0616-z.
Nguyen TH, Liu J, Lombroso PJ: Striatal enriched phosphatase 61 dephosphorylates Fyn at phosphotyrosine 420. J Biol Chem. 2002, 277: 24274-24279. 10.1074/jbc.M111683200.
Resh MD: Fyn, a Src family tyrosine kinase. Int J Biochem Cell Biol. 1998, 30: 1159-1162. 10.1016/S1357-2725(98)00089-2.
Kennedy MB: Signal-processing machines at the postsynaptic density. Science. 2000, 290: 750-754. 10.1126/science.290.5492.750.
Nakazawa T, Komai S, Tezuka T, Hisatsune C, Umemori H, Semba K, Mishina M, Manabe T, Yamamoto T: Characterization of Fyn-mediated tyrosine phosphorylation sites on GluR epsilon 2 (NR2B) subunit of the N-methyl-D-aspartate receptor. J Biol Chem. 2001, 276: 693-699.
Kojima N, Ishibashi H, Obata K, Kandel ER: Higher seizure susceptibility and enhanced tyrosine phosphorylation of N-methyl-D-aspartate receptor subunit 2B in fyn transgenic mice. Learn Mem. 1998, 5: 429-445.
Grant SG, O’Dell TJ, Karl KA, Stein PL, Soriano P, Kandel ER: Impaired long-term potentiation, spatial learning, and hippocampal development in fyn mutant mice. Science. 1992, 258: 1903-1910. 10.1126/science.1361685.
Prybylowski K, Chang K, Sans N, Kan L, Vicini S, Wenthold RJ: The synaptic localization of NR2B-containing NMDA receptors is controlled by interactions with PDZ proteins and AP-2. Neuron. 2005, 47: 845-857. 10.1016/j.neuron.2005.08.016.
Suzuki T, Okumura-Noji K: NMDA receptor subunits epsilon 1 (NR2A) and epsilon 2 (NR2B) are substrates for Fyn in the postsynaptic density fraction isolated from the rat brain. Biochem Biophys Res Commun. 1995, 216: 582-588. 10.1006/bbrc.1995.2662.
Malenka RC, Nicoll RA: Long-term potentiation – a decade of progress?. Science. 1999, 285: 1870-1874. 10.1126/science.285.5435.1870.
Massey PV, Johnson BE, Moult PR, Auberson YP, Brown MW, Molnar E, Collingridge GL, Bashir ZI: Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J Neurosci. 2004, 24: 7821-7828. 10.1523/JNEUROSCI.1697-04.2004.
Isosaka T, Hattori K, Kida S, Kohno T, Nakazawa T, Yamamoto T, Yagi T, Yuasa S: Activation of Fyn tyrosine kinase in the mouse dorsal hippocampus is essential for contextual fear conditioning. Eur J Neurosci. 2008, 28: 973-981. 10.1111/j.1460-9568.2008.06405.x.
Lu YF, Kojima N, Tomizawa K, Moriwaki A, Matsushita M, Obata K, Matsui H: Enhanced synaptic transmission and reduced threshold for LTP induction in fyn-transgenic mice. Eur J Neurosci. 1999, 11: 75-82. 10.1046/j.1460-9568.1999.00407.x.
Babus LW, Little EM, Keenoy KE, Minami SS, Chen E, Song JM, Caviness J, Koo SY, Pak DT, Rebeck GW, Turner RS, Hoe HS: Decreased dendritic spine density and abnormal spine morphology in Fyn knockout mice. Brain Res. 2011, 1415: 96-102.
Umemori H, Sato S, Yagi T, Aizawa S, Yamamoto T: Initial events of myelination involve Fyn tyrosine kinase signalling. Nature. 1994, 367: 572-576. 10.1038/367572a0.
Sperber BR, Boyle-Walsh EA, Engleka MJ, Gadue P, Peterson AC, Stein PL, Scherer SS, McMorris FA: A unique role for Fyn in CNS myelination. J Neurosci. 2001, 21: 2039-2047.
Shirazi SK, Wood JG: The protein tyrosine kinase, fyn, in Alzheimer’s disease pathology. Neuroreport. 1993, 4: 435-437. 10.1097/00001756-199304000-00024.
Luo YQ, Hirashima N, Li YH, Alkon DL, Sunderland T, Etcheberrigaray R, Wolozin B: Physiological levels of beta-amyloid increase tyrosine phosphorylation and cytosolic calcium. Brain Res. 1995, 681: 65-74. 10.1016/0006-8993(95)00282-U.
Bamberger ME, Harris ME, McDonald DR, Husemann J, Landreth GE: A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J Neurosci. 2003, 23: 2665-2674.
Grace EA, Busciglio J: Aberrant activation of focal adhesion proteins mediates fibrillar amyloid beta-induced neuronal dystrophy. J Neurosci. 2003, 23: 493-502.
Klein WL, Krafft GA, Finch CE: Targeting small Aβ oligomers: the solution to an Alzheimer’s disease conundrum?. Trends Neurosci. 2001, 24: 219-224. 10.1016/S0166-2236(00)01749-5.
Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL: Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998, 95: 6448-6453. 10.1073/pnas.95.11.6448.
Chen S, Yadav SP, Surewicz WK: Interaction between human prion protein and amyloid-beta (Aβ) oligomers: role of N-terminal residues. J Biol Chem. 2010, 285: 26377-26383. 10.1074/jbc.M110.145516.
Calella AM, Farinelli M, Nuvolone M, Mirante O, Moos R, Falsig J, Mansuy IM, Aguzzi A: Prion protein and Aβ-related synaptic toxicity impairment. EMBO Mol Med. 2010, 2: 306-314. 10.1002/emmm.201000082.
Balducci C, Beeg M, Stravalaci M, Bastone A, Sclip A, Biasini E, Tapella L, Colombo L, Manzoni C, Borsello T, Chiesa R, Gobbi M, Salmona M, Forloni G: Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc Natl Acad Sci U S A. 2010, 107: 2295-2300. 10.1073/pnas.0911829107.
Kessels HW, Nguyen LN, Nabavi S, Malinow R: The prion protein as a receptor for amyloid-beta. Nature. 2010, 466: E3-E4. 10.1038/nature09217. discussion E4–E5
Cisse M, Sanchez PE, Kim DH, Ho K, Yu GQ, Mucke L: Ablation of cellular prion protein does not ameliorate abnormal neural network activity or cognitive dysfunction in the J20 line of human amyloid precursor protein transgenic mice. J Neurosci. 2011, 31: 10427-10431. 10.1523/JNEUROSCI.1459-11.2011.
Resenberger UK, Harmeier A, Woerner AC, Goodman JL, Muller V, Krishnan R, Vabulas RM, Kretzschmar HA, Lindquist S, Hartl FU, Multhaup G, Winklhofer KF, Tatzelt J: The cellular prion protein mediates neurotoxic signalling of beta-sheet-rich conformers independent of prion replication. EMBO J. 2011, 30: 2057-2070. 10.1038/emboj.2011.86.
Bate C, Williams A: Amyloid-beta-induced synapse damage is mediated via cross-linkage of cellular prion proteins. J Biol Chem. 2011, 286: 37955-37963. 10.1074/jbc.M111.248724.
Alier K, Ma L, Yang J, Westaway D, Jhamandas JH: Aβ inhibition of ionic conductance in mouse basal forebrain neurons is dependent upon the cellular prion protein PrPC. J Neurosci. 2011, 31: 16292-16297. 10.1523/JNEUROSCI.4367-11.2011.
Chung E, Ji Y, Sun Y, Kascsak RJ, Kascsak RB, Mehta PD, Strittmatter SM, Wisniewski T: Anti-PrPC monoclonal antibody infusion as a novel treatment for cognitive deficits in an Alzheimer’s disease model mouse. BMC Neurosci. 2010, 11: 130-10.1186/1471-2202-11-130.
Gimbel DA, Nygaard HB, Coffey EE, Gunther EC, Lauren J, Gimbel ZA, Strittmatter SM: Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J Neurosci. 2010, 30: 6367-6374. 10.1523/JNEUROSCI.0395-10.2010.
Kudo W, Lee HP, Zou WQ, Wang X, Perry G, Zhu X, Smith MA, Petersen RB, Lee HG: Cellular prion protein is essential for oligomeric amyloid-beta-induced neuronal cell death. Hum Mol Genet. 2012, 21: 1138-1144. 10.1093/hmg/ddr542.
You H, Tsutsui S, Hameed S, Kannanayakal TJ, Chen L, Xia P, Engbers JD, Lipton SA, Stys PK, Zamponi GW: Aβ neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-D-aspartate receptors. Proc Natl Acad Sci U S A. 2012, 109: 1737-1742. 10.1073/pnas.1110789109.
Zou WQ, Xiao X, Yuan J, Puoti G, Fujioka H, Wang X, Richardson S, Zhou X, Zou R, Li S, Zhu X, McGeer PL, McGeehan J, Kneale G, Rincon-Limas DE, Fernandez-Funez P, Lee HG, Smith MA, Petersen RB, Guo JP: Amyloid-beta42 interacts mainly with insoluble prion protein in the Alzheimer brain. J Biol Chem. 2011, 286: 15095-15105. 10.1074/jbc.M110.199356.
Freir DB, Nicoll AJ, Klyubin I, Panico S, McDonald JM, Risse E, Asante EA, Farrow MA, Sessions RB, Saibil HR, Clarke AR, Rowan MJ, Walsh DM, Collinge J: Interaction between prion protein and toxic amyloid beta assemblies can be therapeutically targeted at multiple sites. Nat Commun. 2011, 2: 336-
Barry AE, Klyubin I, McDonald JM, Mably AJ, Farrell MA, Scott M, Walsh DM, Rowan MJ: Alzheimer’s disease brain-derived amyloid-beta-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J Neurosci. 2011, 31: 7259-7263. 10.1523/JNEUROSCI.6500-10.2011.
Salter MW, Kalia LV: Src kinases: a hub for NMDA receptor regulation. Nat Rev. 2004, 5: 317-328.
Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL: Aβ oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J Neurosci. 2007, 27: 796-807. 10.1523/JNEUROSCI.3501-06.2007.
Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ: Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008, 14: 837-842. 10.1038/nm1782.
Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL: Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007, 27: 2866-2875. 10.1523/JNEUROSCI.4970-06.2007.
Selkoe DJ: Alzheimer’s disease is a synaptic failure. Science. 2002, 298: 789-791. 10.1126/science.1074069.
Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L: Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science. 2007, 316: 750-754. 10.1126/science.1141736.
Bhaskar K, Hobbs GA, Yen SH, Lee G: Tyrosine phosphorylation of tau accompanies disease progression in transgenic mouse models of tauopathy. Neuropathol Appl Neurobiol. 2010, 36: 462-477. 10.1111/j.1365-2990.2010.01103.x.
Bhaskar K, Yen SH, Lee G: Disease-related modifications in tau affect the interaction between Fyn and Tau. J Biol Chem. 2005, 280: 35119-35125. 10.1074/jbc.M505895200.
Lee G, Thangavel R, Sharma VM, Litersky JM, Bhaskar K, Fang SM, Do LH, Andreadis A, Van Hoesen G, Ksiezak-Reding H: Phosphorylation of tau by fyn: implications for Alzheimer’s disease. J Neurosci. 2004, 24: 2304-2312. 10.1523/JNEUROSCI.4162-03.2004.
Lee G, Newman ST, Gard DL, Band H, Panchamoorthy G: Tau interacts with src-family non-receptor tyrosine kinases. J Cell Sci. 1998, 111: 3167-3177.
Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, Wu T, Hamto P, Devidze N, Yu GQ, Palop JJ, Noebels JL, Mucke L: Amyloid-beta/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J Neurosci. 2011, 31: 700-711. 10.1523/JNEUROSCI.4152-10.2011.
Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wolfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J: Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell. 2010, 142: 387-397. 10.1016/j.cell.2010.06.036.
Cruchaga C, Kauwe JS, Mayo K, Spiegel N, Bertelsen S, Nowotny P, Shah AR, Abraham R, Hollingworth P, Harold D, Owen MM, Williams J, Lovestone S, Peskind ER, Li G, Leverenz JB, Galasko D, Morris JC, Fagan AM, Holtzman DM, Goate AM, Alzheimer’s Disease Neuroimaging Initiative: SNPs associated with cerebrospinal fluid phospho-tau levels influence rate of decline in Alzheimer’s disease. PLoS Genet. 2010, 6: e1001101-10.1371/journal.pgen.1001101.
Bekris LM, Millard S, Lutz F, Li G, Galasko DR, Farlow MR, Quinn JF, Kaye JA, Leverenz JB, Tsuang DW, Yu CE, Peskind ER: Tau phosphorylation pathway genes and cerebrospinal fluid tau levels in Alzheimer’s disease. Am J Med Genet B Neuropsychiatr Genet. 2012, 159B: 874-883. 10.1002/ajmg.b.32094.
Chin J, Palop JJ, Yu GQ, Kojima N, Masliah E, Mucke L: Fyn kinase modulates synaptotoxicity, but not aberrant sprouting, in human amyloid precursor protein transgenic mice. J Neurosci. 2004, 24: 4692-4697. 10.1523/JNEUROSCI.0277-04.2004.
Chin J, Palop JJ, Puolivali J, Massaro C, Bien-Ly N, Gerstein H, Scearce-Levie K, Masliah E, Mucke L: Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer’s disease. J Neurosci. 2005, 25: 9694-9703. 10.1523/JNEUROSCI.2980-05.2005.
Minami SS, Clifford TG, Hoe HS, Matsuoka Y, Rebeck GW: Fyn knock-down increases Abeta, decreases phospho-tau, and worsens spatial learning in 3xTg-AD mice. Neurobiol Aging. 2012, 33: 825-e15–e24
Zhang Y, Kurup P, Xu J, Carty N, Fernandez SM, Nygaard HB, Pittenger C, Greengard P, Strittmatter SM, Nairn AC, Lombroso PJ: Genetic reduction of striatal-enriched tyrosine phosphatase (STEP) reverses cognitive and cellular deficits in an Alzheimer’s disease mouse model. Proc Natl Acad Sci U S A. 2010, 107: 19014-19019. 10.1073/pnas.1013543107.
Dhawan G, Combs CK: Inhibition of Src kinase activity attenuates amyloid associated microgliosis in a murine model of Alzheimer’s disease. J Neuroinflamm. 2012, 9: 117-10.1186/1742-2094-9-117.
Baer AS, Syed YA, Kang SU, Mitteregger D, Vig R, Ffrench-Constant C, Franklin RJ, Altmann F, Lubec G, Kotter MR: Myelin-mediated inhibition of oligodendrocyte precursor differentiation can be overcome by pharmacological modulation of Fyn-RhoA and protein kinase C signalling. Brain. 2009, 132: 465-481.
Appleby MW, Gross JA, Cooke MP, Levin SD, Qian X, Perlmutter RM: Defective T cell receptor signaling in mice lacking the thymic isoform of p59fyn. Cell. 1992, 70: 751-763. 10.1016/0092-8674(92)90309-Z.
Tamura T, Igarashi O, Hino A, Yamane H, Aizawa S, Kato T, Nariuchi H: Impairment in the expression and activity of Fyn during differentiation of naive CD4+ T cells into the Th2 subset. J Immunol. 2001, 167: 1962-1969.
Reddy KB, Smith DM, Plow EF: Analysis of Fyn function in hemostasis and alphaIIbbeta3-integrin signaling. J Cell Sci. 2008, 121: 1641-1648. 10.1242/jcs.014076.
Piette F, Belmin J, Vincent H, Schmidt N, Pariel S, Verny M, Marquis C, Mely J, Hugonot-Diener L, Kinet JP, Dubreuil P, Moussy A, Hermine O: Masitinib as an adjunct therapy for mild-to-moderate Alzheimer’s disease: a randomised, placebo-controlled phase 2 trial. Alzheimers Res Ther. 2011, 3: 16-10.1186/alzrt75.
Hennequin LF, Allen J, Breed J, Curwen J, Fennell M, Green TP, Lambert-van der Brempt C, Morgentin R, Norman RA, Olivier A, Otterbein L, Plé PA, Warin N, Costello G: N-(5-chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5-(tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine, a novel, highly selective, orally available, dual-specific c-Src/Abl kinase inhibitor. J Med Chem. 2006, 49: 6465-6488. 10.1021/jm060434q.
Gangadhar TC, Clark JI, Karrison T, Gajewski TF: Phase II study of the Src kinase inhibitor saracatinib (AZD0530) in metastatic melanoma. Invest New Drugs. 2013, 31: 769-773. 10.1007/s10637-012-9897-4.
Gucalp A, Sparano JA, Caravelli J, Santamauro J, Patil S, Abbruzzi A, Pellegrino C, Bromberg J, Dang C, Theodoulou M, Massague J, Norton L, Hudis C, Traina TA: Phase II trial of saracatinib (AZD0530), an oral SRC-inhibitor for the treatment of patients with hormone receptor-negative metastatic breast cancer. Clin Breast Cancer. 2011, 11: 306-311. 10.1016/j.clbc.2011.03.021.
Mackay HJ, Au HJ, McWhirter E, Alcindor T, Jarvi A, MacAlpine K, Wang L, Wright JJ, Oza AM: A phase II trial of the Src kinase inhibitor saracatinib (AZD0530) in patients with metastatic or locally advanced gastric or gastro esophageal junction (GEJ) adenocarcinoma: a trial of the PMH phase II consortium. Invest New Drugs. 2012, 30: 1158-1163. 10.1007/s10637-011-9650-4.
Fury MG, Baxi S, Shen R, Kelly KW, Lipson BL, Carlson D, Stambuk H, Haque S, Pfister DG: Phase II study of saracatinib (AZD0530) for patients with recurrent or metastatic head and neck squamous cell carcinoma (HNSCC). Anticancer Res. 2011, 31: 249-253.
Renouf DJ, Moore MJ, Hedley D, Gill S, Jonker D, Chen E, Walde D, Goel R, Southwood B, Gauthier I, Walsh W, McIntosh L, Seymour L: A phase I/II study of the Src inhibitor saracatinib (AZD0530) in combination with gemcitabine in advanced pancreatic cancer. Invest New Drugs. 2012, 30: 779-786. 10.1007/s10637-010-9611-3.
Acknowledgements
The authors acknowledge support from the National Institutes of Health to HBN, CHvD and SMS, and from the Falk Medical Research Trust to SMS.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
SMS is a co-founder of Axerion Therapeutics, seeking to develop Nogo-receptor-based and PrP-based therapeutics. In relation to AD therapeutics, CHvD has served as a scientific advisor or consultant to Bristol-Myers Squibb, Janssen AI, Pfizer, Glaxo Smith Kline, Elan Pharmaceuticals, Roche Pharmaceuticals, and Abbott/AbbVie Inc. CHvD has received research support from Bristol-Myers Squibb, Elan Pharmaceuticals, Janssen Alzheimer’s Immunotherapy, Pfizer Inc., Eli Lilly, Merck, Baxter Pharmaceuticals, GlaxoSmithKline, Abbott Laboratories, Medivation, Inc., Biogen Idec, Eisai, Inc., Genentech, Inc., and Roche Pharmaceuticals. HBN declares that he has no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Nygaard, H.B., van Dyck, C.H. & Strittmatter, S.M. Fyn kinase inhibition as a novel therapy for Alzheimer’s disease. Alz Res Therapy 6, 8 (2014). https://doi.org/10.1186/alzrt238
Published:
DOI: https://doi.org/10.1186/alzrt238