
Abstract
Phosphatidylinositol 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) signaling pathway is extensively explored in cancers. It functions as an important regulator of cell growth, survival and metabolism. Activation of this pathway also predicts poor prognosis in numerous human malignancies. Drugs targeting this signaling pathway have been developed and have shown preliminary clinical activity. Accumulating evidence has highlighted the important role of PI3K in non-Hodgkin lymphoma (NHL), especially in the disease initiation and progression. Therapeutic functions of PI3K inhibitors in NHL have been demonstrated both in vivo and in vitro. This review will summarize recent advances in the activation of PI3K signaling in different types of NHL and the applications of PI3K inhibitors in NHL treatment.
Similar content being viewed by others


Introduction
The PI3K/Akt/mTOR pathway plays a critical role in regulating cancer cell growth, survival, motility and metabolism [1]. Phosphatidylinositol 3-kinase (PI3K) is a critical element in this signaling, it is activated in a wide range of human neoplasms and associated with poor outcomes [2, 3]. Our previous studies have demonstrated that down regulation of heat shock protein 70 (HSP70) contributed to the increased sensitivity of Burkitt lymphoma (BL) cells to chemotherapy through blocking this pathway [4]. Targeted inhibitors for PI3K signaling are opening a new paradigm in cancer treatment. Activation of this pathway was identified in different types of NHL [5]. A number of PI3K inhibitors have been developed and displayed preliminary clinical activities in NHL treatment.
The PI3K signaling pathway in cancer
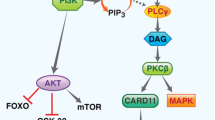
The PI3K signaling pathway is triggered by activation of receptor tyrosine kinase (RTK) in cell membrane. After binding to the growth factors, the intracellular domain of RTK is phosphorylated, and PI3K is activated (Figure 1). There are three classes (I, II, III) of PI3Ks, with class I PI3Ks as the most studied in human cancer [6]. Activated PI3K phosphorylates PI(4,5)P2 (PIP2) to produce PI(3,4,5)P3 (PIP3). The tumor suppressor phosphatase and tensin homolog (PTEN) deleted on chromosome ten could negatively regulate this process through dephosphorylating PIP3. Activated PIP3 could prompt the phosphorylation of Akt and further stimulate the Aktmediated activation of downstream targets, including the Bcl-2 family members, Mdm2 and tuberous sclerosis complex 2 (TSC2) [7]. Activated Akt inhibits the Rheb GTPase activity of TSC1/2 complex through phosphorylating TSC2. Then the activated Rheb promotes mTOR complex 1 (mTORC1) to phosphorylate p70S6 and 4E binding protein1 (4EBP1), resulting in dysregulation of protein synthesis and cell survival [8]. On the other hand, mTORC2, another type of mTOR complex, could phosphorylate Akt on serine 473 and facilitate its complete activation [9].

The PI3K/Akt/mTOR pathway and relative inhibitors in NHL. Once RTKs binding to the growth factors, the PI3K signaling pathway is triggered. Activated PI3K could phosphorylate PI (4,5) P2 (PIP2) to produce PI(3,4,5)P3 (PIP3). This process is negatively regulated by PTEN. Akt inhibits the Rheb GTPase activity of TSC1/TSC2 dimer by phosphorylating TSC2. Then activated Rheb stimulates mTOR to phosphorylate the p70S6 and 4E-binding protein (4EBP-1), resulting in dysregulation of protein synthesis and cell survival. On the other hand, mTORC2, another type of mTOR complex, could phosphorylate Akt and promote the complete activation of it.
The PI3K/Akt/mTOR pathway is constitutively activated in human cancers and is critical for tumor progression and chemo-resistance [10]. Alterations of several components in this pathway have been identified in numerous tumors [11]. Mutation of PI3KA was most commonly recognized in breast, colorectal and endometrial cancers [12]. And the alteration of Akt was found in gastric, pancreatic and ovarian cancers. These alterations promoted the development of PI3K pathway-specific inhibitors [7]. Several PI3K pathway inhibitors have been developed and are being evaluated in preclinical or clinical studies. As PI3K/Akt/mTOR pathway plays a key role in the proliferation and survival of lymphoma cell, various inhibitors targeting this pathway have been studied in different types of NHL (Table 1). In spite of preclinical studies, several PI3K inhibitors for NHL treatment are currently undergoing various stages of clinical trials (Table 2) [13]. Here we will focus on the clinical development of PI3K inhibitors for NHL.
PI3K inhibitors in follicular lymphoma
Follicular lymphoma (FL) is one of the most common types of indolent NHL. In spite of its indolent phase, about 25%-60% of them eventually transform into diffuse large cell lymphoma (DLBCL), a type of aggressive lymphoma. Combination therapy included rituximab cannot significantly decline the relapse rate of FL [14]. Therefore, novel effective therapeutic agents are urgently needed to improve the outcomes of FL patients.
Gulmann C et al. demonstrated the activation of PI3K/Akt/mTOR pathway in FL by proteomic analysis [30]. They provided evidence that activation and phosphorylation of PI3K as well as its downstream effectors, including Akt, mTOR, and S6K, were found in FL. Recently, a PI3K/mTOR module was reported to mediate the invasion and angiogenesis of FL, which further confirmed its potential use in anti-invasive of FL [31]. NVP-BEZ235, a dual PI3K and mTOR inhibitor, was indicated to be effective in inhibiting FL cell proliferation [14]. Proliferation of FL cell line was substantially inhibited by NVP-BEZ235, activation level of caspase-3 increased by 1.6 to 2 fold in NVP-BEZ235-treated cells compared to that treated with vehicle alone [14]. In addition, anti-tumor function and the therapeutic potential of NVP-BEZ235 were also identified in other human malignancies, such as T-cell acute lymphoblastic leukemia (T-ALL), colorectal and lung cancer [15, 32, 33].
The roles in chronic lymphocytic leukemia
Chronic lymphocytic leukemia (CLL) is the most common type of adult leukemia in the western world, with 15,000 new cases and approximately 4,500 deaths per year [34]. It is characterized by accumulation of malignant B cells in the blood, bone marrow and secondary lymphoid tissues [35]. Novel targeted agents and potential therapeutic options have been provided recently [36, 37].
Consistent expressions of PI3K-δ were found in both primary CLL cells and normal B cells, but the CLL cells represented a statistically higher intrinsic PI3K activity compared to normal B cells [18, 38]. CAL-101 (GS-1101) is a specific inhibitor of PI3K-δ isoform. It could prevent the proliferation and induce apoptosis of CLL cells through disrupting multiple external pathways. Activation of Akt, and secretion of cytokines and chemokines were inhibited by CAL-101 in both vitro and vivo [18, 39, 40]. B cells from 16 CLL patients were treated with CAL-101 at different concentrations for 48 hours [18]. The results showed that CAL-101 promoted CLL cells apoptosis in a dose- and time-dependent pattern.
Coutre et al. have reported a phase I study using CAL-101 as a single agent for relapsed/refractory CLL patients [41]. About 80% of them achieved >50% reduction in the size of lymph node and spleen. On the contrary, approximately >50% increase in lymphocytosis of peripheral blood occurred in 58% patients. This trial also provided evidence of limited toxicity of CAL-101 in CLL treatment [41]. A phase I study of CAL-101 in combination with rituximab or bendamustine in 20 patients with relapsed/refractory B-cell malignancies (indolent B-cell NHL n = 12, CLL n = 8) reached the same conclusion as well. The main adverse effects, Grade 3 neutropenia and thrombocytopenia, were found in 22% of patients receiving bendamustine plus CAL-101. Additionally, the peripheral lymphocyte counts were stable or decreased in 8/8 CLL patients after combination treatment [42].
NVP-BKM120 is an orally available pan class I inhibitor of PI3K. It was reported to inhibit the phosphorylation of Akt in primary B-CLL lymphocytes and further inhibit the PI3K signaling [20]. NVP-BKM120 also contributed to the concomitant Mcl-1 downregulation and Bim induction though regulating the Akt/FoxO3a/Bim axis in CLL [43]. It was 3.6 fold more toxic than CAL-101 in malignant B-CLL lymphocytes in vitro. A study on 65 B-CLL patients revealed that NVP-BKM120 was cytotoxic in 78% of the primary B-CLL lymphocytes [20].
The roles in diffuse large B cell lymphoma
DLBCL represents the most common subtype of NHL. It accounts for 40% of newly diagnosed NHL in the world and approximately 40–50% of newly diagnosed lymphoid neoplasms in China [44, 45].
Dysregulation of the PI3K/Akt/mTOR signaling pathway was observed in DLBCL [46, 47]. Xu et al. investigated the activation of PI3K/Akt/mTOR signaling pathway and their clinical significance in 73 DLBCL cases [44]. Activation of this pathway was related to poor treatment response and decreased survival time in DLBCL patients treated with CHOP chemotherapy regimen (cyclophosphamide, doxorubicin, vincristine, and prednisone) but not in those treated with rituximab-CHOP (R-CHOP) [44].
Previous studies have indicated that apoptosis of DLBCL cell lines could be induced by LY294002, a pan-isoform PI3K inhibitor [22]. NVP-BEZ235 is a novel dual inhibitor of PI3K and mTOR. Concurrent inhibition of PI3K and mTOR by NVP-BEZ235 resulted in the down-regulation of Eif4e phosphorylation and MCL-1 expression. It could inhibit the proliferation of DLBCL cells via inhibiting activation of PI3K, mTORC1 and mTORC2 in both central B-cell (GCB) and activated B-cell (ABC) subtype of DLBCL [16]. But when the concentration of NVP-BEZ235 was 0.5 μM or below, the induction response of cell demise in ABC cell lines was less efficient than that in GCB cell lines.
Recent studies have highlighted that NVP-BKM120, a pan-class I inhibitor of PI3K/Akt/mTOR signaling pathway. NVP-BKM120 reduced cell proliferation and increase the apoptosis of DLBCL cells through blocking the autophagy,as well as up-regulating Puma and Bim and inhibiting anti-apoptotic Mcl-1 expression [21]. Additionally, a phase I and dose-escalation study of NVP-BKM120 provided evidence of the feasibility of PI3K inhibitors in patients with advanced solid cancers [48]. Although few of them were moved into clinical application currently, the PI3K inhibitors will bring up new therapeutic options for relapse/refractory DLBCL.
The roles in mantle cell lymphoma
Mantle cell lymphoma (MCL) accounts for about 6% of all NHL and the median age at diagnosis is about 65. It is characterized by chromosomal translocation t(11;14)(q13;q32) resulting in over-expression of cyclin D1, which are regulated by the Akt/mTOR signaling pathway [49, 50]. Despite the relatively good response to first-line chemotherapy, most of the MCL patients relapsed eventually.
Recent studies have revealed the importance of PI3K/Akt/mTOR signaling pathway and clinical application of PI3K inhibitors in MCL [51, 52]. Gene expression profiling of both purified leukemic MCL cells and the naive B cells were performed through oligonucleotide microarrays [53]. 106 genes were found to be differentially expressed at least three fold in MCL cells compared to naive B cells, with 43 downregulated and 63 upregulated. Several genes relating PI3K/Akt signaling pathway were found to be aberrantly expressed in MCL cells compared with naive B cells, such as PIK3CA, PDK2, PDPK1, AKT1, RPS6KB2, FOXO3A, PPP2R2C and PDK1 [53]. Moreover, increased gene copy number ( ≥ 3) of PIK3CA were discovered in 68% of MCL cases and two MCL cell lines(Rec-1 and GRANTA-519) [23]. Mutation of PIK3CA gene resulted in constitutive activation of PI3K and the consequent activation of Akt pathway in MCL. They further investigated the apoptosis of MCL cell lines treated with LY294002. The apoptotic rates increased from 3% to 20% in GRANTA-519 cells and from 7.3% to 20% in Rec-1 cells [23].
RAD001 (everolimus), an mTOR inhibitor, could halt the translation of proteins critical for cell survival and proliferation via inhibiting mTOR phosphorylation [54]. Approximately 40 ~ 65% antiproliferative effects was found in MCL cell lines treated with single agent RAD001 compared with control groups [28]. However, NVP-BEZ235 is more effective than mTOR inhibitors (rapamycin, RAD001) in inhibiting the downstream pathway of mTOR and mediating cell death. Further analysis demonstrated that NVP-BEZ235 could lead to a dose dependent down-regulation of Mcl-1 protein while rapamycin could not [55]. Civallero et al. analyzed the inhibitory effects of NVP-BEZ235 on MCL cell lines and its effects in combination with enzastaurin, everolimus and perifosine [17]. NVP-BEZ235 induced significant increase of cell apoptosis in MCL through both intrinsic and extrinsic pathways. When combined with enzastaurin, everolimus and perifosine, the NVP-BEZ235 triggered cytotoxicity was enhanced significantly [17]. NVP-BEZ235 also showed a much stronger anti-proliferative function in MCL cells compared to single inhibitors of PI3K/mTOR, such as NVP-BKM120 and RAD001 [56]. Additionally, NVP-BEZ235 could synergistically enhance the cytotoxic function of conventional anti-tumor agents and remarkably overcome the acquired bortezomib resistance in MCL [56].
CAL-101 was reported to inhibit constitutive activation of the PI3K/Akt/mTOR pathway and exert potent antitumor effects across a wide range of B-cell malignancies [39]. Previous studies have demonstrated the functions of CAL-101 in PI3K inhibition and pro-apoptosis effect in NHL cell lines. A phase I study focused on the safety and activity of CAL-101 in patients with relapsed/refractory hematologic malignancies was carried out recently [19]. A total of 55 patients (18 MCL patients included) enrolled, CAL-101 was administered orally once or 2 times per day continuously in a 28-day cycle for up to 12 cycles. As a consequence, the overall response rate for MCL was 62% [19]. Nevertheless, GDC-0941, a dual p110α/δ inhibitor, was more active compared to CAL-101 in both MCL samples and cell lines [26].
The roles in Burkitt leukemia/lymphoma
Burkitt leukemia/lymphoma (BL) is a highly proliferative B-cell lymphoma characterized by constitutive MYC expression [24]. In spite of current intensive, short-term chemotherapy regimens in BL treatment, less toxic and more targeted treatment strategies are still needed to improve BL prognosis, especially in high-risk and relapsed/refractory patients [57, 58]. PI3K pathway acts as a vital determinant in the B cell receptor (BCR)-mediated survival signal in mature, resting B cells [59]. It has been indicated that the MYC-driven lymphoma is associated with mTOR activation and an endogenous DNA damage response transduced by PI3K-related kinase [60].
Activation of PI3K pathway has been found in BL tissues and cell lines. When treated BL cell lines with LY-294002, the phosphorylation of Akt kinase was largely diminished [24]. In drug-resistant Ramos and Daudi B-NHL cell lines, LY294002 treatment also accounted for the inhibition of Bcl-(xL) expression and sensitization to drug-induced apoptosis [61]. Our previous study also indicated the existence of PI3K/Akt/HSP70 cascade in Raji cells lines [4]. LY294002 significantly attenuated Akt activation, resulted in induced cell apoptosis and increased ADM and DDP sensitivity [4]. PI-103, a dual PI3K/mTOR inhibitor, was also associated with the caspase-dependent cleavage of PARP and inhibition of c-MYC activity in BL cells [29].
The studies of PI3K inhibitors in T-cell lymphoma
Activation of PTEN-PI3K-Akt pathway in T-ALL has been assessed by array comparative genomic hybridization and sequence analysis [62]. Alterations of PTEN, PI3K, or Akt existed in 47.7% of total 44 cases. Moreover, patients with lymphoblasts harboring PTEN deletions at the time of diagnosis showed significantly adverse therapeutic consequences [62]. Furthermore, the PI3K transgenic mice could develop an infiltrating lymphoproliferative disorder [63]. Lymphomas (67%) and sarcomas (33%) occurred in p53 knockout mice, however, when p53 deletion was combined with PI3K activation, only lymphomas developed. In addition, PTEN, a negative regulator of PI3K pathway, showed decreased expression level in 66.7% of anaplastic large cell lymphoma (ALCL) cases [64]. And increased expression of PIK3CD gene (encoding PI3K δ isoform), was found in peripheral and cutaneous T-cell lymphoma [27].
P110a, p110h, p110g, and p110y isoforms of PI3Ks were expressed in T-ALL cell lines. A dose-dependent decrease in cell survival was obtained with p110a PI3K selective inhibitor. Nevertheless, PI-103 was more efficient in inhibiting T-ALL cell proliferation and inducing cell apoptosis than inhibitors that are selective only for PI3K (Wortmannin and LY294002) [25]. The pan-PI3K inhibitor, GDC-0941, resulted in arrest of all peripheral and cutaneous T-cell lymphoma cell lines in the G1 phase. When cooperated with MEK inhibitors, GDC-0941 showed a highly synergistic effect in enhancing cell cycle arrest in all T-cell lymphoma cell lines [27].
Conclusions
In summary, PI3K signaling pathway was activated in both B-cell and T-cell NHL and involved in the development and progression of these diseases. The PI3K inhibitors revealed significant cytotoxicity either alone or in combination with other agents in lymphocytic cells. They have promised the breakthrough data and provided an attractive treatment option for anticancer therapeutic intervention of NHL. However, further investigations are still required to get a better understanding of the clinical benefits of PI3K inhibitors.
Abbreviations
- PI3K:
-
Phosphatidylinositol 3-kinase
- mTOR:
-
Mammalian target of rapamycin
- NHL:
-
Non-Hodgkin lymphoma
- HSP70:
-
Heat shock protein70
- BL:
-
Burkitt leukemia/lymphoma
- RTK:
-
Receptor tyrosine kinase
- PTEN:
-
Phosphatase and tensin homolog deleted on chromosome ten
- TSC2:
-
Tuberous sclerosis complex 2
- mTORC1:
-
mTOR complex 1
- 4EBP1:
-
4E binding protein 1
- FL:
-
Follicular lymphoma
- T-ALL:
-
T-cell acute lymphoblastic leukemia
- CLL:
-
Chronic lymphocytic leukemia
- DLBCL:
-
Diffuse large cell lymphoma
- GCB:
-
Germinal central B-cell
- ABC:
-
Activated B-cell
- MCL:
-
Mantle cell lymphoma
- BCR:
-
B cell receptor
- ALCL:
-
Anaplastic large cell lymphoma.
References
Laplante M, Sabatini DM: mTOR signaling in growth control and disease. Cell 2012, 149: 274–293. 10.1016/j.cell.2012.03.017
Krause DS, Van Etten RA: Tyrosine kinases as targets for cancer therapy. N Engl J Med 2005, 353: 172–187. 10.1056/NEJMra044389
Ferte C, Andre F, Soria JC: Molecular circuits of solid tumors: prognostic and predictive tools for bedside use. Nat Rev Clin Oncol 2010, 7: 367–380. 10.1038/nrclinonc.2010.84
Fang X, Jiang Y, Feng L, Chen H, Zhen C, Ding M, Wang X: Blockade of PI3K/AKT pathway enhances sensitivity of Raji cells to chemotherapy through down-regulation of HSP70. Cancer Cell Int 2013, 13: 48. 10.1186/1475-2867-13-48
Kawauchi K, Ogasawara T, Yasuyama M, Otsuka K, Yamada O: The PI3K/Akt pathway as a target in the treatment of hematologic malignancies. Anticancer Agents Med Chem 2009, 9: 550–559. 10.2174/187152009788451851
Yuan TL, Cantley LC: PI3K pathway alterations in cancer: variations on a theme. Oncogene 2008, 27: 5497–5510. 10.1038/onc.2008.245
Courtney KD, Corcoran RB, Engelman JA: The PI3K pathway as drug target in human cancer. J Clin Oncol 2010, 28: 1075–1083. 10.1200/JCO.2009.25.3641
Engelman JA, Luo J, Cantley LC: The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet 2006, 7: 606–619.
Sarbassov DD, Guertin DA, Ali SM, Sabatini DM: Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307: 1098–1101. 10.1126/science.1106148
Altomare DA, Testa JR: Perturbations of the AKT signaling pathway in human cancer. Oncogene 2005, 24: 7455–7464. 10.1038/sj.onc.1209085
Liu P, Cheng H, Roberts TM, Zhao JJ: Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov 2009, 8: 627–644. 10.1038/nrd2926
Kang S, Bader AG, Vogt PK: Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci U S A 2005, 102: 802–807. 10.1073/pnas.0408864102
Clinical Trials.gov http://www.clinicaltrials.gov/
Bhende PM, Park SI, Lim MS, Dittmer DP, Damania B: The dual PI3K/mTOR inhibitor, NVP-BEZ235, is efficacious against follicular lymphoma. Leukemia 2010, 24: 1781–1784. 10.1038/leu.2010.154
Chiarini F, Grimaldi C, Ricci F, Tazzari PL, Evangelisti C, Ognibene A, Battistelli M, Falcieri E, Melchionda F, Pession A, et al.: Activity of the novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 against T-cell acute lymphoblastic leukemia. Cancer Res 2010, 70: 8097–8107. 10.1158/0008-5472.CAN-10-1814
Zang C, Eucker J, Liu H, Muller A, Possinger K, Scholz CW: Concurrent inhibition of PI3-Kinase and mTOR induces cell death in diffuse large B cell lymphomas, a mechanism involving down regulation of Mcl-1. Cancer Lett 2013, 339: 288–297. 10.1016/j.canlet.2012.11.013
Civallero M, Cosenza M, Marcheselli L, Pozzi S, Sacchi S: NVP-BEZ235 alone and in combination in mantle cell lymphoma: an effective therapeutic strategy. Expert Opin Investig Drugs 2012, 21: 1597–1606. 10.1517/13543784.2012.719871
Herman SE, Gordon AL, Wagner AJ, Heerema NA, Zhao W, Flynn JM, Jones J, Andritsos L, Puri KD, Lannutti BJ, et al.: Phosphatidylinositol 3-kinase-delta inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood 2010, 116: 2078–2088. 10.1182/blood-2010-02-271171
Kahl B, Byrd JC, Flinn IW, Wagner-Johnston N, Spurgeon S, Benson DM Jr, Furman RR, Brown JR, Coutre S, Lannutti B, et al.: Clinical Safety and Activity In a Phase 1 Study of CAL-101, An Isoform-Selective Inhibitor of Phosphatidylinositol 3-Kinase P110δ, In Patients with Relapsed or Refractory Non-Hodgkin Lymphoma. ASH Annual Meeting Abstracts 2010, 116: 1777.
Amrein L, Shawi M, Grenier J, Aloyz R, Panasci L: The phosphatidylinositol-3 kinase I inhibitor BKM120 induces cell death in B-chronic lymphocytic leukemia cells in vitro. Int J Cancer 2013, 133: 247–252. 10.1002/ijc.27989
Zang C, Eucker J, Liu H, Coordes A, Lenarz M, Possinger K, Scholz CW: Inhibition of pan-class I PI3 kinase by NVP-BKM120 effectively blocks proliferation and induces cell death in diffuse large B cell lymphoma. Leuk Lymphoma in press. Epub 2013 Jul 25
Uddin S, Hussain AR, Siraj AK, Manogaran PS, Al-Jomah NA, Moorji A, Atizado V, Al-Dayel F, Belgaumi A, El-Solh H, et al.: Role of phosphatidylinositol 3'-kinase/AKT pathway in diffuse large B-cell lymphoma survival. Blood 2006, 108: 4178–4186. 10.1182/blood-2006-04-016907
Psyrri A, Papageorgiou S, Liakata E, Scorilas A, Rontogianni D, Kontos CK, Argyriou P, Pectasides D, Harhalakis N, Pappa V, et al.: Phosphatidylinositol 3'-kinase catalytic subunit alpha gene amplification contributes to the pathogenesis of mantle cell lymphoma. Clin Cancer Res 2009, 15: 5724–5732. 10.1158/1078-0432.CCR-08-3215
Sander S, Calado DP, Srinivasan L, Kochert K, Zhang B, Rosolowski M, Rodig SJ, Holzmann K, Stilgenbauer S, Siebert R, et al.: Synergy between PI3K signaling and MYC in Burkitt lymphomagenesis. Cancer Cell 2012, 22: 167–179. 10.1016/j.ccr.2012.06.012
Chiarini F, Fala F, Tazzari PL, Ricci F, Astolfi A, Pession A, Pagliaro P, McCubrey JA, Martelli AM: Dual inhibition of class IA phosphatidylinositol 3-kinase and mammalian target of rapamycin as a new therapeutic option for T-cell acute lymphoblastic leukemia. Cancer Res 2009, 69: 3520–3528. 10.1158/0008-5472.CAN-08-4884
Iyengar S, Clear A, Bodor C, Maharaj L, Lee A, Calaminici M, Matthews J, Iqbal S, Auer R, Gribben J, Joel S: P110alpha-mediated constitutive PI3K signaling limits the efficacy of p110delta-selective inhibition in mantle cell lymphoma, particularly with multiple relapse. Blood 2013, 121: 2274–2284. 10.1182/blood-2012-10-460832
Martin-Sanchez E, Rodriguez-Pinilla SM, Sanchez-Beato M, Lombardia L, Dominguez-Gonzalez B, Romero D, Odqvist L, Garcia-Sanz P, Wozniak MB, Kurz G, et al.: Simultaneous inhibition of pan-phosphatidylinositol-3-kinases and MEK as a potential therapeutic strategy in peripheral T-cell lymphomas. Haematologica 2013, 98: 57–64. 10.3324/haematol.2012.068510
Haritunians T, Mori A, O'Kelly J, Luong QT, Giles FJ, Koeffler HP: Antiproliferative activity of RAD001 (everolimus) as a single agent and combined with other agents in mantle cell lymphoma. Leukemia 2007, 21: 333–339. 10.1038/sj.leu.2404471
Spender LC, Inman GJ: Phosphoinositide 3-kinase/AKT/mTORC1/2 signaling determines sensitivity of Burkitt's lymphoma cells to BH3 mimetics. Mol Cancer Res 2012, 10: 347–359. 10.1158/1541-7786.MCR-11-0394
Gulmann C, Espina V, Petricoin E 3rd, Longo DL, Santi M, Knutsen T, Raffeld M, Jaffe ES, Liotta LA, Feldman AL: Proteomic analysis of apoptotic pathways reveals prognostic factors in follicular lymphoma. Clin Cancer Res 2005, 11: 5847–5855. 10.1158/1078-0432.CCR-05-0637
Fruchon S, Kheirallah S, Al Saati T, Ysebaert L, Laurent C, Leseux L, Fournie JJ, Laurent G, Bezombes C: Involvement of the Syk-mTOR pathway in follicular lymphoma cell invasion and angiogenesis. Leukemia 2012, 26: 795–805. 10.1038/leu.2011.248
Kim A, Lee JE, Lee SS, Kim C, Lee SJ, Jang WS, Park S: Coexistent mutations of KRAS and PIK3CA affect the efficacy of NVP-BEZ235, a dual PI3K/MTOR inhibitor, in regulating the PI3K/MTOR pathway in colorectal cancer. Int J Cancer 2013, 133: 984–996. 10.1002/ijc.28073
Sano T, Takeuchi S, Nakagawa T, Ishikawa D, Nanjo S, Yamada T, Nakamura T, Matsumoto K, Yano S: The novel phosphoinositide 3-kinase-mammalian target of rapamycin inhibitor, BEZ235, circumvents erlotinib resistance of epidermal growth factor receptor mutant lung cancer cells triggered by hepatocyte growth factor. Int J Cancer 2013, 133: 505–513. 10.1002/ijc.28034
Eltom MA, Jemal A, Mbulaiteye SM, Devesa SS, Biggar RJ: Trends in Kaposi's sarcoma and non-Hodgkin's lymphoma incidence in the United States from 1973 through 1998. J Natl Cancer Inst 2002, 94: 1204–1210. 10.1093/jnci/94.16.1204
Chiorazzi N, Rai KR, Ferrarini M: Chronic lymphocytic leukemia. N Engl J Med 2005, 352: 804–815. 10.1056/NEJMra041720
Lu K, Wang X: Therapeutic advancement of chronic lymphocytic leukemia. J Hematol Oncol 2012, 5: 55. 10.1186/1756-8722-5-55
Ge X, Wang X: Role of Wnt canonical pathway in hematological malignancies. J Hematol Oncol 2010, 3: 33. 10.1186/1756-8722-3-33
Ringshausen I, Schneller F, Bogner C, Hipp S, Duyster J, Peschel C, Decker T: Constitutively activated phosphatidylinositol-3 kinase (PI-3K) is involved in the defect of apoptosis in B-CLL: association with protein kinase Cdelta. Blood 2002, 100: 3741–3748. 10.1182/blood-2002-02-0539
Lannutti BJ, Meadows SA, Herman SE, Kashishian A, Steiner B, Johnson AJ, Byrd JC, Tyner JW, Loriaux MM, Deininger M, et al.: CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood 2011, 117: 591–594. 10.1182/blood-2010-03-275305
Hoellenriegel J, Meadows SA, Sivina M, Wierda WG, Kantarjian H, Keating MJ, Giese N, O'Brien S, Yu A, Miller LL, et al.: The phosphoinositide 3'-kinase delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leukemia. Blood 2011, 118: 3603–3612. 10.1182/blood-2011-05-352492
Coutre SE, Byrd JC, Furman RR, Brown JR, Benson DM, Wagner-Johnston ND, Flinn IW, Kahl BS, Spurgeon SEF, Lannutti BJ, et al.: Phase I study of CAL-101, an isoform-selective inhibitor of phosphatidylinositol 3-kinase P110d, in patients with previously treated chronic lymphocytic leukemia. ASCO Meeting Abstracts 2011, 29: 6631.
Flinn IW, Schreeder MT, Coutre SE, Leonard J, Wagner-Johnston ND, De Vos S, Boccia RV, Holes L, Peterman S, Miller LL, Yu AS: A phase I study of CAL-101, an isoform-selective inhibitor of phosphatidylinositol 3-kinase P110δ, in combination with anti-CD20 monoclonal antibody therapy and/or bendamustine in patients with previously treated B-cell malignancies. ASCO Meeting Abstracts 2011, 29: 3064.
Rosich L, Saborit-Villarroya I, Lopez-Guerra M, Xargay-Torrent S, Montraveta A, Aymerich M, Villamor N, Campo E, Perez-Galan P, Roue G, Colomer D: The phosphatidylinositol-3-kinase inhibitor NVP-BKM120 overcomes resistance signals derived from microenvironment by regulating the Akt/FoxO3a/Bim axis in chronic lymphocytic leukemia cells. Haematologica 2013, 98: 1739–1747. 10.3324/haematol.2013.088849
Xu ZZ, Xia ZG, Wang AH, Wang WF, Liu ZY, Chen LY, Li JM: Activation of the PI3K/AKT/mTOR pathway in diffuse large B cell lymphoma: clinical significance and inhibitory effect of rituximab. Ann Hematol 2013, 92: 1351–1358. 10.1007/s00277-013-1770-9
Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES: The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood 2011, 117: 5019–5032. 10.1182/blood-2011-01-293050
Fridberg M, Servin A, Anagnostaki L, Linderoth J, Berglund M, Soderberg O, Enblad G, Rosen A, Mustelin T, Jerkeman M, et al.: Protein expression and cellular localization in two prognostic subgroups of diffuse large B-cell lymphoma: higher expression of ZAP70 and PKC-beta II in the non-germinal center group and poor survival in patients deficient in nuclear PTEN. Leuk Lymphoma 2007, 48: 2221–2232. 10.1080/10428190701636443
Kloo B, Nagel D, Pfeifer M, Grau M, Duwel M, Vincendeau M, Dorken B, Lenz P, Lenz G, Krappmann D: Critical role of PI3K signaling for NF-kappaB-dependent survival in a subset of activated B-cell-like diffuse large B-cell lymphoma cells. Proc Natl Acad Sci U S A 2011, 108: 272–277. 10.1073/pnas.1008969108
Bendell JC, Rodon J, Burris HA, de Jonge M, Verweij J, Birle D, Demanse D, De Buck SS, Ru QC, Peters M, et al.: Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol 2012, 30: 282–290. 10.1200/JCO.2011.36.1360
Jares P, Colomer D, Campo E: Genetic and molecular pathogenesis of mantle cell lymphoma: perspectives for new targeted therapeutics. Nat Rev Cancer 2007, 7: 750–762.
Perez-Galan P, Dreyling M, Wiestner A: Mantle cell lymphoma: biology, pathogenesis, and the molecular basis of treatment in the genomic era. Blood 2011, 117: 26–38. 10.1182/blood-2010-04-189977
Weniger MA, Wiestner A: Molecular targeted approaches in mantle cell lymphoma. Semin Hematol 2011, 48: 214–226. 10.1053/j.seminhematol.2011.05.001
Ghobrial IM, McCormick DJ, Kaufmann SH, Leontovich AA, Loegering DA, Dai NT, Krajnik KL, Stenson MJ, Melhem MF, Novak AJ, et al.: Proteomic analysis of mantle-cell lymphoma by protein microarray. Blood 2005, 105: 3722–3730. 10.1182/blood-2004-10-3999
Rizzatti EG, Falcao RP, Panepucci RA, Proto-Siqueira R, Anselmo-Lima WT, Okamoto OK, Zago MA: Gene expression profiling of mantle cell lymphoma cells reveals aberrant expression of genes from the PI3K-AKT, WNT and TGFbeta signalling pathways. Br J Haematol 2005, 130: 516–526. 10.1111/j.1365-2141.2005.05630.x
Jacinto E, Hall MN: Tor signalling in bugs, brain and brawn. Nat Rev Mol Cell Biol 2003, 4: 117–126. 10.1038/nrm1018
Muller A, Zang C, Chumduri C, Dorken B, Daniel PT, Scholz CW: Concurrent inhibition of PI3K and mTORC1/mTORC2 overcomes resistance to rapamycin induced apoptosis by down-regulation of Mcl-1 in mantle cell lymphoma. Int J Cancer 2013, 133: 1813–1824. 10.1002/ijc.28206
Kim A, Park S, Lee JE, Jang WS, Lee SJ, Kang HJ, Lee SS: The dual PI3K and mTOR inhibitor NVP-BEZ235 exhibits anti-proliferative activity and overcomes bortezomib resistance in mantle cell lymphoma cells. Leuk Res 2012, 36: 912–920. 10.1016/j.leukres.2012.02.010
Blum KA, Lozanski G, Byrd JC: Adult Burkitt leukemia and lymphoma. Blood 2004, 104: 3009–3020. 10.1182/blood-2004-02-0405
Jiang Y, Wang X: Comparative mitochondrial proteomics: perspective in human diseases. J Hematol Oncol 2012, 5: 11. 10.1186/1756-8722-5-11
Srinivasan L, Sasaki Y, Calado DP, Zhang B, Paik JH, DePinho RA, Kutok JL, Kearney JF, Otipoby KL, Rajewsky K: PI3 kinase signals BCR-dependent mature B cell survival. Cell 2009, 139: 573–586. 10.1016/j.cell.2009.08.041
Shortt J, Martin BP, Newbold A, Hannan KM, Devlin JR, Baker AJ, Ralli R, Cullinane C, Schmitt CA, Reimann M, et al.: Combined inhibition of PI3K-related DNA damage response kinases and mTORC1 induces apoptosis in MYC-driven B-cell lymphomas. Blood 2013, 121: 2964–2974. 10.1182/blood-2012-08-446096
Suzuki E, Umezawa K, Bonavida B: Rituximab inhibits the constitutively activated PI3K-Akt pathway in B-NHL cell lines: involvement in chemosensitization to drug-induced apoptosis. Oncogene 2007, 26: 6184–6193. 10.1038/sj.onc.1210448
Gutierrez A, Sanda T, Grebliunaite R, Carracedo A, Salmena L, Ahn Y, Dahlberg S, Neuberg D, Moreau LA, Winter SS, et al.: High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood 2009, 114: 647–650. 10.1182/blood-2009-02-206722
Borlado LR, Redondo C, Alvarez B, Jimenez C, Criado LM, Flores J, Marcos MA, Martinez AC, Balomenos D, Carrera AC: Increased phosphoinositide 3-kinase activity induces a lymphoproliferative disorder and contributes to tumor generation in vivo. Faseb J 2000, 14: 895–903.
Uner AH, Saglam A, Han U, Hayran M, Sungur A, Ruacan S: PTEN and p27 expression in mature T-cell and NK-cell neoplasms. Leuk Lymphoma 2005, 46: 1463–1470. 10.1080/10428190500144813
Acknowledgements
This study was supported by the grants from National Natural Science Foundation (No. 81270598), Natural Science Foundations of Shandong Province (No. Y2007C053, No. 2009ZRB14176 and No. ZR2012HZ003), Technology Development Projects of Shandong Province (No. 2007GG10 and No. 2010GSF10250), Medicine and Health Care Technology Development Project of Shandong Province (2013WS0117), Program of Shandong Medical Leading Talent, and Taishan Scholar Foundation of Shandong Province.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare no competing financial interests.
Authors’ contributions
All authors have contributed to data preparation, drafting and revising the manuscripts. All authors have read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Fang, X., Zhou, X. & Wang, X. Clinical development of phosphatidylinositol 3-kinase inhibitors for non-Hodgkin lymphoma. Biomark Res 1, 30 (2013). https://doi.org/10.1186/2050-7771-1-30
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2050-7771-1-30