
Abstract
Background
Spirocerca lupi is a life-threating parasitic nematode of dogs that has a cosmopolitan distribution but is most prevalent in tropical and subtropical countries. Despite its veterinary importance in canids, the epidemiology, molecular ecology and population genetics of this parasite still remain unexplored.
Methods
The complete mitochondrial (mt) genome of S. lupi was amplified in four overlapping long fragments using primers designed based on partial cox 1, rrn S, cox 2 and nad 2 sequences. Phylogenetic re-construction of 13 spirurid species (including S. lupi) was carried out using Bayesian inference (BI) based on concatenated amino acid sequence datasets.
Results
The complete mt genome sequence of S. lupi is 13,780 bp in length, including 12 protein-coding genes, 22 transfer RNA genes and two ribosomal RNA genes, but lacks the atp 8 gene. The gene arrangement is identical to that of Thelazia callipaeda (Thelaziidae) and Setaria digitata (Onchocercidae), but distinct from that of Dracunculus medinensis (Dracunculidae) and Heliconema longissimum (Physalopteridae). All genes are transcribed in the same direction and have a nucleotide composition high in A and T. The content of A + T is 73.73% for S. lupi, in accordance with mt genomes of other spirurid nematodes sequenced to date. Phylogenetic analyses using concatenated amino acid sequences of the 12 protein-coding genes by BI showed that the S. lupi (Thelaziidae) is closely related to the families Setariidae and Onchocercidae.
Conclusions
The present study determined the complete mt genome sequence of S. lupi. These new mt genome dataset should provide novel mtDNA markers for studying the molecular epidemiology and population genetics of this parasite, and should have implications for the molecular diagnosis, prevention and control of spirocercosis in dogs and other canids.
Similar content being viewed by others


Background
The nematode Spirocerca lupi (Rudolphi, 1809) (at the adult stage) parasitizes the oesophagus and aorta of canids, especially in dogs. S. lupi is responsible for canine spirocercosis with a worldwide distribution but is usually found in tropical and subtropical countries [1, 2]. Canine spirocercosis is usually associated with several clinical signs, such as regurgitation, vomiting and dyspnoea [3, 4]. This disease is also fatal when it causes malignant neoplasms or aortic aneurysms [2, 4, 5]. Fortunately, spirocercosis can be treated efficiently using anthelminthics, such as doramectin [6].
Canine spirocercosis caused by S. lupi is often neglected and underestimated by some veterinary scientists and practitioners. However, S. lupi is most prevalent in dogs in rural areas, such as in Bangladesh (40%) [7], Greece (10%) [8], Grenada (8.8% in owned dogs and 14.2% in stray dogs) [1], India (23.5%) [9], Iran (19%) [10], South Africa (13%) [11] and Kenya (85% in stray dogs and 38% in owned dogs) [12]. S. lupi has been also reported in dogs in China, with a very high prevalence (78.6%) [13]. Although canine spirocercosis is an emerging disease, little is known about the molecular biology and genetics of S. lupi[14]. A previous study has found utility of mitochondrial (mt) cytochrome c oxidase subunit 1 (cox 1) for population genetic and phylogenetic studies of S. lupi[14], yet, there is still a paucity of information on S. lupi mt genomics.
mt genome sequences provide useful genetic markers not only for genetic and epidemiological investigations and molecular identification of parasites, but also for phylogenetic and population studies [15–18] due to its maternal inheritance, rapid evolutionary rate, and lack of recombination [19, 20]. To date, although mt genome sequences have been sequenced for 12 species within the order Spirurida, only one mt genome (for Thelazia callipaeda) is available within the family Thelaziidae [21]. Therefore, the objectives of the present study were to determine the complete mt genome sequence of S. lupi and to assess the phylogenetic position of this nematode in relation to other spirurid nematodes for which complete mt sequence datasets are available.
Methods
Ethics statement
This study was approved by the Animal Ethics Committee of Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences (Approval No. LVRIAEC2010-007). The farmed dog from which S. lupi adults were collected, was handled in accordance with good animal practices required by the Animal Ethics Procedures and Guidelines of the People’s Republic of China.
Parasites and DNA extraction
Adult nematodes representing S. lupi were obtained at post mortem from the oesophagus of an infected farmed dog in Zhanjiang, Guangdong province, China. These specimens were washed in physiological saline, identified morphologically to species according to existing descriptions [22], fixed in 70% (v/v) ethanol and stored at −20°C until use.
Total genomic DNA was isolated from one S. lupi worm using sodium dodecyl sulphate/proteinase K treatment, followed by spin-column purification (TIANamp Genomic DNA kit). In order to independently verify the identity of this specimen, the mt cox 1 gene was amplified by the polymerase chain reaction (PCR) and sequenced according to an established method [14]. The cox 1 sequence of this S. lupi sample had 96.5% similarity with that of S. lupi in dogs in South Africa (GenBank accession no. HQ674759).
Amplification and sequencing of partial cox 1, rrn S, cox 2 and nad 2 genes
Initially, a fragment of cox 1 (346 bp) was amplified by conserved primers JB3/JB4.5 [23], and rrn S (213 bp), cox 2 (300 bp) and nad 2 (1200 bp) were amplified by PCR with primers designed (Table 1) based on sequences well conserved in many related taxa. PCR reactions (25 mL) were performed in 10 mM Tris–HCl (pH 8.4), 50 mM KCl, 4 mM MgCl2, 200 mM each of dNTP, 50 pmol of each primer and 2 U Taq polymerase (Takara) in a thermocycler (Biometra) under the following conditions: after an initial denaturation at 94°C for 5 min, then 94°C for 30 s (denaturation), 55°C (for cox 1) or 48°C (for cox 2) or 50°C (for nad 2 and rrn S) for 30 s (annealing), 72°C for 1 min (extension) for 36 cycles, followed by 72°C for 10 min (final extension). Two microliters (5–10 ng) of genomic DNA was added to each PCR reaction. Each amplicon (5 μL) was examined by agarose gel electrophoresis to validate amplification efficiency. Then, these amplicons were sent to Sangon Company (Shanghai, China) for sequencing from both directions by using primers used in PCR amplifications.
Long-PCR amplification and sequencing
After we had obtained partial cox 1, rrn S, cox 2 and nad 2 sequences for the S. lupi, we then designed four primers (Table 1) in the conserved regions to amplify the entire mt genome of S. lupi from this representative sample in four overlapping long fragments between cox 1 and rrn S (approximately 4.5 kb), between rrn S and cox 2 (approximately 2.5 kb), between cox 2 and nad 2 (approximately 4 kb), and between nad 2 and cox 1 (approximately 3 kb). Long-PCR reactions (25 μl) were performed in 2 mM MgCl2, 0.2 mM each of dNTPs, 2.5 μl 10× LA Taq buffer, 2.5 μM of each primer, 1.25 U LA Taq polymerase (Takara), and 2 μl of DNA sample in a thermocycler (Biometra) under the following conditions: 92°C for 2 min (initial denaturation), then 92°C for 10 s (denaturation), 60°C (for 4.5 kb) or 44°C (for 2.5 kb) or 52°C (for 4 kb) or 48°C (for 3 kb fragment) for 30 s (annealing), and 60°C for 10 min (extension) for 10 cycles, followed by 92°C for 10 s, 60°C (for 4.5 kb) or 44°C (for 2.5 kb) or 52°C (for 4 kb) or 48°C (for 3 kb fragment) for 30 s (annealing), and 60°C for 10 min for 20 cycles, with a cycle elongation of 10 s for each cycle and a final extension at 60°C for 10 min. Each PCR reaction yielded a single band detected in a 0.8% (w/v) agarose gel (not shown). PCR products were sent to Sangon Company (Shanghai, China) for sequencing using a primer-walking strategy.
Sequence analyses
Sequences were assembled manually using the commercial software ContigExpress program of the Vector NTI software package version 6.0 (Invitrogen, Carlsbad, CA), and aligned against the complete mt genome sequences of other spirurid nematodes available using the computer program Clustal X 1.83 [24] and MegAlign procedure within the DNAStar 5.0 [25] to infer gene boundaries. The open-reading frames were analysed with Open Reading Frame Finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html) using the invertebrate mitochondrial code, and subsequently compared with that of T. callipaeda[21]. Protein-coding gene sequences were translated into amino acid sequences using the invertebrate mitochondrial genetic code; amino acid sequences were aligned using default settings with MEGA 5.0 [26]. Translation initiation and termination codons were identified by comparison with those of the spirurid nematodes reported previously [21, 27]. For analyzing ribosomal RNA genes, putative secondary structures of 22 tRNA genes were identified using tRNAscan-SE [28], of the 22 tRNA genes, 14 were identified using tRNAscan-SE, the other 8 tRNA genes were found by eye inspection, and rRNA genes were identified by comparison with that of spirurid nematodes [21, 27].
Phylogenetic analysis
The amino acid sequences conceptually translated from individual genes of the mt genome of S. lupi were concatenated. Selected for comparison were concatenated amino acid sequences predicted from published mt genomes of key nematodes representing the order Spirurida, including the superfamilies Thelazoidea (T. callipaeda[21]), Filarioidea (Acanthocheilonema viteae[29], Brugia malayi[30], Chandlerella quiscali[29], Dirofilaria immitis[31], Loa loa[29], Onchocerca flexuosa[29], O. volvulus[32], S. digitata[27] and Wuchereria bancrofti[18]), Dracunculoidea (Dracunculus medinensis[33]) and Physalopteroidea (Heliconema longissimum[33]) (GenBank accession numbers JX069968, NC_016197, NC_004298, NC_014486, NC_005305, NC_016199, NC_016172, AF015193, NC_014282, JN367461, NC_016019 and NC_016127, respectively), using Ascaris suum[34] (GenBank accession number HQ704901) as the outgroup. The amino acid sequences were aligned using Clustal X 1.83 [24] using default settings, ambiguously aligned regions were excluded using Gblocks online server (http://molevol.cmima.csic.es/castresana/Gblocks_server.html) using the options for a less stringent selection, and then subjected to phylogenetic analysis using Bayesian inference (BI) as described previously [35, 36]. Phylograms were drawn using the Tree View program v.1.65 [37].
Results and discussion
General features of the S. lupi mt genome
The complete mtDNA sequence of S. lupi was 13,780 bp in size (Figure 1), and has been deposited in the GenBank under the accession number KC305876. The mt genome of S. lupi contains 12 protein-coding genes (cox 1-3, nad 1-6, nad 4L, atp 6 and cyt b), 22 transfer RNA genes, two ribosomal RNA genes (rrn L and rrn S) and a non-coding (control or AT-rich) region, but lacks an atp 8 gene (Table 2). All genes are transcribed in the same direction. The gene order is identical to those of T. callipaeda and S. digitata[21, 27], but distinct from those of H. longissimum (rearrangement markedly) and Dracunculus medinensis (tRNA-Met and tRNA-Val change) [33]. The nucleotide compositions of S. lupi mt genome is biased toward A and T, with T being the most favored nucleotide and C being the least favored, in accordance with mt genomes of other spirurid nematodes [27, 31]. The content of A + T is 73.73% for S. lupi, similar to that of mt genomes of other spirurid nematodes sequenced to date, such as that of T. callipaeda (74.57%) [21] and W. bancrofti (74.59%) [18] (Table 3). Furthermore, the S. lupi mt genes overlap a total of 98 bp in 16 locations ranging from 1 to 32 bp (Table 2). The longest is a 32 bp overlap between nad 1 and tRNA-Phe. The mt genome of S. lupi has 150 bp of intergenic regions at 16 locations ranging in size from 1 bp to 59 bp, the longest intergenic region is a 59 bp between tRNA-Pro and tRNA-Asp (Table 2). The mt genome of T. callipaeda has 14 intergenic regions, which range from 1 to 62 bp in length. The longest region is 62 bp between tRNA-Pro and tRNA-Asp [21].

Arrangement of the mitochondrial genome of Spirocerca lupi. Gene scaling is only approximate. All genes are coded by the same DNA strand and are transcribed clockwise. All genes have standard nomenclature except for the 22 tRNA genes, which are designated by the one-letter code for the corresponding amino acid, with numerals differentiating each of the two leucine- and serine-specifying tRNAs (L1 and L2 for codon families CUN and UUR, respectively; S1 and S2 for codon families UCN and, AGN respectively). “AT” refers to the non-coding region.
Protein-coding genes
The S. lupi mt genome encodes 12 protein-coding genes, which are identical to those of T. callipaeda and S. digitata[21, 27]. For S. lupi, the sizes of the protein-coding genes were in the order: cox 1 > nad 5 > nad 4 > cyt b > nad 1 > nad 2 > cox 3 > cox 2 > atp 6 > nad 6 > nad 3 > nad 4L (Table 2). The predicted translation initiation and termination codons for the 12 protein-coding genes of S. lupi mt genome were compared with that of T. callipaeda and S. digitata[21, 27]. The most common initiation codon for S. lupi is TTG (5 of 12 protein genes), followed by ATG (4 of 12 protein genes), ATT (2 of 12 protein genes) and ATA (1 of 12 protein genes) (Table 2). In this mt genome, all protein genes were predicted to have a TAA or TAG as termination codon (Table 2). Although incomplete termination codons (T or TA) are present in some other nematodes, including Anisakis simplex (s. l.) [38], A. suum[39], Caenorhabditis elegans[39], S. digitata[27], Toxocara spp. [40] and Trichinella spiralis[41], they were not identified in the S. lupi mt genome.
Excluding the termination codons, a total of 3,458 amino acids of protein-coding genes are encoded by the S. lupi mt genome. Table 4 shows the codon usage. Condons composed of A and T are predominantly used, which seems to reflect the high A + T content of the mt genome of S. lupi. A strong preference for A + T rich codons usage is found in mtDNA of S. lupi. For example, the most frequently used amino acid was Phe (TTT: 17.03%), followed by Leu (TTG: 6.77%), Tyr (ATA: 6.16%) and IIe (ATT: 6.10%). This result is consistent with a recent study [21].
Transfer RNA genes and ribosomal RNA genes
The sizes of 22 tRNA genes identified in the S. lupi mt genome ranged from 50 to 62 bp in size. Secondary structures predicted for the 22 tRNA genes of S. lupi (not shown) are similar to that of S. digitata[27]. The rrn L and rrn S genes of S. lupi were identified by comparison with the mt genomes of T. callipaeda and S. digitata. The rrn L is located between tRNA-His and nad 3, and rrn S is located between nad 4L and tRNA-Tyr. The lengths of the rrn L and rrn S genes were 988 bp and 686 bp for S. lupi, respectively (Table 2). The A + T contents of the rrn L and rrn S genes for S. lupi are 79.05% and 76.09%, respectively.
Non-coding regions
The majority of nematode mtDNA sequences contain usually two non-coding regions of significant size difference, the long non-coding region and the short non-coding region, including A. lumbricoides and A. suum[34], Contracaecum rudolphii B [42], Oesophagostomum spp. [43], Toxocara spp. [40] and Trichuris spp. [44, 45]. However, there is only one non-coding region (AT-rich region) in the mt genome of S. lupi, which is located between cox 3 and tRNA-Ala (Figure 1 and Table 2), with 88.50% A + T content (Table 3). This region of the mt genome of S. lupi was considered as a non-coding region (or AT-rich region) due to its location and AT rich feature based on comparison with those of spirurid nematodes reported previously [21, 27]. Moreover, in the AT-rich region of S. lupi consecutive sequences [A]13 and [T]12 were found, but there are no AT dinucleotide repeat sequences similar to that of A. simplex s.l. and S. digitata in the this region [27, 38].
Phylogenetic analyses
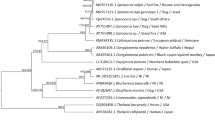
The phylogenetic relationships of 12 spirurid species based on concatenated amino acid sequence datasets, plus the mtDNA sequence of S. lupi obtained in the present study, using BI is shown in Figure 2. The results revealed that S. lupi (Thelaziidae) was a sister taxon to a clade containing S. digitata (Setariidae) and other members of the Onchocercidae, including B. malayi and D. immitis (posterior probability = 1.00), consistent with results of previous studies [14, 21, 46].

Relationship of Spirocerca lupi with other selected spirurid nematodes based on mitochondrial sequence data. The concatenated amino acid sequences of 12 protein-coding genes were subjected to analysis by Bayesian inference (BI) using Ascaris suum as the outgroup. Posterior probability (pp) values are indicated.
Many studies have demonstrated that mtDNA sequences are valuable genetic markers for phylogenetic studies of members within the Nematoda. A recent study analyzed mt sequence variations in human- and pig-derived Trichuris and demonstrated that they represent separate species [44]. In addition, a previous study sequenced and compared the mt genomes of A. lumbricoides and A. suum from humans and pigs and indicted that A. lumbricoides and A. suum may represent the same species [34]. In the present study, the characterization of the mt genome of S. lupi can promote to reassess the systematic relationships within the order Spirurida using mt genomic datasets. For many years, there have been considerable debates about the phylogenetic position of members of spirurid nematodes [47, 48]. Given this utility of mt genomic datasets, thus, further work should sequence more mt genomes of spirurid nematodes and re-construct the phylogenetic relationships of spirurid nematodes using expanded mt datasets.
Conclusions
The present study determined the complete mt genome sequence of S. lupi, and ascertained its phylogenetic position within the Spirurida. These new mtDNA data will provide useful novel markers for studying the molecular epidemiology and population genetics of S. lupi, and have implications for the diagnosis, prevention and control of spirocercosis in canid animals.
References
Chikweto A, Bhaiyat MI, Tiwari KP, de Allie C, Sharma RN: Spirocercosis in owned and stray dogs in Grenada. Vet Parasitol. 2012, 190: 613-616. 10.1016/j.vetpar.2012.07.006.
van der Merwe LL, Kirberger RM, Clift S, Williams M, Keller N, Naidoo V: Spirocerca lupi infection in the dog: a review. Vet J. 2008, 176: 294-309. 10.1016/j.tvjl.2007.02.032.
Mazaki-Tovi M, Baneth G, Aroch I, Harrus S, Kass PH, Ben Ari T, Zur G, Aizenberg I, Bark H, Lavy E: Canine spirocercosis: clinical, diagnostic, pathologic and epidemiologic characteristics. Vet Parasitol. 2002, 107: 235-250. 10.1016/S0304-4017(02)00118-8.
Ranen E, Lavy E, Aizenberg I, Perl S, Harrus S: Spirocercosis associated esophageal sarcomas in dogs. A retrospective study of 17 cases (1997–2003). Vet Parasitol. 2004, 119: 209-221. 10.1016/j.vetpar.2003.10.023.
Rinas MA, Nesnek R, Kinsella JM, DeMatteo KE: Fatal aortic aneurysm and rupture in a neotropical bush dog (Speothos venaticus) caused by Spirocerca lupi. Vet Parasitol. 2009, 164: 347-349. 10.1016/j.vetpar.2009.05.006.
Lavy E, Aroch I, Bark H, Markovics A, Aizenberg I, Mazaki-Tovi M, Hagag A, Harrus S: Evaluation of doramectin for the treatment of experimental canine spirocercosis. Vet Parasitol. 2002, 109: 65-73. 10.1016/S0304-4017(02)00250-9.
Shubhagata D, Abdul A, Mohammad MH, Suchandan S, Muraduzzaman M: Spirocercosis in stray dogs of Chittagong Metropolitan area of Bangladesh: an epidemiological and pathological investigation. Vet World. 2011, 4: 485-491.
Mylonakis ME, Koutinas AF, Liapi MV, Saridomichelakis MN, Rallis TS: A comparison of the prevalence of Spirocerca lupi in three groups of dogs with different life and hunting styles. J Helminthol. 2001, 75: 359-361.
Ramachandran PV, Shakir SA, Ramakrishnan R: Spirocercosis in canines: a necropsy survey. Cheiron-Tamil Nadu J Vet Sci Anim Husbandry. 1984, 13: 132-135.
Oryan A, Sadjjadi SM, Mehrabani D, Kargar M: Spirocercosis and its complications in stray dogs in Shiraz, Southern Iran. Vet Med Czech. 2008, 53: 617-624.
Minnaar WN, Krececk RC, Fourie LJ: Helminthes of dogs from a peri-urban resource-limited community in free state province, South Africa. Vet Parasitol. 2002, 107: 343-349. 10.1016/S0304-4017(02)00155-3.
Brodey RS, Thompson RG, Sayer PD, Eugster B: Spirocerca lupi infection in dogs in Kenya. Vet Parasitol. 1977, 3: 49-59. 10.1016/0304-4017(77)90007-3.
Jiang XL, Dai ZJ: Investigation of parasites in dogs in Sichuan province. Sichuan J Zool. 1988, 7: 9-(in Chinese)
Traversa D, Costanzo F, Iorio R, Aroch I, Lavy E: Mitochondrial cytochrome c oxidase subunit 1 (cox1) gene sequence of spirocerca lupi (nematoda, spirurida): avenues for potential implications. Vet Parasitol. 2007, 146: 263-270. 10.1016/j.vetpar.2007.03.015.
Unnasch TR, Williams SA: The genomes of onchocerca volvulus. Int J Parasitol. 2000, 30: 543-552. 10.1016/S0020-7519(99)00184-8.
Choi KS, Koekemoer LL, Coetzee M: Population genetic structure of the major malaria vector anopheles funestus s.s. and allied species in southern Africa. Parasit Vectors. 2012, 5: 283-10.1186/1756-3305-5-283.
Gasser RB, Jabbar A, Mohandas N, Höglund J, Hall RS, Littlewood DT, Jex AR: Assessment of the genetic relationship between Dictyocaulus species from Bos taurus and Cervus elaphus using complete mitochondrial genomic datasets. Parasit Vectors. 2012, 5: 241-10.1186/1756-3305-5-241.
Ramesh A, Small ST, Kloos ZA, Kazura JW, Nutman TB, Serre D, Zimmerman PA: The complete mitochondrial genome sequence of the filarial nematode Wuchereria bancrofti from three geographic isolates provides evidence of complex demographic history. Mol Biochem Parasitol. 2012, 183: 32-41. 10.1016/j.molbiopara.2012.01.004.
Wolstenholme DR: Animal mitochondrial DNA, structure and evolution. Int Rev Cytol. 1992, 141: 173-216.
Boore JL: Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27: 1767-1780. 10.1093/nar/27.8.1767.
Liu GH, Gasser RB, Otranto D, Xu MJ, Shen JL, Mohandas N, Zhou DH, Wang Y, Zhu XQ: Mitochondrial genome of the eyeworm, thelazia callipaeda (nematoda: spirurida), as the first representative from the family thelaziidae. PLoS Negl Trop Dis. 2013, 7: e2029-10.1371/journal.pntd.0002029.
Skrjabin KI: Key to parasitic nematodes, Spirurata and Filariata. 1969, Moscow: Academy of Sciences USSR
Gasser RB, Zhu XQ, McManus DP: NADH dehydrogenase subunit 1 and cytochrome c oxidase subunit I sequences compared for members of the genus taenia (cestoda). Int J Parasitol. 1999, 29: 1965-1970. 10.1016/S0020-7519(99)00153-8.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG: The Clustal X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 24: 4876-4882.
Burland TG: DNASTAR’s Lasergene sequence analysis software. Meth Mol Biol. 2000, 132: 71-91.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S: MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011, 28: 2731-2739. 10.1093/molbev/msr121.
Yatawara L, Wickramasinghe S, Rajapakse RP, Agatsuma T: The complete mitochondrial genome of Setaria digitata (Nematoda: Filarioidea): Mitochondrial gene content, arrangement and composition compared with other nematodes. Mol Biochem Parasitol. 2010, 173: 32-38. 10.1016/j.molbiopara.2010.05.004.
Lowe TM, Eddy SR: TRNAscan-SE: a program for improved transfer RNA detection in genomic sequence. Nucleic Acids Res. 1997, 25: 955-964.
McNulty SN, Mullin AS, Vaughan JA, Tkach VV, Weil GJ, Fischer PU: Comparing the mitochondrial genomes of Wolbachia-dependent and independent filarial nematode species. BMC Genomics. 2012, 13: 145-10.1186/1471-2164-13-145.
Ghedin E, Wang S, Spiro D, Caler E, Zhao Q, Crabtree J, Allen JE, Delcher AL, Guiliano DB, Miranda-Saavedra D, Angiuoli SV, Creasy T, Amedeo P, Haas B, El-Sayed NM, Wortman JR, Feldblyum T, Tallon L, Schatz M, Shumway M, Koo H, Salzberg SL, Schobel S, Pertea M, Pop M, White O, Barton GJ, Carlow CK, Crawford MJ, Daub J: Draft genome of the filarial nematode parasite Brugia malayi. Science. 2007, 317: 1756-1760. 10.1126/science.1145406.
Hu M, Gasser RB, Abs El-Osta YG, Chilton NB: Structure and organization of the mitochondrial genome of the canine heartworm, dirofilaria immitis. Parasitology. 2003, 127: 37-51. 10.1017/S0031182003003275.
Keddie EM, Higazi T, Unnasch TR: The mitochondrial genome of onchocerca volvulus: sequence, structure and phylogenetic analysis. Mol Biochem Parasitol. 1998, 95: 111-127. 10.1016/S0166-6851(98)00102-9.
Park JK, Sultana T, Lee SH, Kang S, Kim HK, Min GS, Eom KS, Nadler SA: Monophyly of clade III nematodes is not supported by phylogenetic analysis of complete mitochondrial genome sequences. BMC Genomics. 2011, 12: 392-10.1186/1471-2164-12-392.
Liu GH, Wu CY, Song HQ, Wei SJ, Xu MJ, Lin RQ, Zhao GH, Huang SY, Zhu XQ: Comparative analyses of the complete mitochondrial genomes of ascaris lumbricoides and ascaris suum from humans and pigs. Gene. 2012, 492: 110-116. 10.1016/j.gene.2011.10.043.
Jex AR, Waeschenbach A, Hu M, van Wyk JA, Beveridge I, Littlewood DT, Gasser RB: The mitochondrial genomes of ancylostoma caninum and bunostomum phlebotomum-two hookworms of animal health and zoonotic importance. BMC Genomics. 2009, 10: 79-10.1186/1471-2164-10-79.
Liu GH, Wang SY, Huang WY, Zhao GH, Wei SJ, Song HQ, Xu MJ, Lin RQ, Zhou DH, Zhu XQ: The complete mitochondrial genome of galba pervia (gastropoda: mollusca), an intermediate host snail of fasciola spp. PLoS One. 2012, 7: e42172-10.1371/journal.pone.0042172.
Page RD: TREEVIEW: an application to display phylogenetic trees on personal computers. Comput Appl Biosci. 1996, 12: 357-358.
Kim KH, Eom KS, Park JK: The complete mitochondrial genome of anisakis simplex (ascaridida: nematoda) and phylogenetic implications. Int J Parasitol. 2006, 36: 319-328. 10.1016/j.ijpara.2005.10.004.
Okimoto R, Macfarlane JL, Clary DO, Wolstenholme DR: The mitochondrial genomes of two nematodes, caenorhabditis elegans and ascaris suum. Genetics. 1992, 130: 471-498.
Li MW, Lin RQ, Song HQ, Wu XY, Zhu XQ: The complete mitochondrial genomes for three toxocara species of human and animal health significance. BMC Genomics. 2008, 9: 224-10.1186/1471-2164-9-224.
Lavrov DV, Brown WM: Trichinella spiralis mtDNA: a nematode mitochondrial genome that encodes a putative ATP8 and normally structured tRNAS and has a gene arrangement relatable to those of coelomate metazoans. Genetics. 2001, 157: 621-637.
Lin RQ, Liu GH, Zhang Y, D’Amelio S, Zhou DH, Yuan ZG, Zou FC, Song HQ, Zhu XQ: Contracaecum rudolphii B: gene content, arrangement and composition of its complete mitochondrial genome compared with anisakis simplex s.l. Exp Parasitol. 2012, 130: 135-140. 10.1016/j.exppara.2011.11.003.
Lin RQ, Liu GH, Hu M, Song HQ, Wu XY, Li MW, Zhang Y, Zou FC, Zhu XQ: Oesophagostomum dentatum and oesophagostomum quadrispinulatum: characterization of the complete mitochondrial genome sequences of the two pig nodule worms. Exp Parasitol. 2012, 131: 1-7. 10.1016/j.exppara.2012.02.015.
Liu GH, Gasser RB, Su A, Nejsum P, Peng L, Lin RQ, Li MW, Xu MJ, Zhu XQ: Clear genetic distinctiveness between human- and pig-derived trichuris based on analyses of mitochondrial datasets. PLoS Negl Trop Dis. 2012, 6: e1539-10.1371/journal.pntd.0001539.
Liu GH, Wang Y, Xu MJ, Zhou DH, Ye YG, Li JY, Song HQ, Lin RQ, Zhu XQ: Characterization of the complete mitochondrial genomes of two whipworms trichuris ovis and trichuris discolor (nematoda: trichuridae). Infect Genet Evol. 2012, 12: 1635-1641. 10.1016/j.meegid.2012.08.005.
Iorio R, Slapeta J, Otranto D, Paoletti B, Giangaspero A, Traversa D: Phylogenetic relationships of habronema microstoma and habronema muscae (spirurida: habronematidae) within the order spirurida inferred using mitochondrial cytochrome c oxidase subunit 1 (cox1) gene analysis. Parasitol Res. 2009, 104: 979-984. 10.1007/s00436-008-1276-x.
De Ley P, Blaxter M: Systematic position and phylogeny. The Biology of Nematodes. Edited by: Lee DL. 2002, Taylor & Francis, London and New York, 1-30.
Nadler SA, Carreno RA, Mejía-Madrid H, Ullberg J, Pagan C, Houston R, Hugot JP: Molecular phylogeny of clade III nematodes reveals multiple origins of tissue parasitism. Parasitology. 2007, 134: 1421-1442. 10.1017/S0031182007002880.
Acknowledgements
This work was supported in part by the International Science & Technology Cooperation Program of China (Grant No. 2013DFA31840), the Science Fund for Creative Research Groups of Gansu Province (Grant No. 1210RJIA006), the China Postdoctoral Science Foundation (Grant No. 2012 M520353) and the Shanghai Postdoctoral Sustentation Fund (Grant No. 12R21416500).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
XQZ and XLY conceived and designed the study, and critically revised the manuscript. GHL, YW and HQS performed the experiments, analyzed the data and drafted the manuscript. MWL and LA helped in study design, study implementation and manuscript revision. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Liu, GH., Wang, Y., Song, HQ. et al. Characterization of the complete mitochondrial genome of Spirocerca lupi: sequence, gene organization and phylogenetic implications. Parasites Vectors 6, 45 (2013). https://doi.org/10.1186/1756-3305-6-45
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-3305-6-45