
Abstract
Research over the role of Bruton’s agammaglobulinemia tyrosine kinase (BTK) in B-lymphocyte development, differentiation, signaling and survival has led to better understanding of the pathogenesis of B-cell malignancies. Down-regulation of BTK activity is an attractive novel strategy for treating patients with B-cell malignancies. Ibrutinib (PCI-32765), a potent inhibitor of BTK induces impressive responses in B-cell malignancies through irreversible bond with cysteine-481 in the active site of BTK (TH/SH1 domain) and inhibits BTK phosphorylation on Tyr223. This review discussed in details the role of BTK in B-cell signaling, molecular interactions between B cell lymphoma/leukemia cells and their microenvironment. Clinical trials of the novel BTK inhibitor, ibrutinib (PCI-32765), in B cell malignancies were summarized.
Similar content being viewed by others

Introduction
Bruton’s tyrosine kinase (BTK) is a cytoplasmic tyrosine kinase, which is essential in B-lymphocyte development, differentiation, signaling and survival [1]. The BTK gene is located on X-chromosome at Xq21.33-Xq22. The gene has 19 exons spanning 37.5 kb genomic DNA. It encodes a non-receptor tyrosine kinase of the Btk/Tec family. The Tec family kinases (TFKs) have five members (Btk, Tec, Itk, Txk, Bmx), forming the second largest family of cytoplasmic tyrosine kinases in mammalian cells [2]. BTK is expressed in almost all hematopoietic cells, except T-cells and plasma cells. However, its essential functions appear to be limited to B-cells. BTK is required for B-cell receptor (BCR) signaling and implicated in the development of the B-cell malignancies including chronic lymphocytic leukemia (CLL), mantle cell lymphoma (MCL), follicular lymphoma (FL), diffuse large B-cell lymphoma (DLBCL) and acute lymphocytic leukemia (ALL) [3, 4].
Several preclinical and clinical studies have been performed, targeting BTK in B-cell malignancies. Ibrutinib/PCI-32765, a novel potent inhibitor of BTK induces impressive responses in B-cell malignancies and has been approved for therapy of refractory mantle cell lymphoma.
BCR/BTK signaling pathway
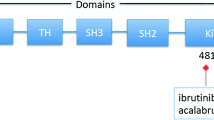
BCR is critical for normal B-cell development and is also associated in the development of the most common B-cell malignancies [5–7]. BCR serves as an antigen receptor and regulates multiple cellular processes, including proliferation, differentiation, apoptosis, and cell migration [2, 8]. The BCR consists of a transmembrane immunoglobulin (Ig) receptor associated with the Ig-alpha (CD79a) and Ig-beta (CD79b) heterodimers [7, 9–11]. Once the antigen binds to the receptor, the tyrosine kinases LYN and SYK initiate a signaling cascade that involves downstream kinases, adapter molecules, and generation of second messengers [9] (Figure 1). BTK is one of the signaling molecules that is essential in the BCR pathway. BTK comprises of several domains from the N-terminus: pleckstrin homology (PH), Tec homology (TH), SH2, SH3, and kinase (SH1) domains [1]. BTK requires Zn2+ for optimal activity and stability. Binding and coordination of BTK to the Zn2+ ion is mediated by a highly conserved zinc finger motif located in the TH domain. Mutations affecting Zn2+ binding lead to the generation of extremely unstable protein [12, 13].

A schematic representation of BCR/BTK signaling pathway. The BCR consists of a transmembrane immunoglobulin (Ig) receptor associated with the Ig-alpha (CD79a) and Ig-beta (CD79b) heterodimers. The activation of BTK and PI3 kinase after antigen binding to BCR prompts calcium release, which leads to activation of the signaling cascade. BCR: B cell receptor; BTK: Bruton’s tyrosine kinase.
Additional signaling molecules in the BCR signaling pathway include mammalian target of rapamycin (mTOR), SYK, LYN, phosphatidylinositol 3- kinase (PI3K) [14, 15], the adaptor protein GrB2. The activation of BTK and PI3 kinase prompts calcium release and activation of mTOR, protein kinase C-beta, AKT kinase, and mitogen-activated protein kinase ERK. These events then result in cell proliferation and survival of B-cells, which is mediated by up-regulation of transcription factors, mainly nuclear factor-kB (NFkB). Upon activation of BTK, PI3K is then activated, which stimulates the production of phosphatidylinositol-3, 4, 5 (PIP3) [14, 15]. Once an adequate amount of PIP3 is produced, BTK is mobilized to the plasma membrane. Phosphorylation of the BTK at the Y551 site is done by the Src family kinases, especially LYN and FYN. The phosphorylated BTK activates phospholipase C2, leading to downstream activation of protein kinases, and finally activation of transcription factor NFkB. The stimulation of NFkB pathway leads to inhibition of apoptosis (Figure 1). This series of events has been associated with the proliferation and survival of B-cell malignancies [16, 17]. BTK was found to be significantly overexpressed in CLL compared with normal B-cells. Although BTK is not always constitutively active in CLL cells, BCR or CD40 signaling is accompanied by effective activation of this pathway.
Preclinical studies
Ibrutinib is an orally active irreversible BTK inhibitor through bonding with cysteine-481 in the active site of BTK (TH/SH1 domain). It inhibits BTK phosphorylation on Tyr223 and has an IC50 of 0.5 nM for more than 24 hours [18]. In an in vitro study, Herman et al. demonstrated dose- and time-dependent cytotoxicity in CLL through caspase-3 dependent apoptotic pathway [19]. Efficacy of ibrutinib was also investigated using a canine model of B-cell lymphoma. There was a 70% reduction in tumor burden. After testing for BTK occupancy, it was determined that a single dose of 2.5 to 20 mg/kg was sufficient to fully occupy BTK [18]. Furthermore, ibrutinib can affect the CLL microenvironment by inhibiting CD40, BAFF, Toll-like receptor and cytokine signaling [20]. The above preclinical findings have led to further clinical trials [21].
Clinical studies
Chronic lymphocytic leukemia (CLL)
A phase 1 dose escalation study of ibrutinib as a single agent enrolled 56 patients with relapsed/refractory B-cell malignancies, including non-Hodgkin’s lymphoma, CLL, and Waldenstrom’s macroglobulinemia [22, 23]. In this study, ibrutinib was well tolerated with substantial activity across B-cell histologies. Fifty-six patients were treated over seven cohorts. Ibrutinib was administered in 1.25, 2.5, 5.0, 8.3, 12.5 and 17.5 mg/kg/day dose orally once per day for 28 days followed by a 7-day rest period to determine maximum tolerated dose (MTD). Pharmacokinetic studies showed that ibrutinib had a mean peak plasma concentration observed at 1–2 hours after administration. Most adverse events were grade 1 and 2 (including neutropenia and thrombocytopenia) and self-limited. Dose limiting events were not observed, even with prolonged dosing. Full occupancy of the BTK active site occurred at 2.5 mg/kg/day, and dose escalation continued to 12.5 mg/kg/day without reaching MTD. The dose of 420 mg daily was chosen for further studies. Objective response rate (RR) in 50 evaluable patients was 60%, including complete response (CR) of 16% and median PFS in all patients was 13.6 months.
With the promising results of phase 1 study in CLL, a phase 1b/II study was done to evaluate the efficacy, safety, pharmacokinetics and pharmacodynamics [24]. A total of 85 patients with relapsed/refractory CLL/SLL received ibrutinib, the first-in-class, oral covalent inhibitor of BTK; Among these 85 patients, 51 received 420 mg and 34 received 840 mg. The overall RR was the same for both groups (71%), and an additional 20% and 15% of patients in the respective group had a partial response with lymphocytosis (PR-L). The response was independent of clinical and genomic risk factors present before treatment, including advanced-stage disease, the number of previous therapies and 17p13.1 deletion. After 26 months of follow-up, the estimated PFS and OS rates were 75% and 83% respectively. Lymphocytosis was found to be treatment related and was not a sign of disease progression. It was thought to be due to lymphocyte mobilization by ibrutinib from bone marrow, lymph nodes, and spleen where stromal elements promote leukemic-cell proliferation, drug resistance, and survival. Another interesting finding from this study was that patients with IgVH unmutated (hence higher risk) had earlier resolution of lymphocytosis and higher response rate (P = 0.02). This was possibly due to the fact that these B cells tend to have higher BCR signaling and therefore more dependence on this pathway [25].
An interim update reported phase Ib/II data from 116 patients who were grouped into three cohorts [26]. First cohort included 31 treatment -naïve patients aged ≥ 65 in which 26 patients received 420 mg/day of Ibrutinib whereas 5 received 840 mg daily. Second cohort comprised relapsed/refractory CLL patients (n = 61), out of which 27 patients received 420 mg of Ibrutinib per day and 34 received 840 mg/day. Third group with high-risk relapsed/refractory (defined as relapse within two years following treatment and/or presence of 17p deletion) received ibrutinib 420 mg (n = 24). For the treatment-naïve CLL patients >65 years old, their median age was 71 years old (age 65–84) and the median follow-up durations for the whole group was 16.6 months (range 1.4-23.2). Unmutated IgVH, 11q deletion, and 17p deletion were found in 55%, 3%, and 7% of patients in this cohort. The overall RR by International Workshop on CLL criteria was 71% (10% CR and 61% PR). Among these, PR-L was 10%. This PR-L, defined as 50% reduction in lymphadenopathy with residual lymphocytosis, is not considered as clinical progression but rather as a form of response called compartmental shift where the drug induces mobilization of leukemic B cells from the bone marrow and spleen into the blood. Overtime, many of the patients had more than 50% decrease in absolute lymphocyte count compared with baseline [26–29]. For the relapsed/refractory CLL cohort, the median age was 64 years old (range 40–81) and the number of prior treatments were four for the whole cohort. There were unmutated IgVH (86%), 17p deletion (37%) and 11q deletion (40%) documented. Median follow up was 17.5 months for patients who received 420 mg and 10.3 months for those who received 840 mg. The overall RR was 67% (CR = 3% and PR 64%). Lastly, for high-risk CLL, all 24 patients received 420 mg of ibrutinib. Median age was 68 years old (range 37–82) with a median of four prior treatments. The documented unmutated IgVH, 17p deletion, and 11q deletion were 83%, 30% and 35%, respectively. The median follow-up was 10.3 months (range 1.1-11.5) and the overall response was 50%, all partial remissions, with 29% achieving PR-L (Table 1). Four percent of patients had progressed while on treatment. The adverse effects reported were grade 1 to 2 including joint pain, nausea, rash, infection, upper respiratory infection, diarrhea and fatigue. Rare hematologic side effects were also observed including neutropenia and thrombocytopenia [26–29]. Lymphocytosis was again found to resolve faster and occur more frequently in CLL patients with unmuted IgVH. The results of these studies also show that although the response rates were high, the CR rate with ibrutinib remained low; therefore, the activity of this drug needs to be evaluated in patients who have received less treatment.
Single agent ibrutinib is being compared with ofatumumab for R/R CLL (RESONATE trial) [30]. RESONATE-2 will investigate single agent ibrutinib versus chlorambucil as frontline therapy for newly diagnosed elderly patients with CLL [31].
Mantle cell lymphoma (MCL)
MCL is characterized by the overexpression of cyclin D1 due to the translocation t (11:14) (q13; q32). Although there are promising treatment modalities, a great majority of patients with MCL remain incurable [32]. Cinar et al. demonstrated a moderate to strong BTK expression in all MCL cases (n = 19) compared to benign lymphoid tissues. Treatment of MCL cell lines (Mino or Jeko-1) with ibrutinib resulted in decreased phospho-BTK-Tyr223 expression. Also, ibrutinib inhibited the viability of both Mino and JeKo-1 cells in concentration- and time-dependent manners. Ibrutinib also induced concentration-dependent apoptosis in both cell lines and decreased the levels of anti-apoptotic Bcl-2, Bcl-xL, and Mcl-1 protein. These findings suggest that BTK signaling plays an integral role in MCL cell survival and targeting of BTK is an encouraging therapeutic modality for this type of disease [32, 33]. In a phase 1 study of ibrutinib, there is an antitumor activity in several types of NHL including mantle cell lymphoma. A Phase 2 study was performed where ibrutinib was investigated at a daily dose of 560 mg in 111 patients with relapsed/refractory mantle-cell lymphoma [34]. Patients were divided into two groups: 1) those who received at least 2 cycles of bortezomib therapy; 2) those that had received less than 2 complete cycles of bortezomib or had no prior bortezomib therapy. The primary end point was the overall response rate (ORR). Secondary end points were duration of response, PFS, OS and safety. The median age was 68 years old, and 86% of patients had intermediate-risk or high-risk mantle-cell lymphoma according to clinical prognostic factors. A response rate of 68% (75 patients) was observed, with a complete remission rate of 21% and a partial response rate of 47%; prior treatment with bortezomib had no effect on the response rate. Median follow-up was 15.3 months and the estimated median response duration was 17.5 months. The median OS was not reached, the estimated rate of OS was 58% at 18 months. The most common treatment-related adverse effects were mild to moderate diarrhea, nausea and fatigue. Grade 3 or higher hematologic events included neutropenia (16%), thrombocytopenia (11%) and anemia (10%). The study shows that ibrutinib is a durable single-agent effective in relapsed/refractory MCL. Ibrutinib was approved by FDA for relapsed/refractory MCL who at least received one prior therapy. However, the complete response rate is lower than that associated with combination regimens, and it is not yet known whether the addition of ibrutinib can improve the molecular response rate.
Diffuse large B cell lymphoma and multiple myeloma
There are early studies of ibrutinib in DLBCL and multiple myeloma. These studies have shown that ibrutinib has cytotoxic effects on these cells and warrants further research. Dasmahapatra et al. examined the interaction of ibrutinib and the proteasome inhibitor (bortezomib) in DLBCL and mantle cell lymphoma cells, including those highly resistant to bortezomib. Co-administration of ibrutinib and bortezomib synergistically increased mitochondrial injury and apoptosis in germinal center or activated B-cell like DLBCL and MCL cells. These events were accompanied by marked AKT and NFkB inactivation, down-regulation of Mcl-1, Bcl-xL and XIAP which enhanced DNA damage and endoplasmic reticulum stress. The same interactions were seen in highly bortezomib-resistant DLBCL and MCL cells and in primary DLBCL cells [35]. On the other hand, Rushworth et al. reported that ibrutinib is cytotoxic to malignant plasma cells from patients with multiple myeloma and the treatment with ibrutinib significantly augments the cytotoxic activity of bortezomib and lenalidomide chemotherapies. It was described that the cytotoxicity of ibrutinib in multiple myeloma is mediated through an inhibitory effect on the NFkB pathway. In addition, ibrutinib blocks the phosphorylation of serine-536 of the p65 subunit of NFkB, preventing its nuclear translocation, leading to down regulation of anti-apoptotic proteins Bcl-xL, FLIP and survivin, culminating in caspase-mediated apoptosis within the malignant plasma cells [36, 37].
It has been shown that activated B cell (ABC) but not germinal center B cell (GCB) subtypes of DLBCL cell lines are driven by "chronic active" B cell receptor (BCR) signaling [5, 38]. BCR subunit CD79B mutations occur in 21% of ABC but only 5% of GCB DLBCL tumors. In a phase 2 study patients with relapsed/refractory DLBCL received ibrutinib 560 mg daily [39]. Seventy subjects were enrolled; median age was 63 yrs (28–92); median prior systemic therapies 3 (1–7); 23% had prior stem cell transplant. Ibrutinib was well tolerated. In the ABC subtype (N = 29), ORR was 40% (10/25), CR 8% (2/25) and PR 32% (8/25). The median PFS was 5.5 months in ABC responders. PR was observed in only one with the GCB subtype (N = 20) and none in unclassifiable cases (N = 16). Therefore preferential response favored ABC subtype of DLBCL (p = 0.0126), but ibrutinib sensitivity did not require a BCR mutation. These results suggest that future clinical trials of ibrutinib in DLBCL should aim for patients with ABC subtype of DLBCL.
Ibrutinib in combination therapy
The encouraging results of ibrutinib in treatment naïve and relapse/refractory(R/R) CLL led the investigators to explore combination therapy to determine. Ibrutinib has been studied in combination with rituximab in R/R CLL (n = 40). This combination has an ORR of 85% [40] (Table 1). This was a phase 2 single-center study of 40 high-risk patients at the time of analysis. Patients were treated with ibrutinib 420 mg daily and weekly rituximab (375 mg/m2) for weeks 1–4 (cycle 1), then daily ibrutinib plus monthly rituximab until cycle 6. This was then followed by ibrutinib daily. The median age was 65 (range 35–82) with a median of 2 prior therapies. Among these, 19 patients had del17p or TP53 mutation (4 without prior therapy), 13 patients had del11q. With a median follow up of 4 months, 17 out of 20 patients evaluable for efficacy achieved a partial remission (PR) for an ORR of 85%, and three achieved a PR with persistent lymphocytosis. In this combination trial, the re-distribution lymphocytosis was noted to peak earlier and the duration was shorter than with single-agent ibrutinib. Treatment was well tolerated and induced very high early response rates.
Ofatumumab was also studied with ibrutinib in relapsed/refractory (R/R) CLL (n = 40) with an ORR of 100% [41]. In this report, 27 patients with CLL/SLL following 2 prior therapies received 420 mg daily, in 28-day cycles. Ofatumumab (O) is added at a dose of 300 mg on day 1 of cycle 2, followed by 2000 mg on day 8, 15, and 22 of cycle 2, Day 1, 8, 15, and 22 of cycle 3, and on day 1 of cycles 5–8. This combination had rapid onset of response, low relapse rate and favorable safety profile.
Ibrutinib was combined with bendamustine (B) and rituximab (R) in a phase I study in patients with relapsed/refractory NHL (FL, MZL, MCL, transformed NHL, and DLBCL) [42]. Treatment consisted of standard R 375 mg/m2 day 1, B 90 mg/m2 days 1 and 2, with escalating doses of ibrutinib (280 mg or 560 mg) every 28 days for 6 cycles. Ibrutinib was continued after cycle 6 in responding patients. Pegfilgrastim was used for patients with severe neutropenia. Eleven patients were enrolled, with a median age of 72 (range 45–84) and a median of 3 prior therapies (range 0–10). Nine patients completed two or more cycles of therapy (median 3, range 1–6), of whom 6 received 280 mg and 3 treated with 560 mg of ibrutinib. No DLTs were observed. Severe lymphopenia (64%) was the most common adverse events. Three patients required dose reductions from 280 mg ibrutinib to 140 mg. Bendamustine dose was reduced to 60 mg/m2 in 1 patient for grade 3 thrombocytopenia. ORR was 38% in 8 evaluable patients. There are phase III clinical trials currently ongoing that test the efficacy of BR +/- ibrutinib in R/R CLL patients. There is also a phase 3 study of BR +/- ibrutinib as front-line therapy for MCL (the SHINE trial, MCL3002).
Phase 1b trial assessing the feasibility of incorporating ibrutinib with R-CHOP was updated at 2013 ASCO [43]. In this study, ibrutinib was given as 280, 420, or 560 mg daily in combination with standard R-CHOP. 17 patients were enrolled. At 560 mg dosage, 1 patient had grade 2 gastritis. The phase 2 dose was established at 560 mg daily. The most common adverse event was neutropenia (77%). The response rate was 100% at the time of analysis in 10 patients (7 CR and 3 PR).
Conclusion and future directions
Novel agents are being studied for CLL and lymphoid malignancies [14, 32, 44–49]. Ibrutinib as a single agent has brought encouraging results for the targeted therapy of B-cell lymphoproliferative malignancies. Ibrutinib targets B-cell receptor signaling in CLL/MCL cells. Lymphocytosis with ibrutinib therapy is a remarkable clinical phenomenon, and can stay for a prolonged period of time. Additional novel BTK inhibitors (GDC-0834, CC-292, ONO-4059, CNX-774, LFM-A13 and HM-71224) are currently under active investigation [1]. Further research is warranted to assess long-term effects of prolonged use, such as hypogammaglobulinemia, unusual side effects and opportunistic infection [50]. It is also interesting to evaluate the use of BTK inhibitors in peri-transplantation as well as possible maintenance therapy on high-risk patients. Low CR rate from ibrutinib single agent therapy remains a concern. Ibrutinib appears to have activity in myeloma [36, 37, 51]. Ibrutinib in combination regimens is an intense focus of research in an attempt to improve the response quality.
References
Akinleye A, Chen Y, Mukhi N, Song Y, Liu D: Ibrutinib and novel BTK inhibitors in clinical development. J Hematol Oncol 2013,6(1):59. 10.1186/1756-8722-6-59
Mohamed AJ, Yu L, Backesjo CM, Vargas L, Faryal R, Aints A, Christensson B, Berglof A, Vihinen M, Nore BF, et al.: Bruton's tyrosine kinase (Btk): function, regulation, and transformation with special emphasis on the PH domain. Immunol Rev 2009,228(1):58–73. 10.1111/j.1600-065X.2008.00741.x
Pan Z, Scheerens H, Li SJ, Schultz BE, Sprengeler PA, Burrill LC, Mendonca RV, Sweeney MD, Scott KC, Grothaus PG, et al.: Discovery of selective irreversible inhibitors for Bruton's tyrosine kinase. Chem Med Chem 2007,2(1):58–61. 10.1002/cmdc.200600221
Cheson BD, Plass RR, Rothstein G: Defective opsonization in multiple myeloma. Blood 1980,55(4):602–606.
Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, Kohlhammer H, Lamy L, Zhao H, Yang Y, et al.: Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 2010,463(7277):88–92. 10.1038/nature08638
Tricot G, Jagannath S, Vesole D, Nelson J, Tindle S, Miller L, Cheson B, Crowley J, Barlogie B: Peripheral blood stem cell transplants for multiple myeloma: identification of favorable variables for rapid engraftment in 225 patients. Blood 1995,85(2):588–596.
Wang K, Wei G, Liu D: CD19: a biomarker for B cell development, lymphoma diagnosis and therapy. Exp Hematol Oncol 2012,1(1):36. 10.1186/2162-3619-1-36
Chavez JC, Sahakian E, Pinilla-Ibarz J: Ibrutinib: an evidence-based review of its potential in the treatment of advanced chronic lymphocytic leukemia. Core Evid 2013, 8: 37–45.
Wiestner A: Targeting B-Cell receptor signaling for anticancer therapy: the Bruton's tyrosine kinase inhibitor ibrutinib induces impressive responses in B-cell malignancies. J Clin Oncol 2013,31(1):128–130. 10.1200/JCO.2012.44.4281
Binet JL, Caligaris-Cappio F, Catovsky D, Cheson B, Davis T, Dighiero G, Dohner H, Hallek M, Hillmen P, Keating M, et al.: Perspectives on the use of new diagnostic tools in the treatment of chronic lymphocytic leukemia. Blood 2006,107(3):859–861.
Woyach JA, Johnson AJ, Byrd JC: The B-cell receptor signaling pathway as a therapeutic target in CLL. Blood 2012,120(6):1175–1184. 10.1182/blood-2012-02-362624
Nisitani S, Satterthwaite AB, Akashi K, Weissman IL, Witte ON, Wahl MI: Posttranscriptional regulation of Bruton's tyrosine kinase expression in antigen receptor-stimulated splenic B cells. Proc Natl Acad Sci USA 2000,97(6):2737–2742. 10.1073/pnas.050583597
Vihinen M, Kwan SP, Lester T, Ochs HD, Resnick I, Valiaho J, Conley ME, Smith CI: Mutations of the human BTK gene coding for bruton tyrosine kinase in X-linked agammaglobulinemia. Hum Mutat 1999,13(4):280–285. 10.1002/(SICI)1098-1004(1999)13:4<280::AID-HUMU3>3.0.CO;2-L
Akinleye A, Avvaru P, Furqan M, Song Y, Liu D: Phosphatidylinositol 3-kinase (PI3K) inhibitors as cancer therapeutics. J Hematol Oncol 2013,6(1):88. 10.1186/1756-8722-6-88
Fang X, Zhou X, Wang X: Clinical development of phosphatidylinositol 3-kinase inhibitors for non-Hodgkin lymphoma. Biomarkers Res 2013,1(1):30. 10.1186/2050-7771-1-30
Nore BF, Vargas L, Mohamed AJ, Branden LJ, Backesjo CM, Islam TC, Mattsson PT, Hultenby K, Christensson B, Smith CI: Redistribution of Bruton's tyrosine kinase by activation of phosphatidylinositol 3-kinase and Rho-family GTPases. Eur J Immunol 2000,30(1):145–154. 10.1002/1521-4141(200001)30:1<145::AID-IMMU145>3.0.CO;2-0
Varnai P, Rother KI, Balla T: Phosphatidylinositol 3-kinase-dependent membrane association of the Bruton's tyrosine kinase pleckstrin homology domain visualized in single living cells. J Biol Chem 1999,274(16):10983–10989. 10.1074/jbc.274.16.10983
Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, Li S, Pan Z, Thamm DH, Miller RA, et al.: The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci 2010,107(29):13075–13080. 10.1073/pnas.1004594107
Herman SE, Gordon AL, Hertlein E, Ramanunni A, Zhang X, Jaglowski S, Flynn J, Jones J, Blum KA, Buggy JJ, et al.: Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood 2011,117(23):6287–6296. 10.1182/blood-2011-01-328484
Virtue A, Wang H, Yang X-F: MicroRNAs and Toll-like Receptor/Interleukin-1 Receptor Signaling. J Hematol Oncol 2012,5(1):66. 10.1186/1756-8722-5-66
Foa R, Guarini A: A mechanism-driven treatment for chronic lymphocytic leukemia? N Engl J Med 2013,369(1):85–87. 10.1056/NEJMe1303054
Advani RH, Sharman JP, Smith SM, Boyd TE, Grant BW, Kolibaba KS, Furman RR, Buggy JJ, Loury DJ, Hedrick E, et al.: The BTK inhibitor PCI-32765 is highly active and well tolerated in patients with relapsed/refractory B cell malignancies: final results from a phase 1 study. Ann Oncol 2011, 22: iv135.
Advani RH, Buggy JJ, Sharman JP, Smith SM, Boyd TE, Grant B, Kolibaba KS, Furman RR, Rodriguez S, Chang BY, et al.: Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol 2013,31(1):88–94. 10.1200/JCO.2012.42.7906
Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, Grant B, Sharman JP, Coleman M, Wierda WG, et al.: Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med 2013,369(1):32–42. 10.1056/NEJMoa1215637
Guarini A, Chiaretti S, Tavolaro S, Maggio R, Peragine N, Citarella F, Ricciardi MR, Santangelo S, Marinelli M, De Propris MS, et al.: BCR ligation induced by IgM stimulation results in gene expression and functional changes only in IgV H unmutated chronic lymphocytic leukemia (CLL) cells. Blood 2008,112(3):782–792. 10.1182/blood-2007-12-127688
Byrd JC, Furman RR, Coutre S, Flinn IW, Burger JA, Blum KA, Sharman J, Grant B, Jones JA, Wierda WG, et al.: The Bruton's tyrosine kinase (BTK) inhibitor ibrutinib (PCI-32765) promotes high response rate, durable remissions, and is tolerable in treatment naive (TN) and relapsed or refractory (RR) chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL) patients including patients with high-risk (HR) disease: new and updated results of 116 patients in a phase Ib/II study. ASH Annu Meet Abstr 2012,120(21):189.
Byrd JC, Blum KA, Burger JA, Coutre SE, Sharman JP, Furman RR, Flinn IW, Grant BW, Richards DA, Zhao W, et al.: Activity and tolerability of the Bruton’s tyrosine kinase (Btk) inhibitor PCI-32765 in patients with chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL): interim results of a phase Ib/II study. J Clin Oncol 2011,29(15):6508.
Byrd JC, Furman RR, Coutre SE, Burger JA, Blum KA, Sharman JP, Flinn IW, Grant BW, Heerema NA, Johnson AJ, et al.: The Bruton's tyrosine kinase(BTK) inhibitor PCI-32765 (P) in treatment-naive (TN) chronic lymphocytic leukemia (CLL) patients (pts): interim results of a phase Ib/II study ASCO . ASH Annu Meet Abstr 2012,30(15):6507.
O'Brien S, Burger JA, Blum KA, Furman RR, Coutre SE, Sharman J, Flinn IW, Grant B, Heerema NA, Johnson AJ, et al.: The Bruton’s tyrosine kinase (BTK) inhibitor PCI-32765 induces durable responses in relapsed or refractory chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL): follow-up of a phase Ib/II study. ASH Annu Meet Abstr 2011,118(21):983.
Byrd JC, Barrientos JC, Devereux S, Brown JR, Kay NE, Reddy NM, O'Brien SM, Kipps TJ, Furman RR, Schuster SJ, et al.: A randomized, multicenter, open-label, phase III study of the Bruton tyrosine kinase (BTK) inhibitor ibrutinib (PCI-32765) versus ofatumumab in patients (pts) with relapsed or refractory (RR) chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL): RESONATE. ASH Annu Meet Abstr 2013,31(15_suppl):TPS8619.
Burger JA, Ghia P, Polliack A, Tam C, Suri D, Clow F, Kraljevic S, James DF, Kipps TJ: Randomized, multicenter, open-label, phase III study of the BTK inhibitor ibrutinib versus chlorambucil in patients 65 years or older with treatment-naive CLL/SLL (RESONATE-2, PCYC-1115-CA). ASH Annu Meet Abstr 2013,31(15_suppl):TPS7130.
Zucca E, Bertoni F: Toward new treatments for mantle-cell lymphoma? N Engl J Med 2013,369(6):571–572. 10.1056/NEJMe1307596
Cinar M, Hamedani F, Mo Z, Cinar B, Amin HM, Alkan S: Bruton tyrosine kinase is commonly overexpressed in mantle cell lymphoma and its attenuation by Ibrutinib induces apoptosis. Leuk Res 2013,37(10):1271–1277. 10.1016/j.leukres.2013.07.028
Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS, Jurczak W, Advani RH, Romaguera JE, Williams ME, et al.: Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med 2013,369(6):507–516. 10.1056/NEJMoa1306220
Dasmahapatra G, Patel H, Dent P, Fisher RI, Friedberg J, Grant S: The Bruton tyrosine kinase (BTK) inhibitor PCI-32765 synergistically increases proteasome inhibitor activity in diffuse large-B cell lymphoma (DLBCL) and mantle cell lymphoma (MCL) cells sensitive or resistant to bortezomib. Br J Haematol 2013,161(1):43–56. 10.1111/bjh.12206
Edwards CM: BTK inhibition in myeloma: targeting the seed and the soil. Blood 2012,120(9):1757–1759. 10.1182/blood-2012-07-439216
Rushworth SA, Bowles KM, Barrera LN, Murray MY, Zaitseva L, MacEwan DJ: BTK inhibitor ibrutinib is cytotoxic to myeloma and potently enhances bortezomib and lenalidomide activities through NF-κB. Cell Signal 2012,25(1):106–112.
Staudt LM: Chronic Active B-Cell Receptor Signaling in Lymphoma. ASH Annu Meet Abstr 2012,120(21):26.
Wilson WH, Gerecitano JF, Goy A, de Vos S, Kenkre VP, Barr PM, Blum KA, Shustov AR, Advani RH, Lih J, et al.: The Bruton's Tyrosine Kinase (BTK) Inhibitor, Ibrutinib (PCI-32765), Has Preferential Activity in the ABC Subtype of Relapsed/Refractory De Novo Diffuse Large B-Cell Lymphoma (DLBCL): Interim Results of a Multicenter, Open-Label, Phase 2 Study. ASH Annu Meet Abstr 2012,120(21):686.
Burger JA, Keating MJ, Wierda WG, Hoellenriegel J, Ferrajoli A, Faderl S, Lerner S, Zacharian G, Huang X, James DF, et al.: The Btk Inhibitor Ibrutinib (PCI-32765) in Combination with Rituximab Is Well Tolerated and Displays Profound Activity in High-Risk Chronic Lymphocytic Leukemia (CLL) Patients. ASH Annu Meet Abstr 2012,120(21):187.
Jaglowski SM, Jones JA, Flynn JM, Andritsos LA, Maddocks KJ, Blum KA, Grever MR, Geyer SM, Woyach JA, Johnson AJ, et al.: A phase Ib/II study evaluating activity and tolerability of BTK inhibitor PCI-32765 and ofatumumab in patients with chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) and related diseases. ASCO Annu Meet Abstr 2012,30(15_suppl):6508.
Blum KA, Christian B, Flynn JM, Jaglowski SM, Jones JA, Maddocks K, Byrd JC: A Phase I Trial of the Bruton's Tyrosine Kinase (BTK) Inhibitor, Ibrutinib (PCI-32765), in Combination with Rituximab (R) and Bendamustine in Patients with Relapsed/Refractory Non-Hodgkin's Lymphoma (NHL). ASH Annu Meet Abstr 2012,120(21):1643.
Younes A, Flinn I, Berdeja JG, Friedberg JW, Alberti S, Thieblemont C, Morschhauser F, Hellemans P, Hall B, Smit J, et al.: Phase Ib study combining ibrutinib with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) in patients with CD20-positive B-cell non-Hodgkin lymphoma (NHL). ASCO Annu Meet Abstr 2013,31(15_suppl):8502.
Cang S, Mukhi N, Wang K, Liu D: Novel CD20 monoclonal antibodies for lymphoma therapy. J Hematol Oncol 2012, 5: 64. 10.1186/1756-8722-5-64
Furqan M, Mukhi N, Lee B, Liu D: Dysregulation of JAK-STAT pathway in hematological malignancies and JAK inhibitors for clinical application. Biomarker Res 2013,1(1):5. 10.1186/2050-7771-1-5
Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Dohner H, Hillmen P, Keating MJ, Montserrat E, Rai KR, et al.: Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood 2008,111(12):5446–5456. 10.1182/blood-2007-06-093906
Lu K, Wang X: Therapeutic advancement of chronic lymphocytic leukemia. J Hematol Oncol 2012,5(1):55. 10.1186/1756-8722-5-55
Wu M, Akinleye A, Zhu X: Novel agents for chronic lymphocytic leukemia. J Hematol Oncol 2013,6(1):36. 10.1186/1756-8722-6-36
Zhao Y, Huang H, Wei G: Novel agents and biomarkers for acute lymphoid leukemia. J Hematol Oncol 2013,6(1):40. 10.1186/1756-8722-6-40
Flinn IW, Kopecky KJ, Foucar MK, Head D, Bennett JM, Hutchison R, Corbett W, Cassileth P, Habermann T, Golomb H, et al.: Long-term follow-up of remission duration, mortality, and second malignancies in hairy cell leukemia patients treated with pentostatin. Blood 2000,96(9):2981–2986.
Rushworth SA, Bowles KM, Barrera LN, Murray MY, Zaitseva L, MacEwan DJ: BTK inhibitor ibrutinib is cytotoxic to myeloma and potently enhances bortezomib and lenalidomide activities through NF-kappaB. Cell Signal 2013,25(1):106–112. 10.1016/j.cellsig.2012.09.008
Funding
Yamei Chen is supported by Xiamen Zhongshan Hospital, Fujian, China; International Scholar Exchange Foundation, US; and NYMC Blood Disease Fund, US.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
DL and AN were responsible for study design, data collection and drafting the manuscript. All authors have participated in manuscript development, revisions and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Novero, A., Ravella, P.M., Chen, Y. et al. Ibrutinib for B cell malignancies. Exp Hematol Oncol 3, 4 (2014). https://doi.org/10.1186/2162-3619-3-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2162-3619-3-4