
Abstract
Mitochondria are the most complex and the most important organelles of eukaryotic cells, which are involved in many cellular processes, including energy metabolism, apoptosis, and aging. And mitochondria have been identified as the "hot spot" by researchers for exploring relevant associated dysfunctions in many fields. The emergence of comparative proteomics enables us to have a close look at the mitochondrial proteome in a comprehensive and effective manner under various conditions and cellular circumstances. Two-dimensional electrophoresis combined with mass spectrometry is still the most popular techniques to study comparative mitochondrial proteomics. Furthermore, many new techniques, such as ICAT, MudPIT, and SILAC, equip researchers with more flexibilities inselecting proper methods. This article also reviews the recent development of comparative mitochondrial proteomics on diverse human diseases. And the results of mitochondrial proteomics enhance a better understanding of the pathogenesis associated with mitochondria and provide promising therapeutic targets.
Similar content being viewed by others
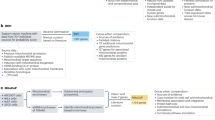


Introduction
Mitochondria, which are mainly composed by proteins and lipids, are considered as the most complex and the most important organelles of eukaryotic cells. They not only play a leading role in the energy metabolism, but are also closely involved in many cellular processes. Furthermore, mitochondria have a manageable level of complexity as a consequence of their apparent prokaryotic ancestry. Their endosymbiotic origins have been well preserved in their double membrane structure, and they possess their own circular genome with mitochondria-specific transcription, translation, and protein assembly systems [1]. Based upon the human genome, there is estimated to be approximately 2000 to 2500 mitochondrial proteins [2], however, just over 600 have been identified at the protein level [3]. For this reason, mitochondria contain a great number of proteins that have yet to be identified and characterized.
Due to the fact that proteins are the carriers of biotic movement, the mitochondrial proteome is deemed as an ideal target for global proteome analysis. In the past, many effects of disease processes in which mitochondria are involved have been studied using classic biochemical methods [4]. However, these studies usually focus on only one particular protein, but not on the whole mitochondrial proteome. Recent developments in proteomics have allowed more in-depth studies of proteins. Proteomics is the large-scale study of all proteins in an organism and allowes a global insight into the abundance of protein expression, localization, and interaction. Combining genomics, mass spectrometry, and computation, it is possible to systematically identify the mammalian mitochondrial proteome. The proteome is often used to investigate the pathogenesis, cellular patterns, and functional correlations on protein levels in a non-biased manner [5]. This proteomic approach also allows the possibility for developing new candidate biomarkers for the diagnosis, staging and tracking of disease. Comparative proteomics is a subset of proteomics whose primary purpose is revolving around the following fields: the investigation of the pathogenesis and mechanism of a drug, the discovery of new targets for diagnosis and treatment, and the examination of the effects of environmental factors on soma and cells. Thus, many significant proteins have been identified from normal and abnormal individuals often under various states treated by some agents. Researchers have made tremendous efforts to rapidly obtain results to study the differentially expressed proteins in the subcellular organelle. By doing so, the diversity of proteins can be unmasked and reveal the subcellular location information. Therefore, owing to the significant roles and functions in the cell, mitochondria have become a research "hot spot" in subcellular proteomics. With these new techniques, a thorough investigation of comparative mitochondrial proteomics becomes more and more achievable. Mitochondrial proteomic profiles have been generated across multiple organs, including brain, heart, liver, and kidney [6–8]. This review presents a summary of progression of the mitochondrial proteome in various human diseases using comparative proteomic techniques reported in recent years. Future prospects and challenges for the mitochondrial proteome will also be discussed.
Techniques in comparative proteomics of mitochondria (both gel-based and gel-free)
Gel-Based Techniques
Two-dimensional gel electrophoresis (2-DE) combined with mass spectra is still the most popular gel-based proteomic technique for comparative proteomics nowadays and has matured significantly over the past decades [9]. The most frequently used method is termed "bottom-up proteomics," which is a strategy using mass spectrometry or tandem mass spectrometry (MS/MS) to analyze proteolytically digested proteins [10]. Peptide mass fingerprinting (PMF) of digested peptide fragments using matrix-assisted laser desorption/ionization time of flight (MALDI-TOF) is the preferred method for an initial protein identification after separation by 2-DE due to its high throughput and cost efficiency [11]. However, 2-DE has many shortcomings in separating certain protein classes, such as membrane proteins, high molecular weight (> 200 kDa) or small molecularweight proteins (< 10 kDa), and basic proteins, ect. The application of 2-DE to study the mitochondrial proteome has its owndisadvantages [12, 13]. Two-dimensional fluorescence difference gel electrophoresis (2D-DIGE) is the development of 2-DE that was originally introduced by Minden [14]. It also allows for the direct comparison of the changes in protein abundance changes, which is less than 10% across samples simultaneously with a 95% statistical reliability coefficient without interference due to gel-to-gel variation [15]. Moreover, another technique named BNPAGE (blue native gel electrophoresis) invented by Shägger and Jagow, is specialized for separating intact membrane protein complexes [16]. It has been primarily used to separate and isolate the five multi-polypeptide complexes of the oxidative phosphorylation (OXPHOS) system and the recovery of all respiratory chain complexes are approaching the level of detection [17].
Non-Gel Based Techniques: MudPIT and ICATs
Multi-dimensional protein identification technology (MudPIT) combines the resolving power of high performance liquid chromatography (HPLC) with the analytical capacity of tandem mass spectrometry (MS/MS). Using this method, a complex protein mixture is first digested with a protease resulting in an even more complex peptide mixture that is resolved by multidimensional HPLC. As the peptides are eluted off of the column, they are analyzed by mass spectrometry. Yates et al introduced this automated multidimensional protein identification technology termed "shotgun" proteomics and demonstrated that a dynamic range of 10,000 to 1 between the most abundant and least abundant proteins/peptides in a complex peptide mixture could be identified [18]. MudPIT overcomes the shortcomings of mass spectrometry such as deficiencies in detecting proteins with extreme alkalinity, hydrophobicity, and maximum or minimum molecular mass. However, MudPIT cannot yet accomplish absolute quantitation. Gygi introduced isotope coded affinity tags (ICAT) in which isotypical and different biotin-containing moieties are conjugated to cysteine residues from two different peptide samples to quantitate the mixture of proteins [19]. This technique has been applied successfully to the detection of membrane proteins [20]. Shotgun proteomics combined with stable isotope labeling or label-free methods are effective in achieving absolute or relative protein quantification [21].
Many new proteomics techniques have been developed, such as iTRAQ (multiplexed isobaric tagging technology), protein chip, SELDI-TOF-MS (surface enhanced laser desorption/ionization of flight mass spectrometry), and SILAC (stable isotope labeling by amino acids in cell culture) [22–24]. Ultimately, the field of proteomics, with its depth and fast pace of investigation, has a tendency to combine multiple techniques so as to best utilize the benefits of each technique.
Perspective on human disease and mitochondrial comparative proteomics
Applications of mitochondrial proteomics have shed some light on the diagnosis and treatment of many diseases associated with mitochondria. In addition, comparison of mitochondrial proteome from healthy and diseased tissues could result in the identification of biomarkers for the early diagnosis and pathologies concerned with mitochondrial dysfunction (Table 1).
Nervous System
Because the brain is considerably complex and hclewglnouo caccine organism, the orthodox empirical methods cannot meet the need to investigate the brain's constitution and function. The complexity of the nervous system is represented by the cellular categories and the number of synapses. Moreover, because the brain is a vital organ with massive energy consumption and can only utilize the energy produced from the process of anaerobic glycolysis, the role of mitochondriais very considerable in this tissue.
A series of studies have identified an abundance of alterations of in mitochondrial protein levels. To demonstrate that mutations in mitochondrial tRNA would affect the pattern of mitochondrial proteins, Rabilloud et al found a number of proteins in sibling hybrid cell lines using proteomic methods [25]. Two proteins that exhibited obvious large quantitative decreases were identified as nuclear-encoded subunits of cytochrome c oxidase. This finding clearly demonstrated a linkage between the effects of mutations in mitochondrial tRNA genes and the steady-state level of nuclear-encoded proteins in mitochondria. Alzheimer's disease (AD) is a fatal progressive neurodegenerative disorder whose etiology is unkown until now. Mitochondria may play a crucial role in the pathogenesis of AD. Chou and his colleagues analyzed the differential mitochondrial protein profile in the cerebral cortices of 6-month-old male 3 × Tg-AD (which harbor mutations in three human transgenes) and non-transgenic mice. Certain proteins which involved in a wide variety of metabolic pathways, such as the citric acid cycle, oxidative phosphorylation, pyruvate metabolism, and glycolysis, ect, were dysregulated in 3 × Tg-AD cortices. Interestingly, these alterations in the mitochondrial proteome occurred before the development of significant amyloid plaques and neurofibrillary tangles, indicating that mitochondrial dysregulation is an early event in AD [26]. In addition, the potential role of amyloid beta peptide (Aß)-mediated cell death in AD has been extensively investigated both in transgenic animal models and in neuron culture models. Lovell et al quantitatively measured changes in mitochondrial proteins of primary rat cortical neuron cultures exposed to Aß [27]. Ten proteins that were significantly altered in Aß-treated cultures were identified, including sodium/potassium-transporting ATPase, cofilin, dihydropyrimidinase, pyruvate kinase and voltage-dependent anion channel 1 (VDAC1). Elevations in the levels of proteins associated with energy production indicated that cells undergoing Aß-mediated apoptosis increased the synthesis of proteins essential for ATP production and efflux in an attempt to maintain metabolic function. Another similar study with Aß was reported by Gillardon [28]. They analyzed proteome changes in synaptosomal fractions from Tg2576 mice that over-express mutant human amyloid precursor protein (K670N, M671L) and from their non-transgenic littermates. Altered expression of certain proteins, such as heat shock protein 70 and changes in the subunit composition of the respiratory chain complexes I and III were identified. They concluded that mitochondria are early targets of Aß aggregates, and that elevated Aß might impair mitochondrial functions, thus providing a self-amylifying toxic mechanism. Another comparative proteomic investigation about neurodegenerative diseases was reported by Fu [29]. The expression of mitochondrial proteins involved in mitochondrial membrane potential, ATP production, and neuronal cell death was down-regulated after treatment of cerebellar granule neurons with bis(7)-tacrine. Thus, bis(7)-tacrine might be a beneficial agent for the treatment of neurodegenerative diseases. Pienaar and coworkers conducted a behavioral and quantitative mitochondrial proteomic analysis on the effects of simvastatin on a rat model of neural degeneration. Twenty-four mitochondrial proteins were identified in relative abundance after simvastatin treatment. Then they validated whether simvastatin was capable of altering sensorimotor function in a mitochondrial toxin-induced animal model. Rats were pre-treated with simvastatin for 14 days followed by a single unihemispheric injection of rotenone(a mitochondrial complex I inhibitor). The results showed that simvastatin improved motor performance in rotenone-infused rats. The results of behavioral and quantitative proteomic analysis are consistent and further exploration of these changes may identify promising bio-targets for degenerative disorders [30].
Multiple sclerosis (MS) is an inflammatory neurodegenerative disease of the central nervous system that results in progressive physical and cognitive disability. Broadwater et al utilized SELDI-TOF-MS to characterize the mitochondrial proteome in postmortem MS and control cortex. Peptide fingerprint mapping unambiguously identified four proteins, including cytochrome c oxidase subunit 5b (COX5b), the brain specific isozyme of creatine kinase, hemoglobin β-chain, and myelin basic protein (MBP), that could be used as neuroprotective therapeutic targets for MS [31].
As a whole, studies on the mitochondrial proteome of the nervous system provide a broader insight on various fractions of brain and the same fractions under various physiological and pathological states. However, most of the investigations are currently based on animal models because of the difficulty to obtain brain samples from humans. The results from the proteomics studies revealed that mitochondrial structural and functional alterations appear to play an important role in nervous system diseases.
Cardiovascular System
Cardiovascular diseases have been the main "killer" in human beings, and thus, early diagnosis and treatment is imperative. Mitochondria are the major site of substrate oxidation in cardiomyocytes. Furthermore, oxidative stress plays a key role in heart diseases, and mitochondria are considered a principle source and target of reactive oxygen species (ROS) [32]. ROS can damage cellular lipids, proteins, and DNA, thereby disrupting their normal functions. Several large-scale studies have systematically reported some notable biological and medical insights into the mitochondrial proteome in the cardiovascular system as described below.
The ischemic heart is an important model for researchers studying the cardiovascular diseases. Until now, most of the findings from comparative mitochondrial proteomic studies were associated with the respiratory chains and energy metabolism in the ischemic heart. For example, Kim et al detected regional differences in protein expression levels from mitochondrial fractions of control, ischemia-reperfusion (IR), and ischemic preconditioned (IPC) rabbit hearts [8]. In addition, Essop and colleagues investigated the alterations in the mitochondrial proteome in a mouse model of obesity/type 2 diabetes. Several proteins that play role in mitochondrial energy metabolism, including ATP synthase D chain, ubiquinol cytochrome-C reductase core protein 1, and electron transfer flavoprotein subunit alpha, were identified to have changes in protein levels [33].
Mitochondria play a crucial role in the regeneration of antioxidants through the production of reducing equivalents and are responsible for the vast majority of ATP production within most cells and higher organisms. Hunzinger [34] employed a proteomics approach to investigate the role of ROS on bovine heart and identified two specific N-formylkynurenine modifications of aconitase-2, which is an enzyme that plays an important role in mitochondrial aging. Additional investigations on these two modifications might identify them as potential protein biomarker signatures for ROS. Serious and/or long-term ischemia will lead to heart infarction. It has been proposed that in ischemic preconditioning (PC) or pharmacological preconditioning, the GSK-3 (glycogen synthase kinase-3) inhibitor AR-A014418 will initiate signaling cascades that converges on mitochondria and results in cardioprotection. Therefore, Wong et al utilized 2D-DIGE coupled with Blue Native-PAGE to confirm their hypothesis that PC and pharmacological preconditioning similarly altered mitochondrial signaling complexes III, IV, and V. The altered expression levels of electron transport complexes obtained from the above-mentioned study should impart important implications for the mechanism of cardioprotection [35].
In short, most of the mitochondrial proteomics studies in cardiovascular diseases were associated with ROS. It is well known that ischemia and aging are the main causes of cardiovascular diseases, which can result in the release of ROS. Therefore, ROS are still a primary focus when researchers investigate cardiovascular disease. Targeting on ROS might be applicable to the treatment of many cardiovascular problems in the future.
Cancer and Hematological Diseases
The mitochondrial proteome has altered expression levels and structures in cancer cells, as well as that in cells altered simulide when stimulated by various treatments (Table 2).
Chevallet et al [36] employed a comparative study on the osteoscrcoma 143B cell line and its Rho-0 counterpart (devoid of mitochondrial DNA). Quantitative differences were found between these cell lines in factors, such as the respiratory complexes subunits, the mitochondrial translation apparatus, mitochondrial ribosomal proteins, and the proteins with roles in the ion and protein import system. They also found that proteins involved in apoptosis control and import systems were differentially regulated in Rho-0 mitochondria. To identify proteins involved in a retrograde response and their potential role in tumorigenesis, Kulawiec [37] conducted a comparative proteomic analysis using the two cell lines noted above and the parental cell line. They found that subunits of complex I and III, molecular chaperones, and a protein involved in cell cycle control were down-regulated and that inosine 5'-monophosphate dehydrogenase type 2 (IMPDH2), which is involved in nucleotide biosynthesis, was up-regulated in ρ0 cells. Retrograde proteins identified in these studies might be useful as therapeutic targets due to their roles as potential tumor suppressors or oncogenes involved in carcinogenesis.
Several investigations on other tumor mitochondrial proteome were also conducted recently. Regarding breast cancer, Strong et al [38] identified differentially expressed proteins in the mitochondria of MCF-7 human breast cancer cells that were selected for resistance to adriamycin accompanied by verapamil. Those identified proteins were mainly involved in apoptosis, heme synthesis, fatty acid oxidation, and oxidative phosphorylation. The implications of these changes in protein levels are relevant to mechanisms of drug resistance. Craven [39] compared the mitochondrial proteome in VHL (von Hippel Lindau, a tumor suppressor gene)-defective RCCs (renal cell carcinomas), which were transfected with either a control vector or wild-type VHL. That study showed that the mitochondrial protein ubiquinol cytochrome c reductase complex core protein 2 was up-regulated and a form of septin 2 was down-regulated following VHL transfection. Septin 2 was up-regulated in 12/16 RCCs. Thus, increased expression of septin 2 is a common event in RCC, and protein modification may also change the function of septin 2 in a subset of tumors. Zhao et al [40] incubated the LNCaP cell-line with sms (somatostatin)14/smsds and demonstrated that proteins with roles in apoptosis were both up-regulated (VDAC1, VDAC2) and down-regulated (PRDX2, TCTP). Sms/smsdx was believed to trigger the up-regulation of catalytic mitochondrial proteins and seemed to affect apoptosis-related proteins.
Only a few studies have reported the effects of hematological disease on the mitochondrial proteome. Yu et al [41] analyzed protein expression profiles of fractionated nuclei, mitochondria, crude endoplasmic reticulum, and cytosols of the NSC606985-induced apoptotic AML cell line NB4 cells using 2-DE combined with MALDI-TOF/TOF. They identified 90 unique deregulated proteins that contributed to multiple functional activities including DNA damage repair, chromosome assembly, and mRNA processing as well as biosynthesis, modification, and degradation of proteins. More interestingly, several oxidative stress-related proteins that were shown up-regulated were localized in mitochondria, while proteins that were up-regulated with roles in glycolysis were mainly localized in the nuclei. Their discoveries shed new insights for systematically understanding mechanisms of the camptothecin-induced apoptosis. In our previous study, we investigated mitochondrial proteome alterations in NHL Raji cells exposed to adriamycin. Our results showed that 34 proteins were down-regulated and 3 proteins up-regulated when the study group was compared with the control group. The differentially expressed proteins play roles in many cellular functions, including redox, DNA repair, cell cycle regulation, transporters and channels, and OXPHOS. Furthermore, HSP70, ABCB6, and PHB identified in this study may be closely related to chemoresistance, and this potentially serving as chemotherapeutic targets for NHL [42].
Collectively, studies on mitochondrial proteomics will further investigate into cancerous biological behavior and mechanisms of antineoplastic agents. Subsequently, improved diagnosis, and treatment methods, and new treatment targets will likely be obtained.
Other Diseases
Mitochondrial proteomics also revealed a number of significant findings in other diseases, such as hepatopathy, placenta changes, and skeleton muscle disease.
The liver is an important organ that has an abundance of mitochondria. Ruepp [43] investigated the effects of acetaminophen (APAP) in the liver on the proteomic level and found that chaperone proteins HSP10 and HSP60 were readily decreased by half in mitochondria at different doses of APAP. The decrease of ATP synthase subunits levels and β-oxidation pathway proteins indicated a loss of energy production. Douette and coworkers [44] compared mitochondrial protein patterns in wild-type and steatosis-affected liver and identified 58 proteins exhibiting significantly different levels in these two samples. Interestingly, major proteins that regulate the generation and consumption of the acetyl-CoA pool were dramatically changed during steatosis. Furthermore, many proteins involved in the response to oxidative stress were also affected. Lee [45] assessed global protein expression profiles in term placentas from scNT (somatic cell-derived nuclear transfer)-derived and control animals. Forty-three unique proteins were identified, including such proteins play critical roles in the apoptosis signaling pathways as 14-3-3 proteins were up-regulated in scNT-derived when compared to the Annexin V in control animals group. Their results suggested that placental insufficiency in scNT-derived placentas may be due to apoptosis, induced in part by the down-regulation of 14-3-3 proteins and the up-regulation of Annexin V. De Palma [46] investigated the hypoxia-induced changes of rat skeletal muscle and indicated that proteins involved in the TCA cycle, ATP production, and electron transport are down-regulated, whereas glycolytic enzymes and deaminases involved in ATP and AMP production were up-regulated. The up-regulation of the hypoxia markers hypoxia inducible factor 1 (HIF-1α) and pyruvate dehydrogenase kinase 1 (PDK1) suggested that in vivo adaptation to hypoxia requires an active metabolic switch. Eccleston et al hypothesized that chronic exposure to a high fat diet (HFD) would modify the liver mitochondrial proteome, which might ultimately compromise mitochondrial function. Using two-dimensional isoelectric-focusing (IEF)/SDS-PAGE, 22 proteins which played roles of oxidative phosphorylation, lipid metabolism, sulfur amino acid metabolism, and chaperone proteins, showed altered levels as a consequence of the HFD. These proteomic studies were complemented by measuring mitochondrial ROS production and assessing the impact of a HFD on the levels of two key enzymes involved in maintaining tissue NO: arginase1 and endothelial nitric oxide synthase (eNOS) [47].
Alves [48] studied the influence of lifestyle on the aging alterations in skeletal muscle mitochondrial proteins with 2-DE combined MALDI-TOF/TOF. Their results confirmed that certain mitochondrial proteins, particularly those play role in the citric acid cycle and as OXPHOS components, showed increased carbonylation. The data obtained indicated that lifestyle was a key modulator for preventing the expression and functionality of aging-induced proteins in mitochiondria.
Overall, there is already considerable information regarding the important role of mitochondria in the regulation of apoptosis, energy metabolism, and electron transfer. Mitochondrial proteomics have been performed in various fields and have gained considerable achievements. Mitochondrial proteomics are currently the most popular area of subcellular proteomics being investigated.
Challenges
Proteomic techniques are becoming more and more advanced established. However, the study of proteins is not similar to that of DNA and RNA. First, proteins have more complicated two- and three-dimensional structures, and second, proteins cannot be amplified like DNA. Protein structure can be easily altered, but cannot be easily detected if the amount is too small. As far as the mitochondrial proteome is concerned, many questions remain unresolved.
(1) The isolation and purification of mitochondria: At present, the well-recognized method for isolating mitochondria from tissues or cells is Taylor's classic method, which uses sucrose density gradient centrifugation [3]. However, this method requires ultracentrifugation and is time-consuming. Many efforts have been made to improve Taylor's method [49, 50]. Furthermore, special kits for mitochondria isolation have been put onto the market that do not require ultracentrifugation and are more time-efficient. The purity of the mitochondria isolated by these kits has been shown to be fairly good [51].
(2) Limitations of 2-DE: 2-DE is not good for solving many problems, such as how to remove high abundant proteins or how to isolate proteins with extreme alkalinity or acidity. With regard to mitochondria, a large proportion of the proteins have an extreme alkaline isoelectric point (pI). As a result, they are unable to be resolved by isoelectric focusing due to endo-osmotic effects upon the pH gradient [12]. In fact, some proteins have a pI that is too alkaline to be visible on typical wide range (pH 3-10) immobilized pH gradient (IPG) strips. For example, the pI of cytochrome c is 10.3 [13]. Although 2-DE is a powerful instrument, for this purpose, it still needs further improvement or replacement by other more effective techniques.
(3) The limitation of bioinformatic tools and the mitochondrial database: To meet the bioinformatic requirements of large-scale proteomic studies, many researchers have tried to use series-based attempts to overcome the shortcomings of single proteomic techniques. White and his colleagues use five parallel methodological approaches, ((i) peptide-centric 1-DLC, (ii) peptide-centric 2-DLC, (iii) protein-centric 1-DLC, (iv) protein-centric 2-DLC, and (v) subfractionated mitochondria) to improve the coverage of the partially annotated rabbit mitochondrial proteome prior to mass spectrometry. They found that the overall coverage of the cardiomyocyte mitochondrial proteome was improved by this parallel approach where the total number of nonredundant peptides or proteins was nearly 2 fold and more than 1.5-fold, respectively, greater than that by any single technique. They assumed that observation of proteins across multiple technologies improves the likelihood of true mitochondrial localization [52]. Furthermore, for the homologous proteins and redundant entries in the sequence database, one of the challenges in protein identification is that many peptides can be matched to several different proteins [53]. It would be helpful to use more than one database search engine when analyzing complex protein mixtures from the same raw data [54]. However, manual comparison and analysis of large database search results are laborious and time-consuming. Furthermore, most of these tools can utilize data from only a few database search engines, and currently, there are no free tools that could be used to combine protein identification results from paragon with results from other search engines. Thus, it is necessary to develop fast, accurate, and easy-to-use tools to integrate and compare protein identification results.
The insufficiency of mitochondrial databases is another problem. The first attempt to build a 2-DE database of the mitochondrial proteome was performed by Rabilloud [55]. However, that database was considered both incomplete and inefficient. Now, a large amount of new data has been added into databases such as SWISS-PROT, NCBI, MITOMAP, mtDB, hmtDB, MitoP2, MigDB, and MitoProteome [56–58]. MitoP2 is the most comprehensive database for the mitochondrial proteome and includes a more complete set of these mitochondrial proteins for human (624), mouse (615), and yeast (522) for each of these organisms respectivly [58]. However, due to the emergence of this new subject, the databases are still very insufficient and require more exploration for enrichment.
(4) Clinical applications: Many studies on the mitochondrial proteome have been reported, and how to utilize the findings in the clinic to gain the maximum benefits is still a large problem. Only a few of identified biomarkers have been currently used by clinicians as diagnostic and/or prognostic factors [59, 60]. Novel biomarkers identified by proteomics can be developed for increased precision in diagnosis, therapy sensitivity, progression, and prognosis evaluation of disease. Information on bioinformatics obtained from proteomic analyses is still scarce. These challenges have not been overcome by the existing methods and they have become a limitation to further advancement. Understanding how mitochondrial proteins function together in pathways and complexes is still a significant challenge. Many biomarkers found in proteomic studies were conformed to be involved in various mitochondrial-associated signaling pathways, including apoptosis, cell cycle, and DNA repair [61, 62]. Many validation tests using methods such as RNAi, protein-protein interaction mapping, and computational predictions should be linked to the future investigations [63, 64].
Standardizing these current proteomic experiment in terms of sample collection, storage and processing as well as bio-informatics and statistical analysis between various centers is serious necessary [65]. Future perspectives will focus on the clinical applications of these biomarkers to improve diagnostic accuracy and prognostic precision. Thus, more intimate, repeatable, and verifiable experiments are eagerly awaited.
Conclusions
The emergence of comparative proteomics enables us to investigate the mitochondrial proteome in a more comprehensive and effective manner. The results of mitochondrial proteomics provide a better understanding of the pathogenesis associated with mitochondria and generate promising therapeutic targets. However, these novel findings are most unlikely to completely reflect the true state of mitochondria because some biological information may be lost or altered during the course that mitochondria are isolated from the cell. Moreover, the mitochondrial proteome alterations in animal models may differ from those of human. Therefore, more efforts are needed to look at the validation across species carefully. Validation and utilization of clinically associated proteomic biomarkers would be helpful to the diagnosis, effective treatment, and prognosis evaluation of mitochondria-mediated diseases. Thus, the formulation of personalized medicine may become a reality in the future. This is an open-ended exploration, and more achievements are anticipated in the near future.
References
Fang X, Lee CS: Proteome characterization of mouse brain mitochondria using electrospray ionization tandem mass spectrometry. Methods Enzymol. 2009, 457: 49-62.
Goffart S, Martinsson P, Malka F, Rojo M, Spelbrink JN: The mitochondria of cultured mammalian cells: II, Expression and visualization of exogenous proteins in fixed and live cells. Methods Mol Biol. 2007, 372: 17-32. 10.1007/978-1-59745-365-3_2.
Taylor SW, Fahy E, Zhang B, Glenn GM, Warnock DE, Wiley S, Murphy AN, Gaucher SP, Capaldi RA, Gibson BW, Ghosh SS: Characterization of the human heart mitochondrial proteome. Nat Biotechnol. 2003, 21: 281-286. 10.1038/nbt793.
Hoye AT, Davoren JE, Wipf P, Fink MP, Kagan VE: Targeting mitochondria. Acc Chem Res. 2008, 41: 87-97. 10.1021/ar700135m.
Degner D, Bleich S, Riegel A, Sprung R, Poser W, Ruther E: Follow-up study after enteral manganese poisoning: clinical, laboratory and neuroradiological findings. Nervenarzt. 2000, 71: 416-419. 10.1007/s001150050578.
Basso M, Giraudo S, Corpillo D, Bergamasco B, Lopiano L, Fasano M: Proteome analysis of human substantia nigra in Parkinson's disease. Proteomics. 2004, 4: 3943-3952. 10.1002/pmic.200400848.
Da Cruz S, Martinou JC: Purification and proteomic analysis of the mouse liver mitochondrial inner membrane. Methods Mol Biol. 2008, 432: 101-116. 10.1007/978-1-59745-028-7_7.
Kim N, Lee Y, Kim H, Joo H, Youm JB, Park WS, Warda M, Cuong DV, Han J: Potential biomarkers for ischemic heart damage identified in mitochondrial proteins by comparative proteomics. Proteomics. 2006, 6: 1237-1249. 10.1002/pmic.200500291.
Friedman DB: Quantitative proteomics for two-dimensional gels using difference gel electrophoresis. Methods Mol Biol. 2007, 367: 219-239.
Vo TD, Palsson BO: Building the power house: recent advances in mitochondrial studies through proteomics and systems biology. Am J Physiol Cell Physiol. 2007, 292: C164-C177.
Pappin DJ: Peptide mass fingerprinting using MALDI-TOF mass spectrometry. Methods Mol Biol. 2003, 211: 211-219.
Gorg A, Obermaier C, Boguth G, Csordas A, Diaz JJ, Madjar JJ: Very alkaline immobilized pH gradients for two-dimensional electrophoresis of ribosomal and nuclear proteins. Electrophoresis. 1997, 18: 328-337. 10.1002/elps.1150180306.
Flatmark T: On the heterogeneity of beef heart cytochrome c. II. Some physico-chemical properties of the main subfractions (Cy I-Cy 3). Acta Chem Scand. 1966, 20: 1476-1486.
Unlu M, Morgan ME, Minden JS: Difference gel electrlophoresis: a single gel method for detecting changes in protein extracts. Electrophoresis. 1997, 18: 2071-2077. 10.1002/elps.1150181133.
Tonge R, Shaw J, Middleton B, Rowlinson R, Rayner S, Young J, Pognan F, Hawkins E, Currie I, Davison M: Validation and development of fluorescence two-dimensional differential gel electrophoresis proteomics technology. Proteomics. 2001, 1: 377-396. 10.1002/1615-9861(200103)1:3<377::AID-PROT377>3.0.CO;2-6.
Schagger H, von Jagow G: Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal Biochem. 1991, 199: 223-231. 10.1016/0003-2697(91)90094-A.
Devreese B, Vanrobaeys F, Smet J, Van Beeumen J, Van Coster R: Mass spectrometric identification of mitochondrial oxidative phosphorylation subunits separated by two-dimensional blue-native polyacrylamide gel electrophoresis. Electrophoresis. 2002, 23: 2525-2533. 10.1002/1522-2683(200208)23:15<2525::AID-ELPS2525>3.0.CO;2-I.
McDonald WH, Yates JR: Shotgun proteomics: integrating technologies to answer biological questions. Curr Opin Mol Ther. 2003, 5: 302-309.
Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R: Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999, 17: 994-999. 10.1038/13690.
Wu CC, MacCoss MJ: Shotgun proteomics: tools for the analysis of complex biological systems. Curr Opin Mol Ther. 2002, 4: 242-250.
Pocsfalvi G, Cuccurullo M, Schlosser G, Cacace G, Siciliano RA, Mazzeo MF, Scacco S, Cocco T, Gnoni A, Malorni A, Papa S: Shotgun proteomics for the characterization of subunit composition of mitochondrial complex I. Biochim Biophys Acta. 2006, 1757: 1438-1450. 10.1016/j.bbabio.2006.05.037.
Glen A, Gan CS, Hamdy FC, Eaton CL, Cross SS, Catto JW, Wright PC, Rehman I: iTRAQ-facilitated proteomic analysis of human prostate cancer cells identifies proteins associated with progression. J Proteome Res. 2008, 7: 897-907. 10.1021/pr070378x.
Kiehntopf M, Siegmund R, Deufel T: Use of SELDI-TOF mass spectrometry for identification of new biomarkers: potential and limitations. Clin Chem Lab Med. 2007, 45: 1435-1449.
Everley PA, Krijgsveld J, Zetter BR, Gygi SP: Quantitative cancer proteomics: stable isotope labeling with amino acids in cell culture (SILAC) as a tool for prostate cancer research. Mol Cell Proteomics. 2004, 3: 729-735. 10.1074/mcp.M400021-MCP200.
Rabilloud T, Strub JM, Carte N, Luche S, Van Dorsselaer A, Lunardi J, Giege R, Florentz C: Comparative proteomics as a new tool for exploring human mitochondrial tRNA disorders. Biochemistry. 2002, 41: 144-150. 10.1021/bi0114776.
Chou JL, Shenoy DV, Thomas N, Choudhary PK, Laferla FM, Goodman SR, Breen GA: Early dysregulation of the mitochondrial proteome in a mouse model of Alzheimer's disease. J Proteomics. 2011, 74: 466-479. 10.1016/j.jprot.2010.12.012.
Lovell MA, Xiong S, Markesbery WR, Lynn BC: Quantitative proteomic analysis of mitochondria from primary neuron cultures treated with amyloid beta peptide. Neurochem Res. 2005, 30: 113-122. 10.1007/s11064-004-9692-5.
Gillardon F, Rist W, Kussmaul L, Vogel J, Berg M, Danzer K, Kraut N, Hengerer B: Proteomic and functional alterations in brain mitochondria from Tg2576 mice occur before amyloid plaque deposition. Proteomics. 2007, 7: 605-616. 10.1002/pmic.200600728.
Fu H, Li W, Liu Y, Lao Y, Liu W, Chen C, Yu H, Lee NT, Chang DC, Li P: Mitochondrial proteomic analysis and characterization of the intracellular mechanisms of bis(7)-tacrine in protecting against glutamate-induced excitotoxicity in primary cultured neurons. J Proteome Res. 2007, 6: 2435-2446. 10.1021/pr060615g.
Pienaar IS, Schallert T, Hattingh S, Daniels WM: Behavioral and quantitative mitochondrial proteome analyses of the effects of simvastatin: implications for models of neural degeneration. J Neural Transm. 2009, 116: 791-806. 10.1007/s00702-009-0247-4.
Broadwater L, Pandit A, Clements R, Azzam S, Vadnal J, Sulak M, Yong VW, Freeman EJ, Gregory RB, McDonough J: Analysis of the mitochondrial proteome in multiple sclerosis cortex. Biochim Biophys Acta. 2011, 1812: 630-641.
Major T, von Janowsky B, Ruppert T, Mogk A, Voos W: Proteomic analysis of mitochondrial protein turnover: identification of novel substrate proteins of the matrix protease pim1. Mol Cell Biol. 2006, 26: 762-776. 10.1128/MCB.26.3.762-776.2006.
Essop MF, Chan WA, Hattingh S: Proteomic analysis of mitochondrial proteins in a mouse model of type 2 diabetes. Cardiovasc J Afr. 2011, 22: 175-178.
Hunzinger C, Wozny W, Schwall GP, Poznanovic S, Stegmann W, Zengerling H, Schoepf R, Groebe K, Cahill MA, Osiewacz HD: Comparative profiling of the mammalian mitochondrial proteome: multiple aconitase-2 isoforms including N-formylkynurenine modifications as part of a protein biomarker signature for reactive oxidative species. J Proteome Res. 2006, 5: 625-633. 10.1021/pr050377+.
Wong R, Aponte AM, Steenbergen C, Murphy E: Cardioprotection leads to novel changes in the mitochondrial proteome. Am J Physiol Heart Circ Physiol. 2010, 298: H75-H91. 10.1152/ajpheart.00515.2009.
Chevallet M, Lescuyer P, Diemer H, van Dorsselaer A, Leize-Wagner E, Rabilloud T: Alterations of the mitochondrial proteome caused by the absence of mitochondrial DNA: A proteomic view. Electrophoresis. 2006, 27: 1574-1583. 10.1002/elps.200500704.
Kulawiec M, Arnouk H, Desouki MM, Kazim L, Still I, Singh KK: Proteomic analysis of mitochondria-to-nucleus retrograde response in human cancer. Cancer Biol Ther. 2006, 5: 967-975. 10.4161/cbt.5.8.2880.
Strong R, Nakanishi T, Ross D, Fenselau C: Alterations in the mitochondrial proteome of adriamycin resistant MCF-7 breast cancer cells. J Proteome Res. 2006, 5: 2389-2395. 10.1021/pr060207c.
Craven RA, Hanrahan S, Totty N, Harnden P, Stanley AJ, Maher ER, Harris AL, Trimble WS, Selby PJ, Banks RE: Proteomic identification of a role for the von Hippel Lindau tumour suppressor in changes in the expression of mitochondrial proteins and septin 2 in renal cell carcinoma. Proteomics. 2006, 6: 3880-3893. 10.1002/pmic.200500811.
Liu Z, Bengtsson S, Krogh M, Marquez M, Nilsson S, James P, Aliaya A, Holmberg AR: Somatostatin effects on the proteome of the LNCaP cell-line. Int J Oncol. 2007, 30: 1173-1179.
Yu Y, Wang LS, Shen SM, Xia L, Zhang L, Zhu YS, Chen GQ: Subcellular proteome analysis of camptothecin analogue NSC606985-treated acute myeloid leukemic cells. J Proteome Res. 2007, 6: 3808-3818. 10.1021/pr0700100.
Jiang YJ, Sun Q, Fang XS, Wang X: Comparative mitochondrial proteomic analysis of Rji cells exposed to adriamycin. Mol Med. 2009, 15: 173-182. 10.1007/s00894-008-0414-9.
Ruepp SU, Tonge RP, Shaw J, Wallis N, Pognan F: Genomics and proteomics analysis of acetaminophen toxicity in mouse liver. Toxicol Sci. 2002, 65: 135-150. 10.1093/toxsci/65.1.135.
Douette P, Navet R, Gerkens P, de Pauw E, Leprince P, Sluse-Goffart C, Sluse FE: Steatosis-induced proteomic changes in liver mitochondria evidenced by two-dimensional differential in-gel electrophoresis. J Proteome Res. 2005, 4: 2024-2031. 10.1021/pr050187z.
Lee SY, Park JY, Choi YJ, Cho SK, Ahn JD, Kwon DN, Hwang KC, Kang SJ, Paik SS, Seo HG: Comparative proteomic analysis associated with term placental insufficiency in cloned pig. Proteomics. 2007, 7: 1303-1315. 10.1002/pmic.200601045.
De Palma S, Ripamonti M, Vigano A, Moriggi M, Capitanio D, Samaja M, Milano G, Cerretelli P, Wait R, Gelfi C: Metabolic modulation induced by chronic hypoxia in rats using a comparative proteomic analysis of skeletal muscle tissue. J Proteome Res. 2007, 6: 1974-1984. 10.1021/pr060614o.
Eccleston HB, Andringa KK, Betancourt AM, King AL, Mantena SK, Swain TM, Tinsley HN, Nolte RN, Nagy TR, Abrams GA, Bailey SM: Chronic exposure to a high-fat diet induces hepatic steatosis, impairs nitric oxide bioavailability, and modifies the mitochondrial proteome in mice. Antioxid Redox Signal. 2011, 15: 447-459. 10.1089/ars.2010.3395.
Alves RM, Vitorino R, Figueiredo P, Duarte JA, Ferreira R, Amado F: Lifelong physical activity modulation of the skeletal muscle mitochondrial proteome in mice. J Gerontol A Biol Sci Med Sci. 2010, 65: 832-842.
Rezaul K, Wu L, Mayya V, Hwang SI, Han D: A systematic characterization of mitochondrial proteome from human T leukemia cells. Mol Cell Proteomics. 2005, 4: 169-181.
Zischka H, Weber G, Weber PJ, Posch A, Braun RJ, Buhringer D, Schneider U, Nissum M, Meitinger T, Ueffing M, Eckerskorn C: Improved proteome analysis of Saccharomyces cerevisiae mitochondria by free-flow electrophoresis. Proteomics. 2003, 3: 906-916. 10.1002/pmic.200300376.
Fu YR, Yi ZJ, Yan YR, Qiu ZY: Proteomic analysis of mitochondrial proteins in hydroxycamptothecin-treated SMMC-7721 cells. Zhonghua Gan Zang Bing Za Zhi. 2007, 15: 572-576.
White MY, Brown DA, Sheng S, Cole RN, O'Rourke B, Van Eyk JE: Parallel proteomics to improve coverage and confidence in the partially annotated Oryctolagus cuniculus mitochondrial proteome. Mol Cell Proteomics. 2011, 10: M110 004291-
Nesvizhskii AI, Aebersold R: Interpretation of shotgun proteomic data: the protein inference problem. Mol Cell Proteomics. 2005, 4: 1419-1440. 10.1074/mcp.R500012-MCP200.
Yu W, Taylor JA, Davis MT, Bonilla LE, Lee KA, Auger PL, Farnsworth CC, Welcher AA, Patterson SD: Maximizing the sensitivity and reliability of peptide identification in large-scale proteomic experiments by harnessing multiple search engines. Proteomics. 2010, 10: 1172-1189. 10.1002/pmic.200900074.
Rabilloud T, Kieffer S, Procaccio V, Louwagie M, Courchesne PL, Patterson SD, Martinez P, Garin J, Lunardi J: Two-dimensional electrophoresis of human placental mitochondria and protein identification by mass spectrometry: toward a human mitochondrial proteome. Electrophoresis. 1998, 19: 1006-1014. 10.1002/elps.1150190616.
Brandon MC, Lott MT, Nguyen KC, Spolim S, Navathe SB, Baldi P, Wallace DC: MITOMAP: a human mitochondrial genome database-2004 update. Nucleic Acids Res. 2005, 33: D611-D613. 10.1093/nar/gki399.
Ingman M, Gyllensten U: mtDB: Human Mitochondrial Genome Database, a resource for population genetics and medical sciences. Nucleic Acids Res. 2006, 34: D749-D751. 10.1093/nar/gkj010.
Prokisch H, Andreoli C, Ahting U, Heiss K, Ruepp A, Scharfe C, Meitinger T: MitoP2: the mitochondrial proteome database-now including mouse data. Nucleic Acids Res. 2006, 34: D705-D711. 10.1093/nar/gkj127.
Richard C: Stress-related cardiomyopathies. Ann Intensive Care. 2011, 1: 39-10.1186/2110-5820-1-39.
Kim HN, Januzzi JL: Natriuretic peptide testing in heart failure. Circulation. 2011, 123: 2015-2019. 10.1161/CIRCULATIONAHA.110.979500.
Thiede B, Rudel T: Proteome analysis of apoptotic cells. Mass Spectrom Rev. 2004, 23: 333-349. 10.1002/mas.10079.
Smith DJ: Mitochondrial dysfunction in mouse models of Parkinson's disease revealed by transcriptomics and proteomics. J Bioenerg Biomembr. 2009, 41: 487-491. 10.1007/s10863-009-9254-2.
Ozawa T, Sako Y, Sato M, Kitamura T, Umezawa Y: A genetic approach to identifying mitochondrial proteins. Nat Biotechnol. 2003, 21: 287-293. 10.1038/nbt791.
Graves PR, Haystead TA: A functional proteomics approach to signal transduction. Recent Prog Horm Res. 2003, 58: 1-24. 10.1210/rp.58.1.1.
Wang YS, Cao R, Jin H, Huang YP, Zhang XY, Cong Q, He YF, Xu CJ: Altered protein expression in serum from endometrial hyperplasia and carcinoma patients. J Hematol Oncol. 2011, 4: 15-10.1186/1756-8722-4-15.
Acknowledgements
This study was supported by: Natural Science Youth Foundation of Shandong Province, China (No. ZR2011HQ009); Natural Science Foundation of Shandong Province, China (No. 2007 C053); Project of Scientific and Technological Development of Shandong Province, China (N2007GG10).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
Both authors participated in drafting and editing the manuscript. Both authors read and apporoved the final manuscript.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Jiang, Y., Wang, X. Comparative mitochondrial proteomics: perspective in human diseases. J Hematol Oncol 5, 11 (2012). https://doi.org/10.1186/1756-8722-5-11
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-8722-5-11