
Abstract
Background and the aim of the study
The objective of the present study was to formulate and optimize nanoparticles (NPs) of sildenafil-loaded poly (lactic-co-glycolic acid) (PLGA) by double emulsion solvent evaporation (DESE) method. The relationship between design factors and experimental data was evaluated using response surface methodology.
Method
A Box-Behnken design was made considering the mass ratio of drug to polymer (D/P), the volumetric proportion of the water to oil phase (W/O) and the concentration of polyvinyl alcohol (PVA) as the independent agents. PLGA-NPs were successfully prepared and the size (nm), entrapment efficiency (EE), drug loading (DL) and cumulative release of drug from NPs post 1 and 8 hrs were assessed as the responses.
Results
The NPs were prepared in a spherical shape and the sizes range of 240 to 316 nm. The polydispersity index of size was lower than 0.5 and the EE (%) and DL (%) varied between 14-62% and 2-6%, respectively. The optimized formulation with a desirability factor of 0.9 was selected and characterized. This formulation demonstrated the particle size of 270 nm, EE of 55%, DL of 3.9% and cumulative drug release of 79% after 12 hrs. In vitro release studies showed a burst release at the initial stage followed by a sustained release of sildenafil from NPs up to 12 hrs. The release kinetic of the optimized formulation was fitted to Higuchi model.
Conclusions
Sildenafil citrate NPs with small particle size, lipophilic feature, high entrapment efficiency and good loading capacity is produced by this method. Characterization of optimum formulation, provided by an evaluation of experimental data, showed no significant difference between calculated and measured data.
Similar content being viewed by others


Explore related subjects
Find the latest articles, discoveries, and news in related topics.Avoid common mistakes on your manuscript.
Background
Sildenafil is a selective inhibitor of phosphodiesterase enzyme type 5 (PDE-5) that effectively inactivates cyclic guanosine monophosphate (cGMP) and enhances the effect of nitric oxide [1]. This drug was primarily prescribed for angina pectoris and now is widely used for the treatment of erectile dysfunction [2]. Recently, PDE-5 inhibitors have been proposed to protect the endothelial function in human by selectively improving local blood flow [3]. Moreover, other complications like wound healing, diabetic gastropathy [4], Reynaud’s phenomenon, respiratory disorders with ventilation/perfusion mismatch, congestive cardiac failure, hypertension and stroke have been widely studied with the hope that PDE-5 inhibitors can serve as novel promising treatment in such conditions. In addition, the selective and potent vasodilatory and antiproliferative effects of sildenafil on pulmonary vascular smooth muscle cells emphasize the importance of this drug in control of pulmonary artery pressure [4–6]. When sildenafil citrate (SC) is given orally, its bioavailability is relatively low (approximately 40%) in healthy subjects [7] because of the first pass metabolism. In addition, it exhibits a very short physiological half-life (about 3–4 hrs). Therefore repeated doses are required to sustain drug plasma level [8] that causes various side effects such as headache, flushing, dyspepsia and epistaxis [9].
Specially designed dosage forms that sustain levels of drug in the therapeutic window or local delivery of this drug into the site of action can be thus helpful [8, 10]. Some published patents have reported that application of nano sized SC powder in formulation, shows faster onset of action, higher bioavailability and absorption than conventional dosage form [11–13]. Thus, nanoparticles (NPs) might improve the efficacy; reduce side effects and dosage of therapeutic agents [14]. Generally, nanocarrier systems provide advantages over conventional drug delivery systems such as protection of the entrapped drug from enzymatic destruction, sustained drug release, reduction of daily drug doses and side effects and cell targeting [15]. Consequently, biodegradable polymeric NPs have engrossed remarkable consideration as potential drug delivery devices in view of their applications in the controlled release of drugs [16]. poly(lactic-co-glycolic acid) (PLGA) is the most successfully used available biodegradable polymer due to its long clinical experience, desirable degradation characteristics and possibilities for sustained drug delivery [17]. This polymer is widely used in the production of NPs [18]. PLGA consists two endogenous monomers that are easily metabolized via the Krebs cycle and therefore, negligible systemic toxicity is associated with the use of PLGA for drug delivery [19]. Also, study of in vitro and in vivo cytotoxicity of PLGA nanoparticles highlighted the safety of biodegradable PLGA nanoparticles [20, 21]. Selection of a particular method for preparation of NPs is usually determined by the solubility properties of the drug [22]. Double Emulsion Solvent Evaporation (DESE) technique is an effective method for encapsulation of hydrophilic compounds [23] and thus we selected this method to fabricate NPs, because of the polar properties of SC [24]. There are several variables in the DESE process that can affect the properties of the product. Response surface methodology (RSM) is a statistical method employed for the modeling and analysis of problems in which a response of concern is influenced by several variables and the goal is to optimize this response [25]. Application of such optimizing technique may be an efficient and economical method to gain the essential information and thus to understand the relationship between controllable independent variables and dependent variables or responses in terms of performance and quality [26].
Methods
Materials
PLGA, Resomer® RG503H, was acquired from Boehringer Ingelheim (Germany). Polyvinyl alcohol (PVA) (87–90% hydrolysis degree and molecular mass 30,000-70,000 g/mol) was purchased from Sigma Chemical Co. (USA). SC was purchased from Selleck Chemicals (USA). The organic solvents were supplied by Duksan (Korea).
Preparation of SC-loaded Nanoparticles
At the first, the drug was dissolved in warm distilled water (3 mg/ml) and emulsified in methylene chloride containing different amounts of PLGA. The emulsification was carried out using a probe sonicator set (Hielscher, Germany) at 80% of the energy output for 3 min. Then, the primary emulsion was added to 20 ml of double distilled water containing PVA and homogenized for 3 min in 20,000 rpm (IKA, Germany). Methylene chloride was eliminated by evaporation under reduced pressure using a rotary evaporator (Buchi, Switzerland). NPs were recovered by ultracentrifugation (Beckman Instruments, USA) at 100,000 g for 60 min at 25°C.
Experimental design
The effects of formulation variables on the NPs characteristics and optimization procedure were examined by employing a Box-Behnken design. The design and statistical analysis were performed by Design-Expert® V8 (DX8) Software for design of experiments (DOE). Experimental factors and factor levels were determined in preliminary studies. Studied responses which evaluated in this investigation, were the mass ratio of drug to PLGA (X1), the volumetric ratio of water to solvent in primary emulsion (X2) and the concentration of PVA (X3) that classified to low, medium, and high values for the chosen variables as are described in Table 1. The evaluated studied responses were size (nm), entrapment efficiency (EE), drug loading (DL) and drug release in 1 hr and 8 hrs. The Box-Behnken design and observational data are shown in Table 2.
The quadratic non-linear model generated by design is in this form:
In which Y is the measured response associated with each factor level combination; A0 is an intercept; A1-A9 are the regression coefficients; X1, X2 and X3 are the studied factors; X12, X22, X32 are quadratic effects, X1X2 + X2X3 + X1X3 are interaction between variables and E is the error term [27].
Physicochemical characteristics of NPs
Particle size
Mean hydrodynamic size (called z-average) and polydispersity index of the NPs were measured by photon correlation spectroscopy (Malvern, UK) at 25C. All the samples were diluted with double distilled water to create a suitable obscuration before analysis.
Determination of entrapped SC
The supernatant part of the centrifuged NP sample was carefully removed and examined to determine the amount of non-encapsulated drug. The precipitant was lyophilized, weighted, and then dissolved in a mixture of 3:2 of chloroform (a common solvent for PLGA) and water (a solvent for SC) by sonicating for one hour. Then, the undissolved fraction was removed by centrifugation. After that, a sample was taken from the aqueous phase to determine the amount of encapsulated SC. The drug incorporation efficiency was defined by the following formulas:
A reverse phase chromatography method was used for evaluation of SC using isocratic HPLC system (Waters, USA) and NucleoDur (5 μm, 25 cm) C18 column. The mobile phase consisted of acetonitrile and water (35:65, pH 4.0) at a flow rate of 1 ml/min with UV detection at 291 nm. The retention time was 5.6 min.
Differential scanning calorimetry (DSC)
A differential scanning calorimeter (Mettler Toledo, Switzerland) was used to evaluate the thermal behavior of all materials used in the NP formulations. The equipment was calibrated using indium. The samples (8 mg) were heated ranging 5–280°C at a scanning rate of 10°C/min in aluminum pans under nitrogen gas.
Scanning electron microscopy (SEM)
The surface morphology of NPs was assessed by a scanning electron microscope (Mira Tescan, Czech Republic). The Nanoparticles were spread on a stub and dried at 25°C and then spattered with gold using a sputter coater (BAL-TEC, Switzerland).
In vitro drug release studies
To predict the optimal formulation of NPs, the drug release of each formulation was studied in phosphate buffer (PBS) at pH 7.4 as dissolution medium. Briefly, 10 mg of each lyophilized NP formulation was dispersed in a screw-capped glass vial (50 ml) containing 40 ml of medium by shaking at 200 rpm and 37 ± 0.5°C in shaker incubator (LABOTEC, Germany). At predetermined time intervals (0, 0.5, 1, 2, 4, 8, 10, 12, 24 hrs) 1 ml of the dispersion was taken away and replaced with 1 ml of fresh PBS. The sample was centrifuged (Eppendorf, Germany) at 14,000 g for 30 min, and the supernatant was analyzed. All of the experiments were done in triplicate.
The release kinetics from optimal NPs was fitted on zero order, first order, Higuchi model, Korsmeyer–Peppas model and Hixson–Crowell model [28].
Results
NPs were successfully prepared by DESE method. The effects of formulation variables on the NP properties were evaluated and finally optimal NPs were proposed by design expert software. The characteristics of this formulation were compared to predicted values. In addition, release profile and release kinetics from optimal NPs were studied.
Particle size
SC loaded NPs sizes varied between 240 to 316 nm. Formulations displayed polydispersity index (PDI) of <0.5 which showed the narrow NP size distribution.
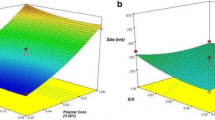
Analysis of data from ANOVA test exhibited that D/P and W/O ratios had significant effects on particle size (p < 0.05). Briefly, decrease in the D/P and increasing the W/O ratios resulted in the production of larger particles (Figure 1).

Surface plot showing the effect of different variables on particle size of SC nanoparticles.
In size response, the reduced model showed better adjusted correlation coefficients than the primary model (0.72 > 0.62) with an F value of 8 (p < 0.05), which clearly indicated that the particle size and some variables were related. The reduced model for predicting size is presented in equation 1.
The minimum particle size of 240 nm was achieved by operating the experiment at the midpoint of each independent variable. Analysis of independent factors showed that D/P and W/O ratios had effective impacts on particle size. There was no significant interaction between the studied factors.
Determination of entrapped SC
The EE of NPs in different formulations is represented in Table 2. Data analysis of this response proved the significant effect of all independent variables (p < 0.05) and an interaction between W/O ratio and the amount of PVA (p < 0.05). The quadratic model of Entrapment Efficiency followed equation 2.
Surface plots indicated that higher EE occurred in formulation with a W/O ratio about 0.38 and D/P ratios between 0.05-0.09. Also, PVA concentration was less effective than two other variables (Figure 2). The predictive model for DL is given in equation 3:

Three dimensional surface plots showing the effect of different variables on the %Entrapment Efficiency (EE) of SC nanoparticles.
DL in NPs ranged between 2% and 6.3% (Figure 3); where, analysis of data by ANOVA showed that the D/P Ratio with an F value of 71.36 (p < 0.05) had the most important impact on DL and W/O ratios had a significant effect on this response (p < 0.05). Comparison of different formulations and surface plots revealed that formulations with the highest DL had the maximum D/P ratio of 0.2 and a W/O ratio of approximately 0.38.

Three dimensional surface plots showing the effect of different variables on the %drug loading of SC nanoparticles.
In vitro drug release studies
To develop a formulation with acceptable release profile, drug release of each formulation was studied. Data of release over the first hour of experiments were considered as a marker of burst effect and the amount of drug releasing in 8 hrs showed retardation efficiency over the time. Analysis of the data demonstrated that W/O and D/P ratios had significant influence on release profiles.
For predicting the release in 1 and 8 hrs, the linear model fitted as an effective one with data (p < 0.05) (equations 4–5).
Data analysis showed that the D/P ratios with F values of 17.08 and 9.10 (p < 0.05) were the most important factors on the release of SC from NPs during 1 and 8 hrs, respectively (Figure 4). When the D/P ratio was minimized and W/O ratio was about 0.38, the formulation had the least burst release. On the other hand, this formulation could provide sustained release profile over the time.

Three dimensional surface plots showing the effect of different variables on the release of drug from SC nanoparticles during 1 hr and 8 hrs.
Optimization
After confirming the polynomial equations relating the response and independent factors, in consequence of acceptable size of all formulations, the optimization model was constructed by combining the DL, EE and drug release in 1 hr responses. Optimization was performed by using a desirability function to obtain the levels of X1, X2 and X3, which maximized EE, while minimizing drug release in 1 hr and targeting DL at 4%. Coefficients with p-value < 0.05 had a significant effect on the prediction efficacy of the model for the measured responses. Simultaneously, the formulation with W/O about 0.40, D/P of 0.06 and PVA about 0.50 conformed higher desirability. This formulation prepared and evaluated. Predicted and actual amounts of responses are compared and shown in Table 3. As seen in Table 3, excluding release in 1 h, the amount of responses for optimized formulation have lower than 10% difference with the predicted amount of Box-behnken design.
Differential scanning calorimetry (DSC)
Thermal analysis is a supportive tool for determining the dispersion of the drug in polymeric materials. DSC thermograms of the pure drug, PVA, PLGA and SC-NPs are represented in Figure 5. The pure drug showed high endothermic peak indication of its melting peak at ∼ 200°C which was absent in NPs.

DSC thermogram of PLGA, PVA, Sildenafil citrate and Sildenafil-nanoparticles.
Scanning electron microscopy (SEM)
SEM micrographs showed that uniform PLGA NPs were successfully prepared by using the DESE method. As shown in Figure 6, the PLGA nanoparticles were in spherical shapes and a smooth surface.

SEM micrograph showing the morphology of optimized PLGA-Sildenafil nanoparticles.
Release kinetics of optimal nanoparticles
Profile of release from optimal NPs is presented in Figure 7. In vitro drug release profiles of SLD from optimal PLGA NPs showed that the cumulative percentage of drug release was about 79% of drug content of the formulation in 12 hrs. The results support a burst release in the first one hour that followed by a sustained release over 12 hrs.

Profile of drug release from optimized SC nanoparticles.
Release kinetics from the optimum formulation of NPs was compared to different kinetic models which showed that the best model fitted with data is the Higuchian equation (R2: 0.95). This model explains the release of drug from an insoluble matrix time-dependently based on Fickian diffusion [29]. The release constant was computed from the slope of the suitable plots, and the regression coefficient was determined (Table 4). The plots and regression coefficient proved that after Higuchian model, the best linearity followed by first-order kinetic (R2: 0.91). The first order kinetics model can be described the drug dissolution of water-soluble drugs in porous matrices [30].
Discussion
Although the most applied method for encapsulation of hydrophilic drugs is DESE method, the low EE is usually a major problem. Experimental design methodology is an economic approach for extracting the maximum useful information from data. Applying this technique reduces the costs of experiments by saving time, materials and energy. Of course, optimization of NPs formulation is a complex procedure, which involves considering various parameters and their interactions. Due to increasing use of sildenafil in the treatment of pulmonary diseases and new indications proposed for this drug, preparation of optimum loaded NPs that release drug over the time can be potentially beneficial in the treatment of different pathological conditions.
Particle size and size distribution are important physicochemical properties that determine both uptake and biological fate of the particulate systems [31]. In the results, important effects of D/P and W/O ratios on size of NPs were confirmed. Production of NPs with higher polymer concentrations resulted in the formation of larger particles. In this manner, changing the diffusion rate of organic solvent through the interface could be proposed as a fundamental mechanism. In fact, increasing the amount of polymer or decreasing the volume of organic phase can potentially hinder the diffusion of solvent molecules through the polymeric chains [32]. Hence, formation of larger particles can be on account of two main factors: (a) the number of polymer chains per volume unit of solvent and (b) the viscosity of the solution [33]. As a result of applying a greater number of the polymeric chains per volume unit of solvent, diffusion of solvent into the aqueous phase becomes hard and causes the formation of aggregated and larger NPs, which is in agreement with previous reports [22, 34]. On the other hand, it is more difficult for the viscous polymer solution to be broken up into smaller droplets during the formation of a second emulsion [35]. In this model, particle size reached to the minimum point when D/P and W/O ratios were about 0.13 and 0.36. Data analysis defined no significant effect of various PVA concentrations and this factor showed the minor influence on the particle size in this study. Of course, the presence of PVA as a surfactant is necessary to form stabilized NPs.
As mentioned in DESE method, the low EE of small and hydrophilic drug molecules into the polymer is an important challenge [36]. The water soluble nature of SC may be the cause of lower EE in higher D/P or W/O ratios. The higher EE values gained by the higher polymer contents can be explicated by the better coverage of drug molecules within the polymeric matrix [37]. In addition the initial amount of dissolved drug in the inner phase showed a great influence on EE. As the difference of drug concentrations between internal and external aqueous phase increases, the drug diffuses faster to the bulk aqueous phase during particle formation. In other words, higher DL values resulted in lower encapsulation efficiencies because of rapid partitioning of the drug between phases. So it can be claimed that DL and EE are strongly related responses [22, 38]. In the formulations that the only changed parameter was W/O ratio, it seems that this factor had a dual effect. Although decreasing this ratio resulted in more efficiently covered aqueous droplets during first emulsion preparation, this larger amount of organic phase required much more time to evaporate [38]. So the drug molecules had greater opportunity to escape from the inner to outer phase in formulations with higher W/O ratios (>midpoint). While the aqueous volume was kept constant, the enhanced viscosity of the polymer solution led to the formation of larger polymer/solvent droplets. Consequently, slower solidification of larger particles allowed more drug diffusion to the external phase, which again resulted in the lower entrapment of the drug into the NPs [39]. Moreover, employing higher concentrations of stabilizer in the external aqueous phase induced higher EE values.
The endothermic peak of SC disappeared in the thermogram of optimized drug loaded NPs, which indicated absence of crystalline drug in the NPs. So, it can be assumed that encapsulated drug was in an amorphous state or a molecular dispersion throughout the polymer matrix after fabrication of NPs [40].
SEM micrographs showed smooth surface and spherical shape of SC-NPs which can be explained by stability of primary emulsion that the polymer had adequate time to form the condense matrix around the drug molecules before particle formation [41].
All formulations were subjects of in vitro release studies. The release profiles exhibited an initial burst release during the first hour, followed by a sustained release pattern over 12 hrs. Surface response plots showed the influence of D/P and W/O ratios on drug release from NPs. The initial burst release was related to the degree of DL, where the minor burst effects were observed in lower DL values. It appears that at higher loading levels, more drug molecules might adsorb onto the surface of NPs, which contribute to the greater initial release. Actually during the first hour of release study, the large concentration gradient of the drug serves as the driving force for the diffusion. But during the next hours not only this gradient decreases but also it takes more time for SC molecules to diffuse via a path constructed from a series of interconnected pores and channels within the polymeric matrix. It is supposed that in formulations with lower polymer concentrations, the internal water droplets have a greater tendency to coalescence and thus more likely to make larger pores and less tortuous network. However, when the higher polymer concentrations are applied, a tighter structure is formed as a result of faster droplet coagulation during second emulsion formation and subsequent polymeric chain entanglement. In another research published recently on the formation of sildenafil-NPs, the sildenafil was incorporated as its water insoluble base into the PLGA-NPs by means of solvent evaporation method, whereas the most of the entrapped drug released during the first 90 min [42]. Fortunately, in the present study, the release profile of sildenafil citrate as a hydrophilic salt with better bioavailability [1], improved up to 12 hrs.
Conclusion
Comparing the actual and predicted responses indicated that surface response methodology is suitable to make optimization of SC NPs to produce a biphasic release pattern. These NPs can be utilized in the form of tablets or processed in the presence of inhalable sugars to form a dry powder for inhalation purposes. Further in vivo studies of SC NPs are recommended to determine whether oral, topical, transdermal and respiratory efficacies are created.
Although, providing SC in the form PLGA nanoparticles brings some advantages such as improvement in reaching many organs, tissues, and cells, the increased entrance into cells might fundamentally rises the chance of toxic effects. It is emphasized that changed size and surface area of the present nano form of sildenafil makes it prone to interact with various cellular components in various tissues. Therefore, future studies to collect the relationships between structure-size-efficacy-toxicity of the present nano form of sildenafil with special regard to portal of entry and target organ is crucial [43]. Additionally it would be nice to prove the safety of the new nano form of sildenafil in an appropriate biological system specially if the expectation is to use the new form of sildenafil for longer duration of time rather than its present single-dose indication in erectile dysfunction [44].
References
Jung SY, Seo YG, Kim GK, Woo JS, Yong CS, Choi HG: Comparison of the solubility and pharmacokinetics of sildenafil salts. Arch Pharm Res. 2011, 34: 451-454. 10.1007/s12272-011-0313-y.
Webb DJ, Freestone S, Allen MJ, Muirhead GJ: Sildenafil citrate and blood-pressure–lowering drugs: results of drug interaction studies with an organic nitrate and a calcium antagonist. The Am J cardiol. 1999, 83: 21-28.
Rosano GM, Aversa A, Vitale C, Fabbri A, Fini M, Spera G: Chronic treatment with tadalafil improves endothelial function in men with increased cardiovascular risk. Eur Urol. 2005, 47: 214-220. 10.1016/j.eururo.2004.10.002. discussion 220–212
Sharma R: Novel phosphodiesterase-5 inhibitors: current indications and future directions. Indian J Med Sci. 2007, 61: 667-679. 10.4103/0019-5359.37789.
Yildiz P: Molecular mechanisms of pulmonary hypertension. Clin Chim Acta. 2009, 403: 9-16. 10.1016/j.cca.2009.01.018.
Farsaie S, Khalili H, Karimzadeh , Dashti-Khavidaki S: An old drug for a new application: potential benefits of Sildenafil in wound healing. J Pharm Pharm Sci. 2012, 15: 483-498.
Fraisse A, Butrous G, Taylor MB, Oakes M, Dilleen M, Wessel DL: Intravenous sildenafil for postoperative pulmonary hypertension in children with congenital heart disease. Intensive Care Med. 2011, 37: 502-509. 10.1007/s00134-010-2065-4.
Elnaggar YS, El-Massik MA, Abdallah OY: Fabrication, appraisal, and transdermal permeation of sildenafil citrate-loaded nanostructured lipid carriers versus solid lipid nanoparticles. Int J Nanomedicine. 2011, 6: 3195-3205.
McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR, Mathier MA, McGoon MD, Park MH, Rosenson RS: ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation. 2009, 119: 2250-2294.
Shah V, Sharma M, Parmar V, Upadhyay U: Formulation of sildenafil citrate loaded nasal microsphers: an in vitro, ex vivo characterization. Int J Drug Del. 2010, 2: 213-220. 10.5138/ijdd.2010.0975.0215.02031.
Ryde TA, Hovey DC, Bosch HW: Novel compositions of sildenafil free base. In Book Novel compositions of sildenafil free base (Editor ed.^eds.). 2004, City: Google Patents
Filipcsei G, Otvos Z, Pongrácz K, Darvas F: Nanostructured Sildenafil base, its pharmaceutically acceptable salts and co-crystals, compositions of them, process for the preparation thereof and pharmaceutical compositions containing them. Book Nanostructured Sildenafil base, its pharmaceutically acceptable salts and co-crystals, compositions of them, process for the preparation thereof and pharmaceutical compositions containing them. 2010, City: Google Patents
Ryde TA, Hovey DC, Bosch WH: Novel compositions of Sildenafil free base. Book Novel compositions of Sildenafil free base. 2008, City: EP Patent, 1,658,053
Chidambaram M, Manavalan R, Kathiresan K: Nanotherapeutics to overcome conventional cancer chemotherapy limitations. J Pharm Pharm Sci. 2011, 14: 67-77.
Ungaro F, d’Angelo I, Miro A, La Rotonda MI, Quaglia F: Engineered PLGA nano- and micro-carriers for pulmonary delivery: challenges and promises. J Pharm Pharmacol. 2012, 64: 1217-1235. 10.1111/j.2042-7158.2012.01486.x.
Soppimath KS, Aminabhavi TM, Kulkarni AR, Rudzinski WE: Biodegradable polymeric nanoparticles as drug delivery devices. J Control Release. 2001, 70: 1-20. 10.1016/S0168-3659(00)00339-4.
Makadia HK, Siegel SJ: Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers (Basel). 2011, 3: 1377-1397. 10.3390/polym3031377.
Zou W, Liu C, Chen Z, Zhang N: Studies on bioadhesive PLGA nanoparticles: a promising gene delivery system for efficient gene therapy to lung cancer. Int J Pharm. 2009, 370: 187-195. 10.1016/j.ijpharm.2008.11.016.
Danhier F, Ansorena E, Silva JM, Coco R, Le-Breton A, Preat V: PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012, 161: 505-522. 10.1016/j.jconrel.2012.01.043.
Mura S, Hillaireau H, Nicolas J, Le-Droumaguet B, Gueutin C, Zanna S, Tsapis N, Fattal E: Influence of surface charge on the potential toxicity of PLGA nanoparticles towards Calu-3 cells. Int J Nanomedicine. 2011, 6: 2591-2605.
Semete B, Booysen L, Lemmer Y, Kalombo L, Katata L, Verschoor J, Swai HS: In vivo evaluation of the biodistribution and safety of PLGA nanoparticles as drug delivery systems. Nanomedicine. 2010, 6: 662-671.
Lamprecht A, Ubrich N, Hombreiro Perez M, Lehr C, Hoffman M, Maincent P: Influences of process parameters on nanoparticle preparation performed by a double emulsion pressure homogenization technique. Int J Pharm. 2000, 196: 177-182. 10.1016/S0378-5173(99)00422-6.
Cohen-Sela E, Chorny M, Koroukhov N, Danenberg HD, Golomb G: A new double emulsion solvent diffusion technique for encapsulating hydrophilic molecules in PLGA nanoparticles. J Control Release. 2009, 133: 90-95. 10.1016/j.jconrel.2008.09.073.
Badwan AA, Nabuls L, Al-Omari MM, Daraghmeh N, Ashour M: Sildenafil Citrate. Analytical Profiles of Drug Substances and Excipients. Volume 27. Edited by: Harry GB. 2001, Amman, Jordan: Academic Press, 339-376.
Prakobvaitayakit M, Nimmannit U: Optimization of polylactic-co-glycolic acid nanoparticles containing itraconazole using 2(3) factorial design. AAPS PharmSciTech. 2003, 4: E71-
Neumann D, Merkwirth C, Lamprecht A: Nanoparticle design characterized by in silico preparation parameter prediction using ensemble models. J Pharm Sci. 2010, 99: 1982-1996.
Nazzal S, Khan MA: Response surface methodology for the optimization of ubiquinone self-nanoemulsified drug delivery system. AAPS PharmSciTech. 2002, 3: E3-
Basu S, Mukherjee B, Chowdhury SR, Paul P, Choudhury R, Kumar A, Mondal L, Hossain CM, Maji R: Colloidal gold-loaded, biodegradable, polymer-based stavudine nanoparticle uptake by macrophages: an in vitro study. Int J Nanomedicine. 2012, 7: 6049-6061.
Higuchi T: Mechanism of sustained-action medication. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J Pharm Sci. 1963, 52: 1145-1149. 10.1002/jps.2600521210.
Dash S, Murthy PN, Nath L, Chowdhury P: Kinetic modeling on drug release from controlled drug delivery systems. Acta Pol Pharm. 2010, 67: 217-223.
Shavi GV, Kumar AR, Karthik A, Naseer M, Aravind G, Praful BD, Reddy MS, Udupa N: Novel paclitaxel nanoparticles: development, in vitro anti-tumor activity in BT-549 cells and in vivo evaluation. J Control Release. 2010, 148: e119-e121. 10.1016/j.jconrel.2010.07.091.
Zhang J, Fan Y, Smith E: Experimental design for the optimization of lipid nanoparticles. J Pharm Sci. 2009, 98: 1813-1819. 10.1002/jps.21549.
Galindo-Rodriguez S, Allemann E, Fessi H, Doelker E: Physicochemical parameters associated with nanoparticle formation in the salting-out, emulsification-diffusion, and nanoprecipitation methods. Pharm Res. 2004, 21: 1428-1439.
Beck-Broichsitter M, Kleimann P, Gessler T, Seeger W, Kissel T, Schmehl T: Nebulization performance of biodegradable sildenafil-loaded nanoparticles using the Aeroneb Pro: formulation aspects and nanoparticle stability to nebulization. Int J Pharm. 2012, 422: 398-408. 10.1016/j.ijpharm.2011.10.012.
Mahboubian A, Hashemein S, Moghadam S, Atyabi F, Dinarvand R: Preparation and in-vitro evaluation of controlled release PLGA microparticles containing triptoreline. Iranian J Pharm Res. 2010, 9: 369-378.
Tewes F, Munnier E, Antoon B, Ngaboni Okassa L, Cohen-Jonathan S, Marchais H, Douziech-Eyrolles L, Souce M, Dubois P, Chourpa I: Comparative study of doxorubicin-loaded poly(lactide-co-glycolide) nanoparticles prepared by single and double emulsion methods. Eur J Pharm Biopharm. 2007, 66: 488-492. 10.1016/j.ejpb.2007.02.016.
Guhagarkar SA, Malshe VC, Devarajan PV: Nanoparticles of polyethylene sebacate: a new biodegradable polymer. AAPS PharmSciTech. 2009, 10: 935-942. 10.1208/s12249-009-9284-4.
Yang Y, Chung T, Ng N: Morphology, drug distribution, and in vitro release profiles of biodegradable polymeric microspheres containing protein fabricated by double-emulsion solvent extraction/evaporation method. Biomaterials. 2001, 22: 231-241. 10.1016/S0142-9612(00)00178-2.
Das MK, Rao KR: Evaluation of zidovudine encapsulated ethylcellulose microspheres prepared by water-in-oil-in-oil (w/o/o) double emulsion solvent diffusion technique. Acta Pol Pharm. 2006, 63: 141-148.
Jelvehgari M, Nokhodchi A, Rezapour M, Valizadeh H: Effect of formulation and processing variables on the characteristics of tolmetin microspheres prepared by double emulsion solvent diffusion method. Indian J Pharm Sci. 2010, 72: 72-78. 10.4103/0250-474X.62251.
Meng FT, Ma GH, Liu YD, Qiu W, Su ZG: Microencapsulation of bovine hemoglobin with high bio-activity and high entrapment efficiency using a W/O/W double emulsion technique. Colloids Surf B Biointerfaces. 2004, 33: 177-183. 10.1016/j.colsurfb.2003.10.003.
Beck-Broichsitter M, Schmehl T, Gessler T, Seeger W, Kissel T: Development of a biodegradable nanoparticle platform for sildenafil: formulation optimization by factorial design analysis combined with application of charge-modified branched polyesters. J Control Release. 2012, 157: 469-477. 10.1016/j.jconrel.2011.09.058.
Mostafalou S, Mohammadi H, Ramazani A, Abdollahi M: Different biokinetics of nanomedicines linking to their toxicity; an overview. Daru. 2013, 22: 21 (1): 14-
Pourmand A, Abdollahi M: Current opinion on nanotoxicology. Daru. 2012, 15: 20 (1): 95-
Acknowledgments
Authors wish to thank Dr Atieh Sadat Tajalli Bakhsh who edited the article linguistically. This study was a PhD thesis of the first author and the study was financially supported by TUMS.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contribution
All authors have contributed significantly to the research and preparation, design and final production of the manuscript and approve its submission.
Elham Ghasemian, Alireza Vatanara, Abdolhossein Rouholamini Najafabadi, Mohammad Reza Rouini, Kambiz Gilani and Majid Darabi contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Ghasemian, E., Vatanara, A., Rouholamini Najafabadi, A. et al. Preparation, characterization and optimization of sildenafil citrate loaded PLGA nanoparticles by statistical factorial design. DARU J Pharm Sci 21, 68 (2013). https://doi.org/10.1186/2008-2231-21-68
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2008-2231-21-68