
Abstract
Background
Cockroaches are omnivorous animals that can incorporate in their diets food of different composition, including lignocellulosic materials. Digestion of these compounds is achieved by the insect’s own enzymes and also by enzymes produced by gut symbiont. However, the influence of diet with different fiber contents on gut bacterial communities and how this affects the digestion of cockroaches is still unclear. The presence of some bacterial phyla on gut tract suggests that cockroaches could be an interesting model to study the organization of gut bacterial communities during digestion of different lignocellulosic diets. Knowledge about the changes in diversity of gut associated bacterial communities of insects exposed to such diets could give interesting insights on how to improve hemicellulose and cellulose breakdown systems.
Methodology/principal findings
We compared the phylogenetic diversity and composition of gut associated bacteria in the cockroach P. americana caught on the wild or kept on two different diets: sugarcane bagasse and crystalline cellulose. For this purpose we constructed bacterial 16S rRNA gene libraries which showed that a diet rich in cellulose and sugarcane bagasse favors the predominance of some bacterial phyla, more remarkably Firmicutes, when compared to wild cockroaches. Rarefaction analysis, LIBSHUFF and UniFrac PCA comparisons showed that gene libraries of wild insects were the most diverse, followed by sugarcane bagasse fed and then cellulose fed animals. It is also noteworthy that cellulose and sugarcane bagasse gene libraries resemble each other.
Conclusion/significance
Our data show a high bacterial diversity in P. americana gut, with communities composed mostly by the phyla Bacteroidetes, Firmicutes, Proteobacteria and Synergistetes. The composition and diversity of gut bacterial communities could be modulated by font of diet composition. The increased presence of Firmicutes in sugarcane bagasse and crystalline cellulose-fed animals suggests that these bacteria are strongly involved in lignocellulose digestion in cockroach guts.
Cockroaches are insects that can accommodate diets of different composition, including lignocellulosic materials. Digestion of these compounds is achieved by the insect’s own enzymes and also by enzymes produced by gut symbionts. The presence of different and modular bacterial phyla on the cockroach gut tract suggests that this insect could be an interesting model to study the organization of gut bacterial communities associated with the digestion of different lignocellulosic diets. Thus, changes in the diversity of gut associated bacterial communities of insects exposed to such diets could give useful insights on how to improve hemicellulose and cellulose breakdown systems. In this work, through sequence analysis of 16S rRNA clone libraries, we compared the phylogenetic diversity and composition of gut associated bacteria in the cockroach Periplaneta americana collected in the wild-types or kept on two different diets: sugarcane bagasse and crystalline cellulose. These high fiber diets favor the predominance of some bacterial phyla, such as Firmicutes, when compared to wild-types cockroaches. Our data show a high bacterial diversity in P. americana gut, with communities composed mostly by the phyla Bacteroidetes, Firmicutes, Proteobacteria and Synergistetes. Our data show that the composition and diversity of gut bacterial communities could be modulated by diet composition. The increased presence of Firmicutes in sugarcane bagasse and crystalline cellulose-fed animals suggests that these bacteria are strongly involved in lignocellulose digestion in cockroach guts.
Similar content being viewed by others


Introduction
Lignocellulosic materials, such as sugarcane bagasse, have been considered promising materials in the biofuel industry for the synthesis of second generation ethanol. However, the affordable production of these biofuels derived from plant biomass is currently dependent on the discovery of new enzymes to increase the efficiency of cellulose hydrolysis. In this context, animals such as insects which feed on plant material or wood may possess interesting biochemical pathways to promote an efficient hydrolysis of plant polymers, solving inherent problems to the digestion of lignocellulose, including detoxification of secondary plant phytochemicals and enzyme inhibitors (Morrison et al. 2009). In insects, the digestion of cellulose and hemicellulose has been observed in several orders such as Thysanura, Plecoptera, Orthoptera, Isoptera, Coleoptera, Trichoptera, Hymenoptera, Phasmida, Blattodea and Diptera (Sun and Scharf 2010).
Several cockroaches (including P. americana) are omnivorous insects that feed and survive on different food sources including cellulose-rich compounds. In this insect, the production of endogenous glucanases is predominantly associated to salivary glands and the midgut (Genta et al. 2003; Bignell 1981). The cockroaches, as well as low termites (Inward et al. 2007), digest these polymers by cooperation between two systems: endogenous insect enzymes and enzymes secreted by a variety of gut microorganisms, including protozoa and bacteria (Sun and Scharf 2010). The digestion of lignocellulosic compounds in insect guts, in a general view, comprises three basic stages involving insect and microbial enzymes: Stage I: hydrolysis, Stage II: oxidation and/or fermentation and Stage III acetogenesis and/or methanogenesis (including the participation of Archaea) (Hongo et al. 2005; Gill et al. 2006; Konig 2006; Hongoh 2010; Watanabe and Tokuda 2010). In insects that digest cellulosis, the trituration (grinding) of fiber material is arguably Stage I, for which both cockroaches and termites have a well-developed gizzard (Hongoh 2010). In termites, the anatomical adaptations of the gut form specialized micro-environmental chambers with different pH levels and redox potential that promote an important increase in concentrations and functionality of the intestinal enzymes. These chambers allow the effective breakdown of lignocellulosic biomass and a concomitant release of sugar monomers (Watanabe and Tokuda 2010). These microorganisms are absolutely essential to the digestion of many different animals, from insects to humans, and their diversity has been mainly studied by 16S rRNA sequence analysis. This approach has revealed the presence of several bacterial species that are mostly affiliated, but not restricted, to the phyla Firmicutes, Actinobacteria, Bacterioidetes, and Spirochetes (Hongo et al. 2005; Gill et al. 2006) that are strongly associated to the stage I of digestion.
Spatial characteristics of insect guts may harbor a significant population of specialized resident bacteria in these different microenvironments (Brune 1998; Dillon and Dillon 2004). Several other factors can influence microbiota composition of animal guts such as the host immune system, environmental microbial inputs, and the presence of specialized intestinal anatomical structures, the pH of distinct segments, the redox potential during food passage and also the diet. It has been shown that diet components alter gut microbiota composition in several organisms such as humans, pigs, dogs, snails and others (Leser et al. 2000; Konstantinov et al. 2002; Middelbos et al. 2010; Cardoso et al. 2012). Changing the foraging source from grain to hay in the diet, for example, can significantly change the bacterial population of bovine rumen (Tajima et al. 2001). Furthermore, in P. americana, a cellulose-rich diet induces a specific growth of the hindgut microbiota, mainly of methanogens and the anaerobic ciliate protozoan Nyctotherus ovalis, that could be involved in increasing cellulolytic activity and methane production (Gijzen and Barugahare 1992; Gijzen et al. 1994). Thus, the adaptation of the gut microbiota to dietary changes could be an important mechanism for digestion, especially for displaying new specific enzymes associated with the degradation of particular types of polymers.
Thus, the guts of termites and cockroaches may represent a large source of untapped microbial diversity and, in this sense, culture-independent methods provide a powerful opportunity to study the species richness of insect gut bacteria involved in plant cell wall deconstruction and possibly help to discover new microbial enzymes for biofuel production (Warnecke et al. 2007). Considering that there is little information on how diet affects the composition of gut microbiota in cockroaches, this work describes for the first time the influence of sugarcane bagasse and crystalline cellulose diets on P. Americana gut bacterial communities.
Materials and methods
Experimental design
Adult male and female cockroaches (Figure 1A) were selected from an established colony and kept under a natural light regime and fed with different diets. The animals were separated into individual containers and exclusively fed with dried finely mowed sugarcane bagasse or cellulose (Avicel® PH 101 Sigma Aldrish code product 11365, PA, USA) ad libitum for at least two weeks.

Digestive system of P. americana. (A) Adult insect and (B) Whole gut. FG–Foregut; MG–Midgut; HG; Hindgut.
The industrial sugarcane bagasse used in this study has around 42% cellulose, 27% hemicellulose, 20% lignin, ashes (2%) and other substances (9%). All animals used in the experiments were able to consume the food provided and survived at least one month in these conditions. Water was offered in wet pieces of glass wool. Adult cockroaches (males and females) from wild-types were collected from sewage drains at Universidade Federal do Rio de Janeiro and immediately used.
DNA extraction
Ten cockroaches of each of the three groups (wild-types, bagasse or cellulose) were dissected and had their gut contents (Figure 1B) removed in sterile PBS (phosphate-buffered saline 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4 pH 7.4 10 mM) and pooled. Animals that, eventually, showed no gut content on dissection were discarded. Gut content was homogenized and centrifuged at 14,000 g for 10 minutes at 4°C. Supernatants were discarded and pellets were stored at-70°C until DNA extraction. Nucleic acids were isolated from the pellets by cell lysis with proteinase K and SDS, followed by phenol-chloroform extraction (Vieira et al. 2007). DNA integrity was checked on a 1% (w/v) agarose gel.
Bacterial 16S rRNA gene library construction
PCR was performed in 50 μl reaction mixtures (2.5 mM MgCl2, 0.2 mM deoxynucleoside triphosphates, 50 ng of each primer, 0.25 U of High Fidelity Taq DNA polymerase (Promega), PCR buffer and 200 ng of DNA sample, using the universal bacterial primers 27BF (59-AGAGTTTGATCCTGGCTCAG-39) (Lane 1991) and 907RAB (59-TTTGAGTTT MCTTAACTGCC-39) 23-(Weisburg et al. 1991). PCR amplification was performed with a 5 min denaturing step at 94°C followed by 45 cycles of 94°C for 90 seconds, 55°C for 90 seconds, and 72°C for 2 min. The final cycle was an extension at 72°C for 10 min. PCR products were concentrated and purified with a GFx PCR DNA and Gel Band Purification Kit (GE Healthcare) after electrophoresis on a 1% (w/v) agarose gel. PCR products were cloned into the pGEM-T cloning vector (Promega) and used to transform competent E. coli DH10B cells. Positive colonies in the blue-white screen used for this vector were picked and frozen at -70°C.
Sequence analyses and taxa identification
Approximately 96 clones from each of the three libraries were subjected to sequence analysis. Plasmid DNA from each clone (400 ng) was prepared and PCR sequencing reactions with primer 27BF were carried out using the DYEnamic ET terminator cycle-sequencing kit (GE Healthcare). Partial 16S rRNA sequences were obtained by capillary electrophoresis on a MegaBace1000 DNA analysis system (GE Healthcare). Chromatograms were transformed into Fasta format with Phred software (Edwing et al. 1998) and sequences with less than 300 bp and chimeras were removed prior to further analysis using MOTHUR (Schloss et al. 2009). A total of 216 (75/Bagasse; 79/cellulose; 62/Wild-types) valid sequences were compared with sequences in the Ribosomal Database Project II 26-(Cole et al. 2003). The Sequences were also analyzed by BLAST 27-(Altschul et al. 1990) searches in the GenBank database (http://www.ncbi.nlm.nih.gov) and were aligned with representative bacterial sequences obtained from the public databases using ClustalX software (Thompson et al. 1997). The partial 16S rRNA gene sequences generated in this study has been deposited in GenBank under accession numbers file barataseqin2: JX887210-JX887422.
Biodiversity and phylogenetic analyses
Resampling and adjustment of the total number of sequence reads to identical sequencing depth was done before analysis (Gilbert et al. 2010). Sequences were clustered as OTUs at an overlap identity cutoff of 97% or 80% by MOTHUR software (Schloss et al. 2009). The diversity of OTUs and community overlaps were also examined using rarefaction analysis and Venn diagrams. The rarefaction analysis related the number of OTUs discovered to the number of samples taken, to discover whether additional sequences would discover additional taxa. The identity of each 16S rRNA sequence was determined by BLAST-n searches against the NCBI GenBank database. Phylogenetic trees were constructed for P. americana gut bacterial libraries with reference sequences from GenBank by the neighbor-joining algorithm based on distances calculated by the Kimura-2 method. This analysis was performed with the MEGA4 program (Kumar et al. 2001) and bootstrap analysis with 1000 replications was used. Tree topology and distribution of hits along the tree were uploaded to the UniFrac computational platform (Lozupone et al. 2007). UniFrac is a beta diversity metric analysis that quantifies community similarities based on phylogenetic relatedness. In order to visualize distribution patterns of bacterial communities we used the UniFrac metric to perform PCA highlighted by significance.
Statistical comparison between 16S rRNA libraries
In an attempt to determine the differences between clone libraries, we applied LIBSHUFF statistics (Schloss et al. 2004) that uses Monte Carlo methods to generate homologous and heterologous coverage curves. Sequences were randomly shuffled 999 times between samples prior to the distance between curves being calculated using Cramer-von Mise statistic test. The DNADIST program of the PHYLIP package, using the Jukes-Cantor model for nucleotide substitution was used to generate the distance matrix analyzed by LIBSHUFF.
Results
A total of 216 valid sequences of approximately 700-800 bp were obtained from three 16S rRNA gene libraries, 62 from wild-types insects and 75 and 79 from sugarcane and cellulose fed cockroaches, respectively. The sequences were assigned to distinct taxonomic phyla with the RDP classifier tool (http://rdp.cme.msu.edu/classifierr/classifier.jsp). In wild-type animals, the Proteobacteria and Bacteroidetes were the predominant sequences of these phyla. Most sequences from the sugarcane and cellulose fed libraries were represented by the Firmicutes and Bacteroidetes phyla, as observed in the microbiota of other animals submitted to high fiber content diets (Ley et al. 2008). In the gut of cellulose fed animals, Firmicutes was by far the most abundant phylum (Figure 2), comprising more than 80% of the sequences. In wild-types animals, BLAST searches (http://blast.ncbi.nlm.nih.gov/Blast) performed with the bacterial sequences that could not be classified with the RDP tool (described as unclassified in Figure 2) retrieved only low similarity sequences suggesting that these microorganisms possibly represent new bacterial groups.

Distribution bacterial phyla in 16S rRNA gene sequences from cockroaches submitted to different diets. Gut bacterial DNA was obtained from cockroaches fed on sugarcane bagasse or cellulose (both for at least one week) and from wild-types cockroaches (collected from sewage). Clone libraries from amplified 16S rRNA genes were prepared and sequenced. Nucleotide sequences were submitted to the RDP Classifier tool at 80% bootstrap cutoffs.
Rarefaction analysis indicates that the libraries exhibit different levels of diversity. Insects fed cellulose and sugarcane bagasse showed lower bacterial diversity than wild-types cockroaches. Rarefaction curves clustered at 97% (Figure 3A) or 80% (Figure 3B) similarity showed higher bacterial species diversity within the wild-types insects when compared with the insects fed cellulose-rich diets. The decline in the rate of OTUs detection at 80% cut-off denotes that only the most predominant sequences of these bacterial phyla have been observed in the animals fed higher fiber diets. Furthermore, at 97% the cut-off curves show that diversity at genus/species level was not entirely detected, mainly for wild-types animals. The number of clones sequenced from wild-types insects was not enough to cover the whole bacterial diversity, although diversity has been achieved almost completely in sugarcane bagasse or cellulose fed animals. Table 1 shows a comparison by LIBSHUFF statistics showing that bacterial community composition differed significantly between wild-types animals and higher fiber diet-fed (cellulose and sugarcane bagasse groups) cockroaches (p < 0.0001). On the other hand, when the libraries of insects fed a fiber-rich diet were compared between themselves, no significant differences were observed (p = 0.9355 and p = 0.0030).

Rarefaction analysis of 16S rRNA gene sequences from gut contents of wild-types cockroaches and those fed on sugarcane bagasse or cellulose. The total number of sequences was plotted against unique OTUs defined by using a distance level of 97% (A) or 80% (B) using the furthest neighbor assignment algorithm in MOTHUR.
No OTUs were shared among the three groups analyzed in this work (Figure 4). Venn diagram shows that only one OUT from the wild-types insect library was shared with the cellulose fed group while none was shared between wild-types and sugarcane fed insects (Figure 4B). Six OTUs were shared between insects fed sugarcane bagasse and cellulose (mainly bacilli). Libraries were randomly sub-sampled and then community similarity was quantified based on phylogenetic relatedness by unweight UniFrac in a PCA plot (Figure 4A). In the scatter plot, the first two principal coordinates, PC1 and PC2, explained 13.4% and 7.1% of the data variation, respectively, clearly separating each community. These results suggest that there is a possible specialization of intestinal microbiota associated with a fiber-rich diet in both groups.

Match between bacterial communities in cockroaches fed different diets. (A) Similarity between bacterial communities. Principal coordinates plots (PCA) were generated using the pair wise unweighted UniFrac distances. □ Wild-types insects; Δ Insects fed sugarcane bagasse; ○ Insects fed cellulose; (B) Venn diagram with OTUs grouped at 97% similarity in the phylogenetic tree of bacterial clones obtained from cockroaches fed with different diets.
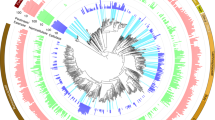
The phylogenetic tree showing the bacterial phylotypes retrieved from our clone libraries are shown in Figure 5. The wild-types cockroach group (W) shows OTUs mainly related to three groups: Proteobacteria, Bacteroidetes and Synergistetes. Clones corresponding to Proteobacteria are assigned to several different genera such as Brucella, Rhodobacter, Acinetobacter, Aeromonas and Escherichia. Clones representing the Bacteroidetes are mainly distributed through several genera such as Blattabacterium, Elizabethkingia and Bacteroidales while clones from Synergistetes are represented by three clones whose sequences are very similar to bacteria retrieved from the termite Macrotermes gilvus.

Neighbor-joining phylogenetic tree of 16S rRNA gene sequences. Cockroach bacterial clones from SB (fed on sugarcane bagasse), C (fed on cellulose) or W (wild-types). Reference sequences were retrieved from Genbank (in bold). Phylogenetic trees were constructed for P. americana gut 16S rRNA bacterial sequences with reference sequences from GenBank by the neighbor-joining algorithm based on distances calculated by the Kimura-2 method.
Among sugarcane bagasse (SB) fed insects, the Firmicutes and Bacteroidetes were the main phyla observed. Firmicutes are predominantly represented by the two genera Lactobacillus and Enterococcus. Bacteroidetes are exclusively represented by the genus Elizabethkingia. Among the Proteobacteria, few clones are assigned to the genera Rhodobacter and Massilia. Regarding cellulose (C) fed animals, the predominant sequences retrieved phyla were also Firmicutes and Bacteroidetes. In this group the Firmicutes are largely represented by the genera Lactobacillus, Paralactobacillus and Enterococcus. Five clones are similar to a bacterium from humus-feeding larva of Pachnoda ephippiata. The Bacteroidetes are principally represented by two genera, Blattabacterium and Elizabethkingia. Among the Proteobacteria, few clones are assigned to the genera Acinetobacter, Achromobacter and Stenotrophomonas.
Discussion
This work studied the influence of diet on gut bacterial communities of P. americana. Here the 16S rDNAs sequencing approach was used to explore the plasticity of the intestinal flora of Periplaneta americana, comparing wild-types insects with others fed on different diets. The analysis of 16S rRNA sequences showed that the diet had a significant influence on gut microbial communities in this insect. It is important to note that the conditioning of these insects to different diets had no effect on animal survival rate during the study period (data not shown). Our results show clearly that there is a significant increase in Firmicutes presence and also an important decrease in Proteobacteria and Synergistetes in the gut of animals fed cellulose-rich diet (Figure 2).
An association between bacteria and insects during biomass degradation is observed in several models. The leaf-cutter ant Atta colombicai, for example, uses fresh leaves to cultivate a fungus and also bacteria (mainly γ-Proteobacteria of the family Enterobacteriaceae) that produce high numbers of different cellulases and hemicellulases in specialized biodegrading gardens (Suen et al. 2010). In higher termites (that do not have symbiotic protists in the gut), such as Cornitermes cumulans and Nasutitermes sp, a high bacterial diversity is observed, basically composed by the phyla Proteobacteria, Spirochaeta, Bacteroidetes, Firmicutes, Actinobacteria, Fibrobacter and Treponema (Gijzen et al. 1994; Grieco et al. 2012). Phylogenetic analysis of gut bacteria from the low termite Reticulitermes flavipes (that harbors protist symbionts in the gut) and Cryptocercus (wood-feeding cockroach) showed a diverse range of members of major bacterial phyla, such as Proteobacteria, Spirochaetes, Bacteroidetes, Firmicutes, Actinobacteria, Synergistetes and the newly proposed Endomicrobia (Fisher et al. 2007; Berlanga et al. 2009). The gut microbiota of the cockroach Shelfordella lateralis was dominated by members of the Bacteroidetes and Firmicutes (mainly Clostridia), however, Deltaproteobacteria, Spirochaetes and Fibrobacteres, which are abundant members of termite gut communities, were absent in this insect (Schauer et al. 2012).
The impact of diet on intestinal microbiota is also observed in other models such as dogs (Middelbos et al. 2010), snails (Cardoso et al. 2012), cattle (Kong et al. 2010; Hess et al. 2011), sheep (Cunha et al. 2011) and humans (Ley et al. 2006) for example. In the higher termite Nasutitermes takasagoensis, it was demonstrated that the intestinal bacterial community structure is not so stable, varying depending on diet composition. The Spirochaetes was predominant sequences in the wood-feeding termites, whereas Bacteroidetes was more abundant in the gut of xylophagous termites. Firmicutes was predominant sequences in xylose fed termites (Miyata et al. 2007). The analysis of the termite Reticulitermes flavipes gut microbiome submitted to different diets showed that diet, environment and host genetics have important effects over microbiome composition (Boucias et al. 2013). Our results showed that the main Proteobacteria clones associated to wild-types cockroaches are distributed along several genera, including typical bacteria from sludge such as Brucella and Alphaproteobacteria.
The presence of microorganisms in the gut of American cockroach P. americana was initially described by light, scanning and transmission electron microscopy. The results suggested the presence of a complex community including protozoa, bacteria and archaea (Gijzen and Barugahare 1992; Bignell et al. 1977). Previous studies showed that a cellulose rich diet induces an increase in the population of protozoa and also in methanogenesis in the hindgut of this insect 19-(Gijzen et al. 1994; Kane and Breznak 1991). In cockroaches and termites, the protists involved in lignocellulose degradation use not only their own enzymes, but also could use the enzymes originated from its endo and/or ecto symbiotic bacteria (Todaka et al. 2010). The greater complexity of nutrients within the wild-types and sugarcane bagasse diets compared to the simple diet (only cellulose) suggests that a greater repertoire of bacteria may be required to efficiently utilize all of the nutritional components of more complex foods. Results from our laboratory show that these bacterial community alterations associated with changes in dietary composition triggered some changes in the intestinal enzyme profile. For example, there is an important increase on Endo-1,3(4)-β-glucanase and Endo-beta-1,3-1,4 glucanase II activities of anterior intestine of insect fed sugarcane bagasse compared to other two groups (data not shown). An interesting hypothesis is that these specialized bacteria could help to digest complex dietary polymers during the passage through the first segments of the insect’s intestinal tract. After that, host enzymes could digest these bacteria as a nutrient source in the last gut segments as observed in flies with a strong participation of cathepsin-D-like proteases and lysozyme (Lemos and Terra 1991).
The exact role of the cockroach microbiota in biomass degradation still remains unknown. Bacteria from the phylum Bacteroidetes (formerly known as Cytophaga-Flavobacteria-Bacteroides-CFB group) are involved in associations with a wide variety of gut protist species as either intracellular endosymbionts or surface-attached ectosymbionts. These bacteria digest a wide variety of substrates, including complex polymers, such as cellulose and chitin, using various glycosyl hydrolases (Noda et al. 2006; Mahowald et al. 2009). It is important to note that all Bacteroidetes related clones retrieved from sugarcane bagasse fed insects are affiliated with Elizabethkingia miricola. These bacteria are Gram-negative, non-motile, that can utilize several glycosidic substrates such as D-fructose, D-glucose, D-maltose, 2-naphthyl-alpha-D-glucopyranoside, 1-naphthyl-N-acetyl-beta-D-glucosaminide and 2-naphthyl-alpha-L-fucopyranoside (Kim et al. 2005).
In the intestine (including in humans), Firmicutes are strongly involved in fermentation processes and may be partners in many catabolic activities such as those observed in the degradation of glucose to generate several catabolites as lactate, ethanol, H2 and CO2 (Ley et al. 2006; Wüst et al. 2011). These bacteria could also reduce sulfate, degrade volatile fatty acids, such as butyrate and its analogs, and provide H2 to archeal methanogens (Ley et al. 2008; Rivière et al. 2009). Most clones from cockroaches fed on a cellulose-rich diet (Sugarcane Bagasse and Cellulose groups) are distributed in two main distantly related species, Lactobacillus dextrinicus and Enterococcus asini that are examples of typical fecal bacteria (Furet et al. 2009). The Synergistetes phyla includes Gram-negative, rod-shaped bacteria isolated from humans, animals and terrestrial and oceanic bacteria that metabolize amino acids and proteins to provide short-chain fatty acids and sulfate for methanogens and sulfate-reducing bacteria (Vartoukian et al. 2007). Our data do not show the presence of bacteria from the genus Clostridium, which is an important group in the rumen of cattle involved in fiber degradation by the enzymatic complex called cellulosome (Ley et al. 2006). This result is also different from Nasutitermes takasagoensis where there is an important presence of Clostridia on the mixed segments of this higher termite (Tokuda et al. 2000).
The enzymatic repertoire involved in digestion of lignocelullosis in insects could include glycoside hydrolases, laccase, peroxidases and detoxification proteins such as superoxide dismutase and catalase (Scharf and Boucias 2010). Considering that insects like cockroaches and termites perform the pretreatment and hydrolysis under mild conditions within a few millimeters of intestinal tissue, our data demonstrate that there is an important specialization of the microbiota in fiber digestion. Thus, the knowledge about gut microorganisms and their enzymes involved in the pretreatment and hydrolysis of biomass could be useful for new insights related to the development of bioethanol or other high-value products.
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol 1990, 5: 403-410.
Berlanga M, Bruce J, Paster BJ, Guerrero R: The taxophysiological paradox: changes in the intestinal microbiota of the xylophagous cockroach Cryptocercus punctulatus depending on the physiological state of the host. Intern Microbiol 2009, 12: 227-236. Doi: 10.2436/20.1501.01.102
Bignell DE: Some observations on the distribution of gut flora in the American cockroach periplaneta Americana. J Invert Pathol 1977, 29: 338-343.
Bignell DE: Nutrition and digestion. In The American Cockroach. Edited by: Bell WJ, Adiyodi KG. London and New York: Chapman and Hall; 1981:57-86.
Boucias DG, Cai Y, Sun Y, Lietze VU, Sen R, Raychoudhury R, Scharf ME: The hindgut lumen prokaryotic microbiota of the termite Reticulitermes flavipes and its responses to dietary lignocellulose composition. Molec Ecol 2013. doi: 10.1111/mec.12230
Brune A: Termite guts: the world’s smallest bioreactors. Trends Biotechnol 1998, 16: 16-21. Termite guts: the world’s smallest bioreactor. DOI.org/10.1016/S0167-7799(97)01151-7
Cardoso AM, Cavalcante JJV, Vieira RP, Lima JL, Grieco MAB, Clementino MM, Vasconcelos ATR, Garcia ES, De Souza W, Albano RM, Gut MOB: Bacterial communities in the Giant Land Snail Achatina fulica and their modification by sugarcane-based diet. PLoS ONE 2012, 7(3):E33440. DOI: 10.1371/journal.pone.0033440
Cole JR, Chai B, Marsh TL, Farris RJ, Wang Q, Kulam SA, Chandra S, McGarrell DM, Schmidt TM, Garrity GM, Tiedje JM: The Ribosomal Database Project (RDP-II): previewing a new auto aligner that allows regular updates and the new prokaryotic taxonomy. Nucleic Acids Res 2003, 31: 442-443. 10.1093/nar/gkg039
Cunha IS, Barreto CC, Costa OU, Bomfim MA, Castro AP, Kruger RH, Quirino BF: Bacteria and Archaea community structure in the rumen microbiome of goats ( Capra hircus ) from the semiarid region of Brazil. Anaerobe 2011, 17(3):118-124. DOI: 10.1016/j.anaerobe.2011.04.018
Dillon RJ, Dillon VM: The gut bacteria of insects: nonpathogenic interactions. Annual Rev Entomol 2004, 49: 71-92. Doi: 10.1146/annurev.ento.49.061802.123416
Edwing B, Hillier L, Wendl M, Green P: Base-calling of automated sequencer traces using phred, I: accuracy assessment. Gen Res 1998, 8: 175-185. DOI: 10.1101/gr.8.3.175
Fisher M, Miller D, Brewster C, Husseneder C, Dickerman A: Diversity of Gut Bacteria of Reticulitermes flavipes as examined by 16S rRNA Gene Sequencing and Amplified rDNA Restriction Analysis. Curr Microbiol 2007, 55: 254-259. Doi: 10.1007/s00284-007-0136-8
Furet JP, Firmesse O, Gourmelon M, Bridonneau C, Tap J, Mondot S, Doré J, Corthier G: Comparative assessment of human and farm animal faecal microbiota using real-time quantitative PCR. FEMS Microbiol Ecol 2009, 68(3):351-362. Doi: 10.1111/j.1574-6941.2009.00671.x
Genta FA, Terra WR, Ferreira C: Action pattern, specificity, lytic activities, and physiological role of five digestive β-glucanases isolated from Periplaneta americana . Insect Biochem Molec Biol 2003, 33: 1085-1097. DOI.org/10.1016/S0965-1748(03)00121-8
Gijzen HJ, Barugahare M: Contribution of Anaerobic Protozoa and Methanogens to hindgut metabolic activities of the american cockroach, Periplaneta americana . Appl Environ Microbiol 1992, 58(8):2565-2570.
Gijzen HJ, Drift CVD, Barugahare M, Camp HJM: Effect of host diet and hindgut microbial composition on cellulolytic activity in the Hindgut of the American Cockroach, Periplaneta americana . Appl Environ Microbiol 1994, 60(6):1822-1826.
Gilbert JA, Field D, Swift P, Thomas S, Cummings D, Temperton B, Weynberg K, Huse S, Hughes M, Joint I, Somerfield PJ, Mühling M: The taxonomic and functional diversity of microbes at a temperate coastal site: a ‘multi-omic’ study of seasonal and diel temporal variation. PLoS ONE 2010, 5(11):e15545. Doi: 10.1371/journal.pone.0015545
Gill SR, Pop M, DeBoy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, Gordon JI, Relman DA, Fraser-Ligget CM, Nelson KE: Metagenomic analysis of the human distal gut microbiome. Science 2006, 312: 1355-1359. Doi: 10.1126/science.1124234
Grieco MA, Cavalcante JJ, Cardoso AM, Vieira RP, Machado EA, Clementino MM, Medeiros MN, Albano RM, Garcia ES, De Souza W, Constantino R, Martins OB: Microbial community diversity in the gut of the South American Termite Cornitermes cumulans (Isoptera: Termitidae). Microb Ecol 2012. Doi: 10.1007/s00248-012-0119-6
Hess M, Sczyrba A, Egan R, Kim T, Chokhawala H, Schroth LS, Clark DS, Chen F, Zhang T, Mackie RI, Pennachio LA, Tringe SG, Visel A, Woyke T, Wang Z, Rubin EM: Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science 2011, 331(28):463-467. Doi: 10.1126/science.1200387
Hongo Y, Deevong P, Inoue T, Moriya S, Trakulnaleamsai S, Ohkuma M, Vongkaluang C, Noparatnaraporn N, Kudo T: Intra-and interspecific comparisons of bacterial diversity community structure support co-evolution of gut microbiota and termite host. Appl Environ Microbiol 2005, 71: 6590-6599. Doi: 10.1128/AEM.71.11.6590-6599.2005
Hongoh Y: Diversity and genomes of uncultured microbial symbionts in the termite gut. Biosc Biotechnol Biochem 2010, 74(6):1145-1151. DOI 10.271/bbb.10009
Inward D, Beccaloni G, Eggleton P: Death of an order: a comprehensive molecular phylogenetic study confirms that termites are eusocial cockroaches. Biol Lett 2007, 3: 331-335. Doi: 10.1098/rsbl.2007.0102
Kane MD, Breznak JA: Effect of host diet on production of organic acids and methane by cockroach gut bacteria. Appl Environ Microbiol 1991, 57: 2628-2634.
Kim KK, Kim MK, Lim JH, Park HY, Lee ST: Transfer of Chryseobacterium meningosepticum and Chryseobacterium miricola to Elizabethkingia gen. nov. as Elizabethkingia meningoseptica comb. nov. and Elizabethkingia miricola comb. nov. Int J System Evol Microbiol 2005, 55: 1287-1293. Doi: 10.1099/ijs.0.63541-0
Kong Y, Teather R, Forster R: Composition, spatial distribution, and diversity of the bacterial communities in the rumen of cows fed different forages. FEMS Microbiol Ecol 2010, 74: 612-622. Doi: 10.1111/j.1574-6941.2010.00977.x
Konig H: Bacillus species in the intestine of termites and other soil invertebrates. J Appl Microbiol 2006, 101: 620-627. Doi: 10.1111/j.1365-2672.2006.02914.x
Konstantinov SR, Fitzsimons N, Vaughan EE, Akkermans ADL: From composition to functionality of the intestinal microbial communities. In Probiotics and Prebiotics: where are we going?. Edited by: Tannock GW. London, UK: Caister Academic Press; 2002:59-84.
Kumar S, Tamura K, Jakobsen IB, Nei M: MEGA2: molecular evolutionary genetics analysis software. Bioinformatics 2001, 17: 1244-1245. Doi: 10.1093/bioinformatics/17.12.1244
Lane DJ: 16S/23S rRNA sequencing. In Nucleic acid techniques in bacterial systematic. Edited by: Stackebrandt E, Goodfellow M. New York: Willey; 1991:115-175. http://dx.doi.org/10.1016/j.ijfoodmicro.2007.06.023
Lemos FJA, Terra WR: Digestion of bacteria and the role of midgut lysozyme in some insect larvae. Comp Biochem Physiol B 1991, 100: 265-268.
Leser TD, Lindecrona RH, Jensen TK, Jensen BB, Moller K: Changes in bacterial community structure in the colon of pigs fed different experimental diets and after infection with Brachyspira hyodysenteriae . Appl Environ Microbiol 2000, 66(8):3290-3296.
Ley RE, Peterson DA, Jeffrey I, Gordon JI: Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 2006, 124(24):837-848. Doi: http://dx.doi.org/10.1016/j.cell.2006.02.017
Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, Schlegel ML, Tucker TA, Schrenzel MD, Knight R, Gordon JI: Evolution of mammals and their gut microbes. Science 2008, 320(20):1647-1651. Doi: 10.1126/science.1155725
Lozupone CA, Hamady M, Kelley ST, Knight R: Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microbiol 2007, 73: 1576-1585. Doi: 10.1128/AEM.01996-06
Mahowald MA, Rey FE, Seedorf H, Turnbaugh PJ, Fulton RS, Wollam A, Shah N, Wang C, Magrini V, Wilson RK, Cantarel BL, Coutinho PM, Henrissat B, Crock LW, Russell A, Verberkmoes NC, Hettich RL, Gordon JI: Characterizing a model human gut microbiota composed of members of its two dominant bacterial phyla. Proc Natl Acad Sci U S A 2009, 7(106):5859-5864. Doi: 10.1073/pnas.0901529106
Middelbos IS, Vester Boler BM, Qu A, White BA, Swanson KS, Fahey GC, Jr GC: Phylogenetic characterization of fecal microbial communities of dogs fed diets with or without supplemental dietary fiber using 454 pyrosequencing. PLoS ONE 2010, 5(3):1-9. Doi: 10.1371/journal.pone.0009768
Miyata R, Noda N, Tamaki H, Kinjyo K, Aoyagi H, Uchiyama H, Tanaka H: Influence of feed components on symbiotic bacterial community structure in the gut of the wood-feeding higher termite Nasutitermes takasagoensis . Biosc Biotech Bioch 2007, 71(5):1244-1251. DOI: http://dx.doi.org/10.1271/bbb.60672
Morrison M, Pope PB, Denman SE, McSweeney CM: Plant biomass degradation by gut microbiomes: more of the same or something new? Curr Opin Biotech 2009, 20: 358-363. Doi: 10.1016/j.copbio.2009.05.004
Noda S, Inoue T, Hongoh Y, Kawai M, Nalepa CA, Vongkaluang C, Kudo T, Ohkuma M: Identification and characterization of ectosymbionts of distinct lineages in Bacteroidales attached to flagellated protists in the gut of termites and a wood-feeding cockroach. Environ Microbiol 2006, 8: 11-20. Doi: 10.1111/j.1462-2920.2005.00860.x
Rivière D, Desvignes V, Pelletier E, Chaussonnerie S, Guermazi S, Weissenbach J, Li T, Camacho P, Sghir A: Towards the definition of a core of microorganisms involved in anaerobic digestion of sludge. ISME J 2009, 3: 700-714. Doi: 10.1038/ismej.2009.2
Scharf ME, Boucias DG: Potential of termite-based biomass pre-treatment strategies for use in bioethanol production. Insect Science 2010, 17: 166-174. Doi: 10.1111/j.1744-7917.2009.01309.x
Schauer C, Thompson CL, Brune A: The Bacterial Community in the Gut of the Cockroach Shelfordella lateralis Reflects the Close Evolutionary Relatedness of Cockroaches and Termites. Applied and Environmental Microbiology 2012, 78: 2758-2767.
Schloss PD, Larget BR, Handelsman J: Integration of microbial ecology and statistics: a test to compare gene libraries. Appl Environ Microbiol 2004, 70(9):5485-5492. Doi: 10.1128/AEM.70.9.5485-5492.2004
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF: Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 2009, 75: 7537-7541. DOI10.1128/AEM.01541-09
Suen G, Scott JJ, Aylward FO, Adams SM, Tringe SG, Pinto-Tomás AA, Foster CE, Pauly M, Weimer PJ, Barry KW, Goodwin LA, Bouffard P, Li L, Osterberger J, Harkins TT, Slater SC, Donohue TJ, Cameron R, Currie CR: An Insect herbivore microbiome with high plant biomass-degrading capacity. PLoS Genet 2010, 6(9):1-14. DOI: 10.1371/journal.pgen.1001129
Sun JZ, Scharf ME: Exploring and integrating cellulolytic systems of insects to advance biofuel technology. Insect Science 2010, 17: 163-165. Doi: 10.1111/j.1744-7917.2010.01348.x
Tajima K, Aminov RI, Nagamine T, Matsui H, Nakamura M, Benno Y: Diet-dependent shifts in the bacterial population of the rumen revealed with real-time PCR. Appl Envir Microb 2001, 67(6):2766-2774. Doi: 10.1128/AEM.67.6.2766-2774.2001
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG: The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 1997, 24: 4876-4882. Doi: 10.1093/nar/25.24.4876
Todaka N, Inoue T, Saita K, Ohkuma M, Nalepa CA, Lenz M, Kudo T, Moriya S: Phylogenetic analysis of cellulolytic enzyme genes from representative lineages of termites and a related cockroach. PLoS ONE 2010, 5(1):1-10. Doi: 10.1371/journal.pone.0008636
Tokuda G, Yamaoka I, Noda H: Localization of symbiotic clostridia in the mixed segment of the termite Nasutitermes takasagoensis (Shiraki). Appl Environ Microbiol 2000, 66(5):2199-2207.
Vartoukian SR, Palmer RM, Wade WG: The division ‘Synergistes’. Anaerobe 2007, 13(99):106. DOI: http://dx.doi.org/10.1016/j.anaerobe.2007.05.004
Vieira RP, Clementino MM, Cardoso AM, Oliveira DN, Albano RM, Gonzalez AM, Paranhos R, Martins OB: Archaeal communities in a tropical estuarine ecosystem: Guanabara Bay, Brazil. Microb Ecol 2007, 54: 460-468. Doi: 10.1007/s00248-007-9261-y
Warnecke F, Luginbühl P, Ivanova N, Ghassemian M, Richardson TH, Stege JT, Cayouette M, McHardy AC, Djordjevic G, Aboushadi N, Sorek R, Tringe SG, Podar M, Martin HG, Kunin V, Dalevi D, Madejska J, Kirton E, Platt D, Szeto E, Salamov A, Barry K, Mikhailova N, Kyrpides NC, Matson EG, Ottesen EA, Zhang X, Hernández M, Murillo C, Acosta LG, et al.: Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature 2007, 450(22):560-565. Doi: 10.1038/nature06269
Watanabe H, Tokuda G: Cellulolytic systems in insects. Annu Rev Entomol 2010, 55: 609-632. Doi: 10.1146/annurev-ento-112408-085319
Weisburg WG, Barns SM, Pelletier DA, Lane DJ: 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol 1991, 173: 697-703.
Wüst PK, Horn MA, Drake HL: Clostridiaceae and Enterobacteriaceae as active fermenters in earthworm gut content. ISME J 2011, 5: 92-106. Doi: 10.1038/ismej.2010.99
Funding
This work was supported by grants from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, INCT de Entomologia Molecular), Fundação de Coordenação de Aperfeiçoamento do Pessoal de nível Superior (CAPES) and Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ). The funders had no role in study design, data collection and analysis, decision to publish or in the preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
DBG, She conceived of the study, realized the main experiments of the article, participated in the sequence alignment and also drafted the manuscript. MNM, He participated in the design of the study and also drafted the manuscript. RPV, He participated in the design of the study, participated in the sequence alignment and also drafted the manuscript. AMC, He participated in the design of the study, participated in the sequence alignment, performed the statistical analysis and also drafted the manuscript. AST, She realized some experiments of the article, and also drafted the manuscript. CBS, She realized some experiments of the article, and also drafted the manuscript. RMA, He participated in the design of the study, performed the statistical analysis and also drafted the manuscript. SBN, She participated in the design of the study and also drafted the manuscript. ESG, He participated in coordination and also drafted the manuscript. WDS, He participated in coordination and also drafted the manuscript. OBM, He participated in the design of the study, coordination and also drafted the manuscript. EAM, He conceived of the study, participated in the design of the study, coordination and also drafted the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Bertino-Grimaldi, D., Medeiros, M.N., Vieira, R.P. et al. Bacterial community composition shifts in the gut of Periplaneta americana fed on different lignocellulosic materials. SpringerPlus 2, 609 (2013). https://doi.org/10.1186/2193-1801-2-609
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2193-1801-2-609